
Repositioning FDA Drugs as Potential Cruzain Inhibitors from Trypanosoma cruzi: Virtual Screening, In Vitro and In Vivo Studies

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
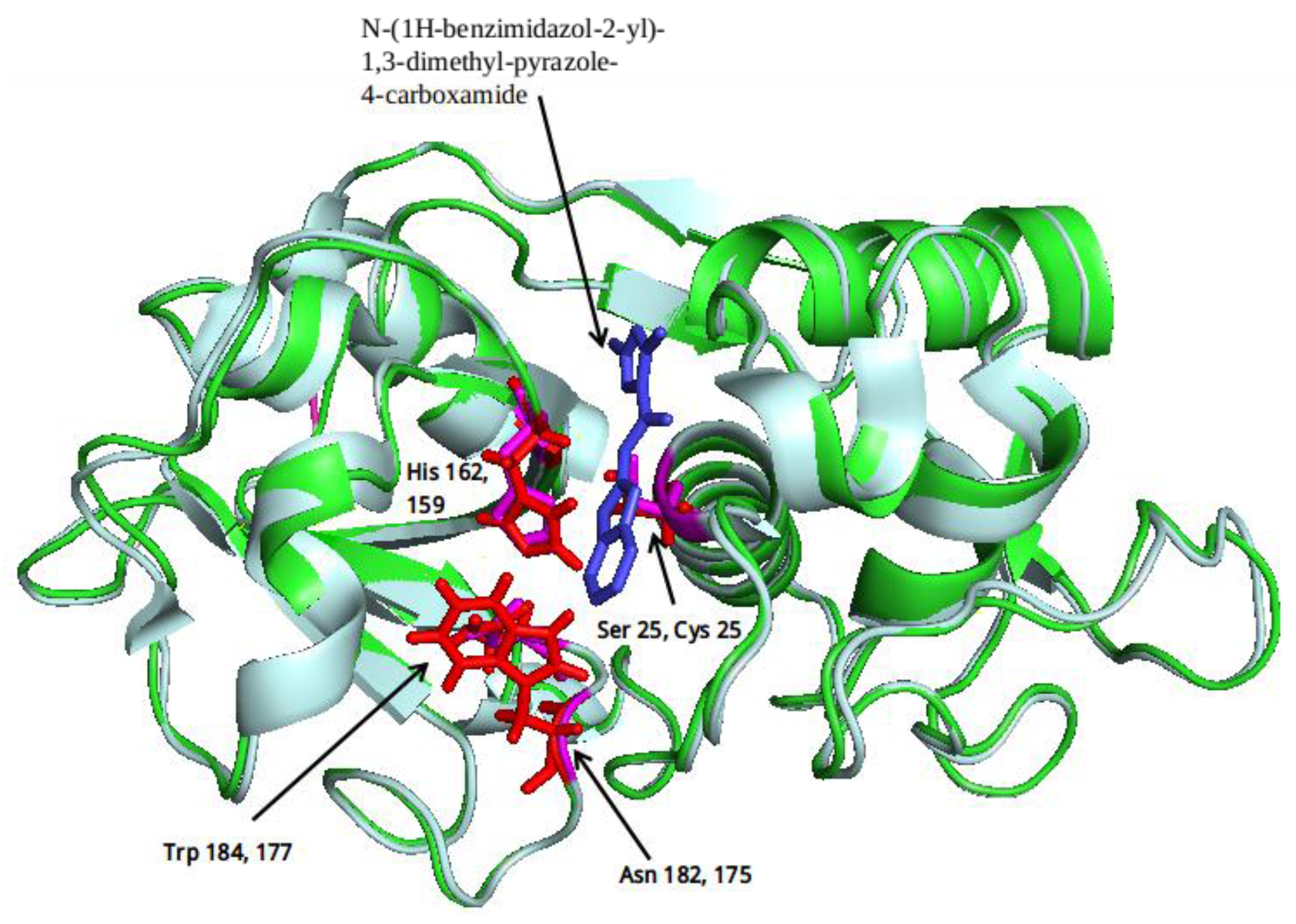
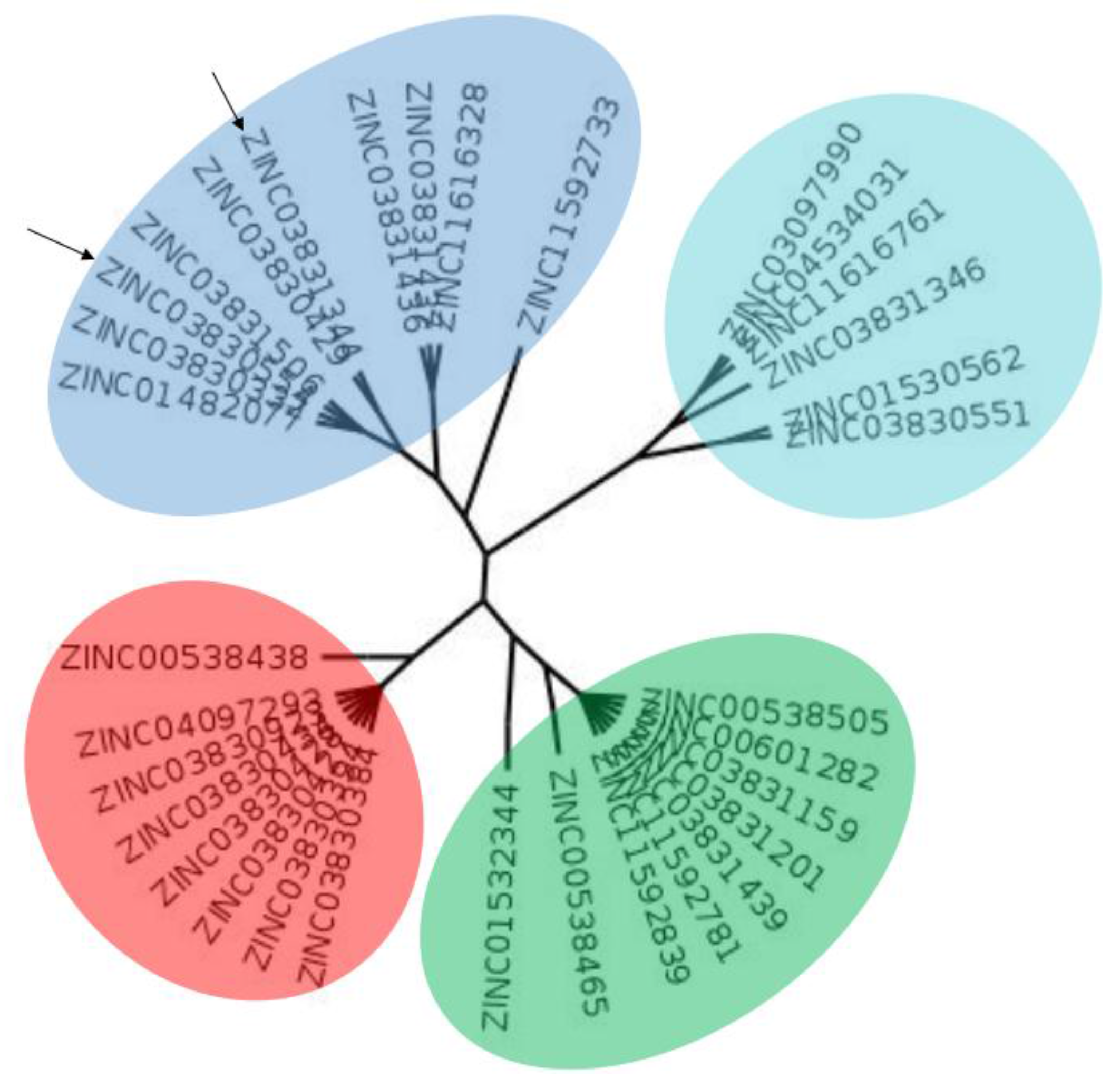
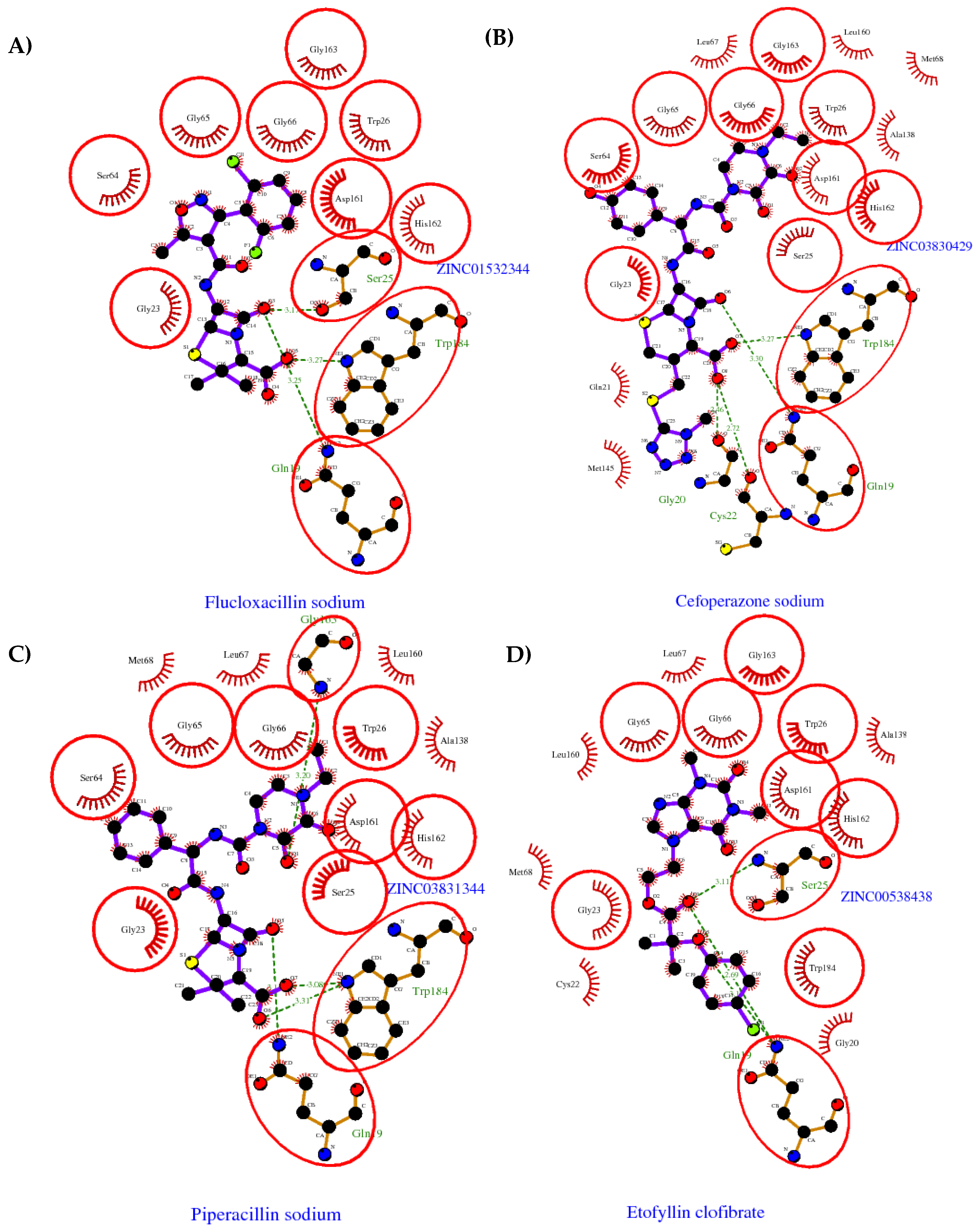
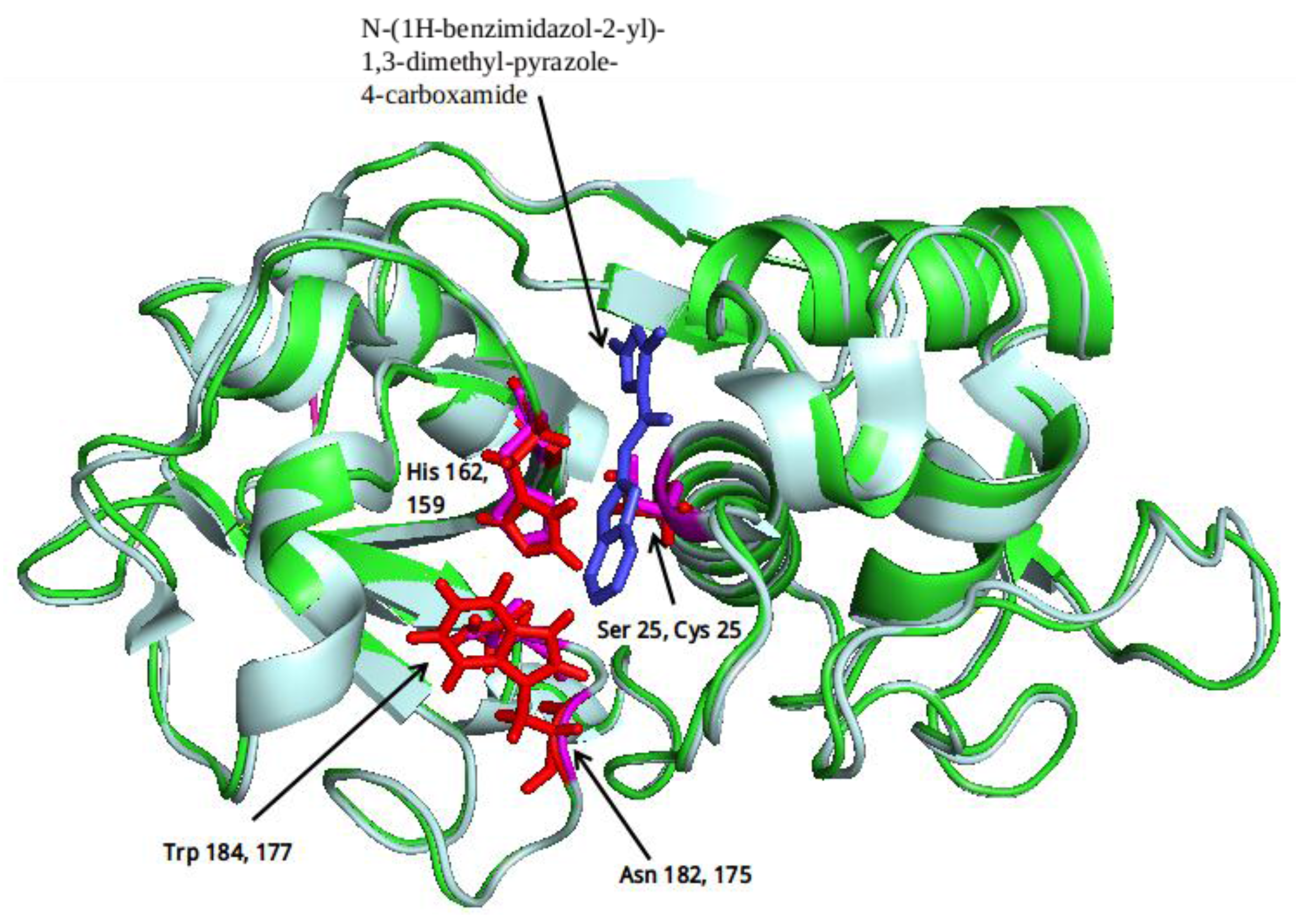
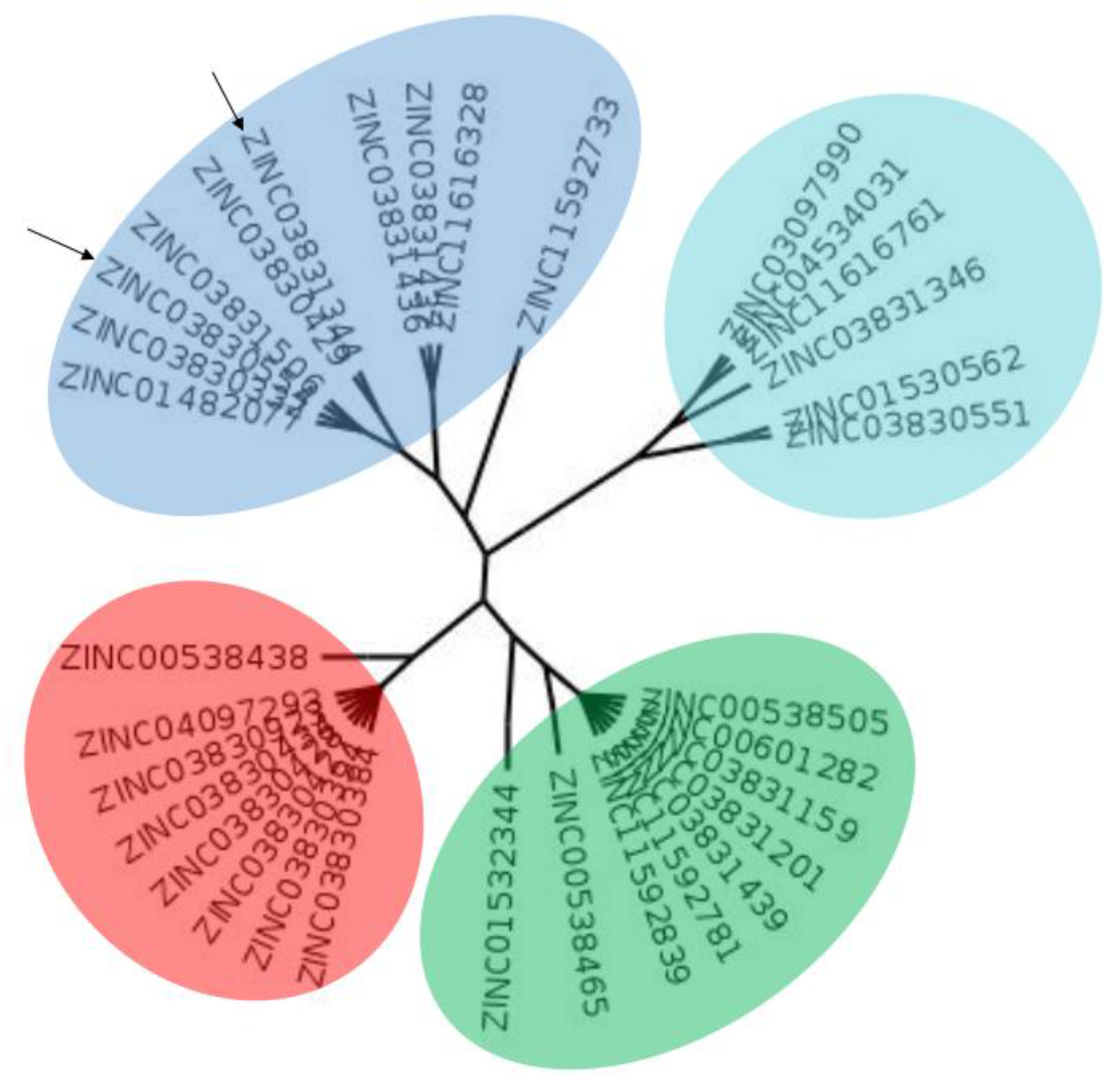
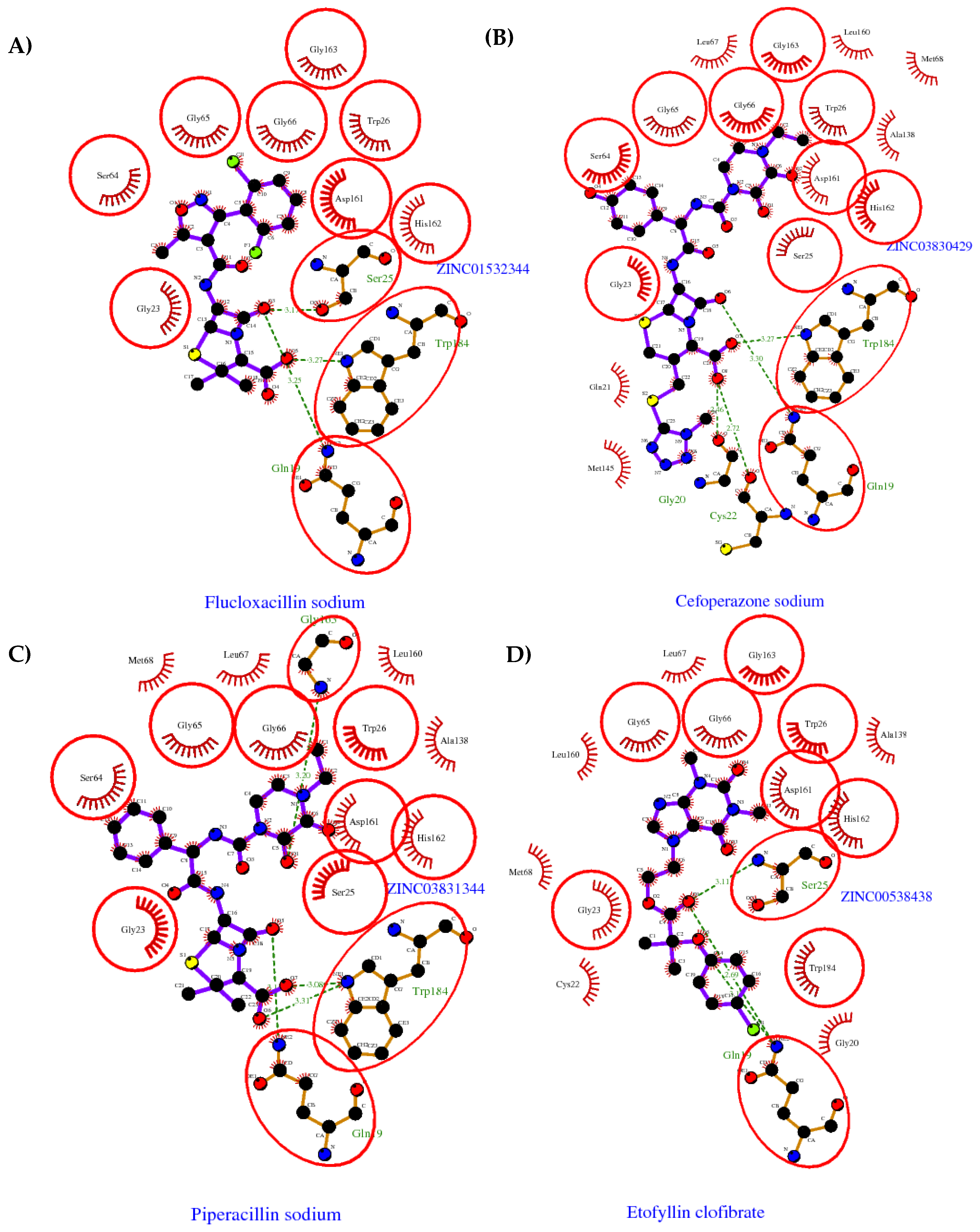
2.1. Computational Analysis
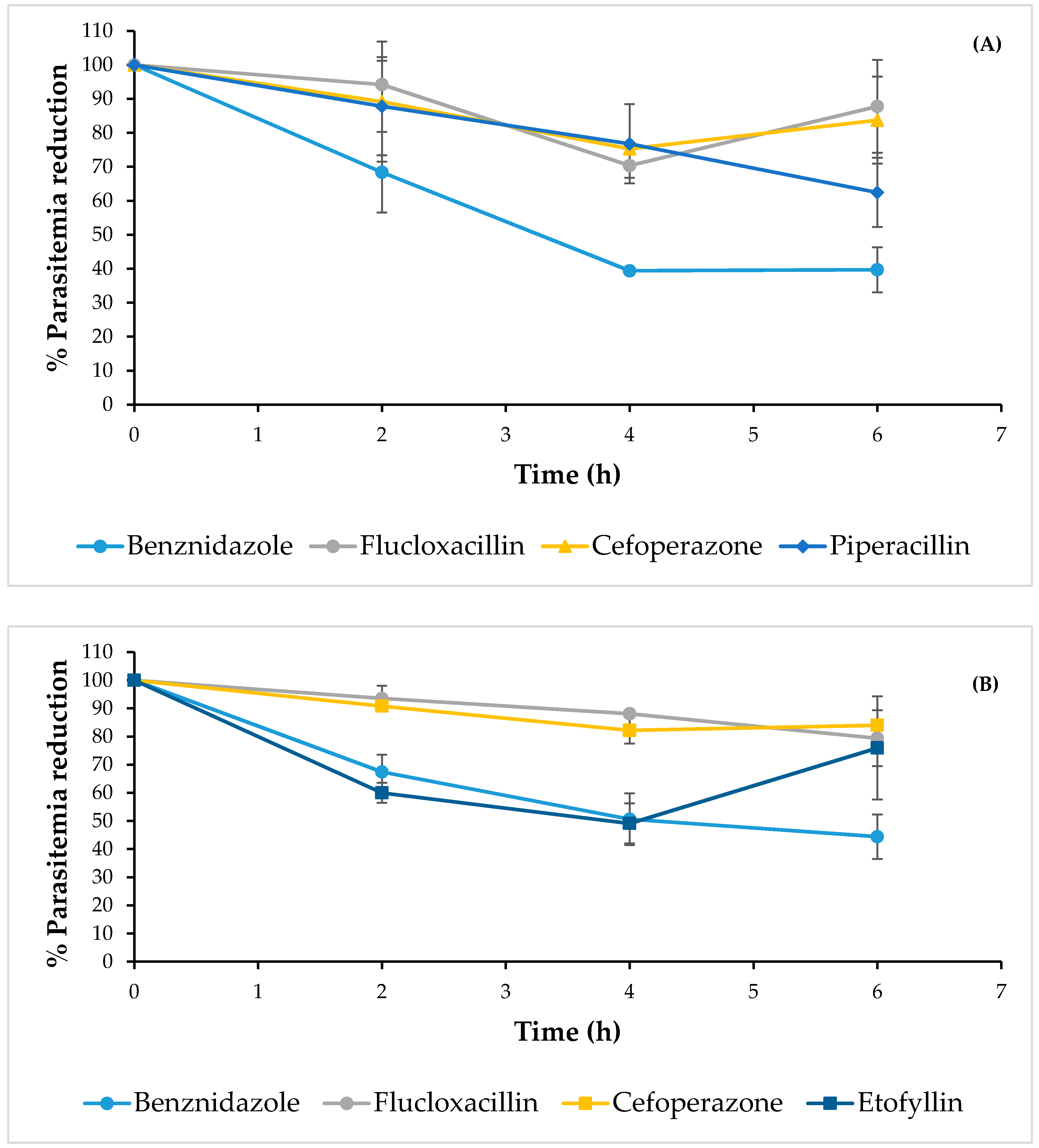
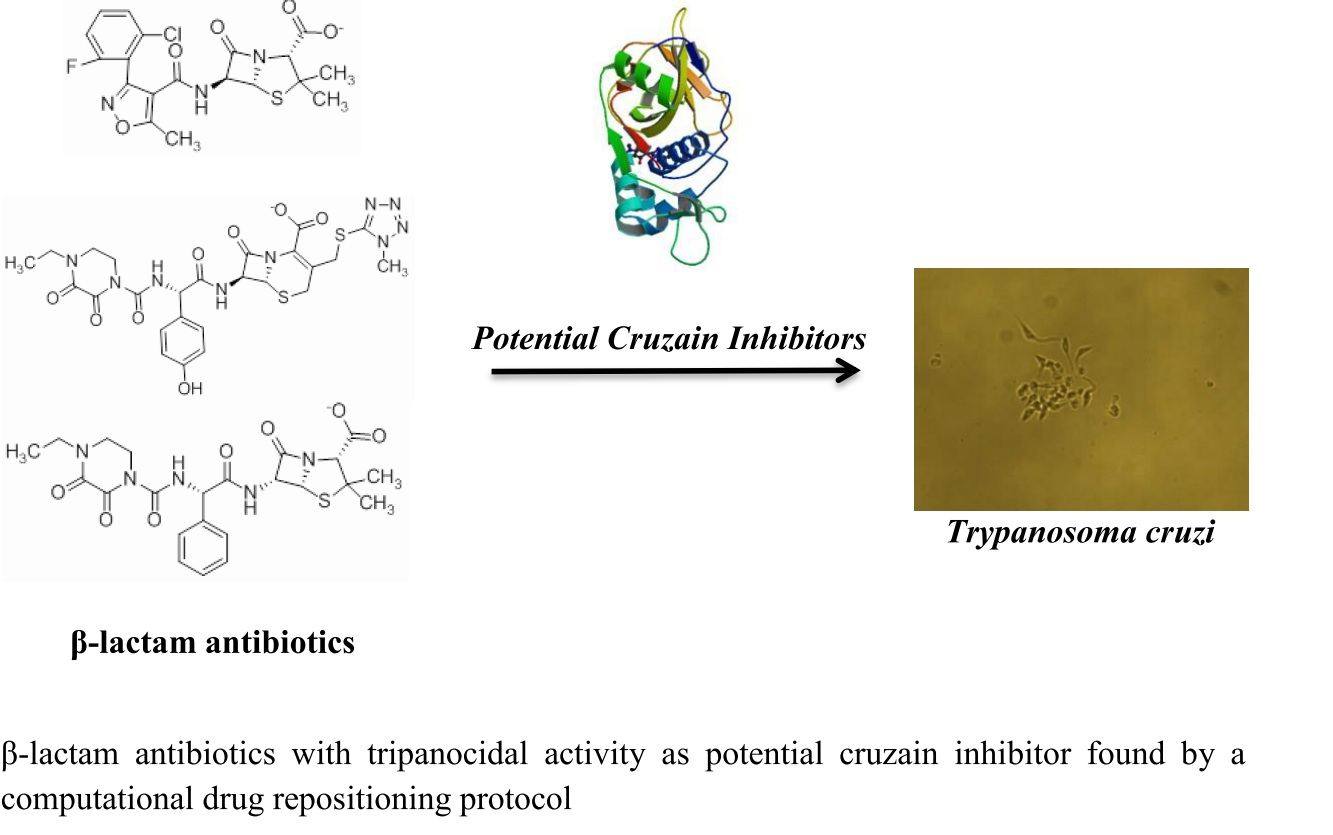
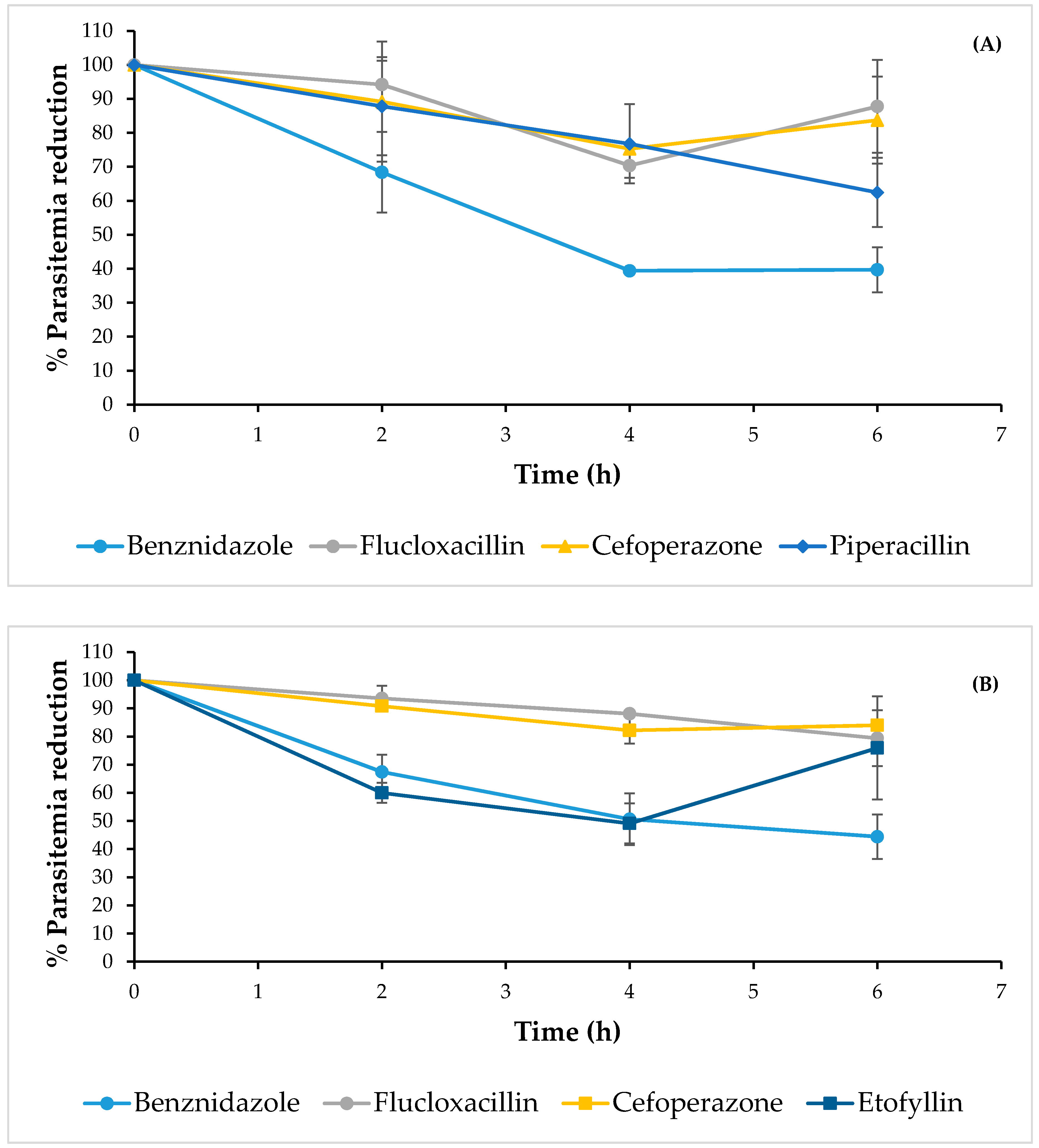
2.2. Anti-Trypanosoma cruzi Activity
3. Materials and Methods
3.1. Database Creation and Docking Protocol
3.2. Clustering and Ligand-Amino Acid Contact Analysis
3.3. In Vitro Evaluation
3.4. In Vivo Study of the Effects of the Compounds on T. cruzi
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Chagas Disease (American Trypanosomiasis); World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Teixeira, A.R.L.; Hecht, M.M.; Guimaro, M.C.; Sousa, A.O.; Nitz, N. Pathogenesis of Chagas’ Disease: Parasite Persistence and Autoimmunity. Clin. Microbiol. Rev. 2011, 24, 592–630. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M.; Engman, D.M. Chagas Heart Disease Pathogenesis: One Mechanism or Many? Curr. Mol. Med. 2008, 8, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Duschak, V.G.; Couto, A.S. Cruzipain, the major cysteine protease of Trypanosoma cruzi: A sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. A review. Curr. Med. Chem. 2009, 16, 3174–3202. [Google Scholar] [CrossRef] [PubMed]
- Bellera, C.L.; Balcazar, D.E.; Alberca, L.; Labriola, C.A.; Talevi, A.; Carrillo, C. Application of computer-aided drug repurposing in the search of new cruzipain inhibitors: Discovery of amiodarone and bromocriptine inhibitory effects. J. Chem. Inf. Model. 2013, 53, 2402–2408. [Google Scholar] [CrossRef] [PubMed]
- Bellera, C.L.; Balcazar, D.; Alberca, L.; Labriola, C.A.; Talevi, A.; Carrillo, C. Identification of Levothyroxine Antichagasic Activity through Computer-Aided Drug Repurposing. Sci. World J. 2014, 2014, 279618. [Google Scholar] [CrossRef] [PubMed]
- Bellera, C.L.; Balcazar, D.E.; Vanrell, M.C.; Casassa, A.F.; Palestro, P.H.; Gavernet, L.; Labriola, C.A.; Gálvez, J.; Bruno-Blanch, L.E.; Romano, P.S.; et al. Computer-guided drug repurposing: Identification of trypanocidal activity of clofazimine, benidipine and saquinavir. Eur. J. Med. Chem. 2015, 93, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.A.; Diaz de Toranzo, E.G. Toxic effects of nifurtimox and benznidazole, two drugs used against American trypanosomiasis (Chagas’ disease). Biomed. Environ. Sci. 1988, 1, 19–33. [Google Scholar] [PubMed]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Mäser, P.; Tadoori, L.P.; Ioset, J.-R.; Brun, R. Antiprotozoal Activity Profiling of Approved Drugs: A Starting Point toward Drug Repositioning. PLoS ONE 2015, 10, e0135556. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Harrison, R. Drug Repositioning: The Business Case and Current Strategies to Repurpose Shelved Candidates and Marketed Drugs. In Drug Repositioning; Barratt, M.J., Frail, D.E., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 7–32. [Google Scholar]
- Baber, J.C.; Shirley, W.A.; Gao, Y.; Feher, M. The use of consensus scoring in ligand-based virtual screening. J. Chem. Inf. Model. 2006, 46, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Brinen, L.S.; Hansell, E.; Cheng, J.; Roush, W.R.; McKerrow, J.H.; Fletterick, R.J. A target within the target: Probing cruzain’s P1’ site to define structural determinants for the Chagas’ disease protease. Struct. Lond. Engl. 2000, 8, 831–840. [Google Scholar] [CrossRef]
- Păduraru, I.; Nechifor, M.; Dănilă, G.; Jercă, L.; Filip, M.; Iacobovici, A.; Teslaru, E.; Filip, C.; Saramet, A. The mechanisms of action of etophylline-clofibrate in hyperlipidemia. Rev. Med. Chir. Soc. Med. Nat. Iaşi 1993, 97, 445–449. [Google Scholar] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.N.; Barry, A.L. Cefoperazone: A review of its antimicrobial spectrum, beta-lactamase stability, enzyme inhibition, and other in vitro characteristics. Rev. Infect. Dis. 1983, 5 (Suppl. 1), S108–S126. [Google Scholar] [CrossRef] [PubMed]
- Lara-Ramirez, E.E.; López-Cedillo, J.C.; Nogueda-Torres, B.; Kashif, M.; Garcia-Perez, C.; Bocanegra-Garcia, V.; Agusti, R.; Uhrig, M.L.; Rivera, G. An in vitro and in vivo evaluation of new potential trans-sialidase inhibitors of Trypanosoma cruzi predicted by a computational drug repositioning method. Eur. J. Med. Chem. 2017, 132, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- PDB. Available online: http://www.rcsb.org/pdb/explore.do?structureId=4w5b (accessed on 5 February 2016).
- Velec, H.F.G.; Gohlke, H.; Klebe, G. DrugScore (CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005, 48, 6296–6303. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fu, R.; Zhou, L.-H.; Chen, S.-P. Application of Consensus Scoring and Principal Component Analysis for Virtual Screening against β-Secretase (BACE-1). PLoS ONE 2012, 7, e38086. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, G.; Evrard-Todeschi, N.; Girault, J.-P.; Bertho, G. Automatic clustering of docking poses in virtual screening process using self-organizing map. Bioinformatics 2010, 26, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Chiguer, D.L.; Márquez-Navarro, A.; Nogueda-Torres, B.; de la Luz León-Ávila, G.; Pérez-Villanueva, J.; Hernández-Campos, A.; Castillo, R.; Ambrosio, J.R.; Nieto-Meneses, R.; Yépez-Mulia, L.; et al. In vitro and in vivo trypanocidal activity of some benzimidazole derivatives against two strains of Trypanosoma cruzi. Acta Trop. 2012, 122, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Brener, Z. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev. Inst. Med. Trop. São Paulo 1962, 4, 389–396. [Google Scholar] [PubMed]
- Filardi, L.S.; Brener, Z. A rapid method for testing in vivo the susceptibility of different strains of Trypanosoma cruzi to active chemotherapeutic agents. Mem. Inst. Oswaldo Cruz 1984, 79, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Romanha, A.J.; Castro, S.L.; Soeiro, M.N.; Lannes-Vieira, J.; Ribeiro, I.; Talvani, A.; Bourdin, B.; Blum, B.; Olivieri, B.; Zani, C.; et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem. Inst. Oswaldo Cruz 2010, 105, 233–238. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are commercially available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZINC ID | Z-Mean | Compound Structure | FDA Indication * |
|---|---|---|---|
| ZINC03830554 | −1.391 |  | NAI |
| ZINC03831439 | −0.864 | 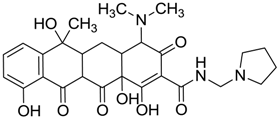 | Antibiotic (rolitetracycline) |
| ZINC03831201 | −0.846 | 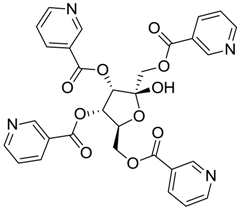 | NAI (analogue fructofuranose tetranicotinate) |
| ZINC03831346 | −0.786 | 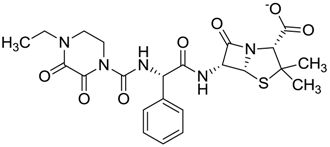 | Antibiotic (piperacillin sodium) |
| ZINC03831506 | −0.768 | 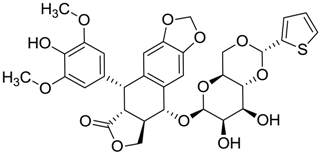 | Antineoplastic (analogue teniposide) |
| ZINC00538438 | −0.609 | 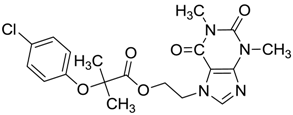 | Antilipemic (etofylline clofibrate) |
| ZINC03830384 | −0.596 |  | Antineoplastic (carubicin) |
| ZINC03830923 | −0.504 | 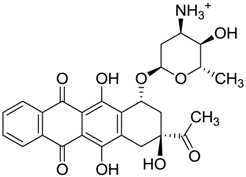 | Antineoplastic (idarubicin) |
| ZINC11592781 | −0.472 |  | Anabolic steroid (analogue methylpredinisoloe) |
| ZINC00538505 | −0.402 |  | Antypsychotic (trifluperidol) |
| ZINC03831344 | −0.352 |  | Antibiotic (piperacillin sodium) |
| ZINC11592839 | −0.346 |  | Anabolic steroid (analogue nandrolone phenilpropionate) |
| ZINC11592733 | −0.274 |  | Antibiotic (analogue ampicillin) |
| ZINC03830427 | −0.171 |  | Antibiotic (analogue cefonicid) |
| ZINC03830429 | −0.137 |  | Antibiotic (cefoperazone) |
| ZINC00601282 | −0.071 | 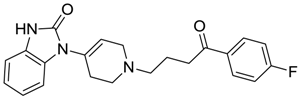 | NAI |
| ZINC01482077 | −0.039 |  | Antidiabetic (gliquidone) |
| ZINC03830428 | −0.033 |  | Antibiotic (cefonicid sodium) |
| ZINC03830332 | −0.013 | 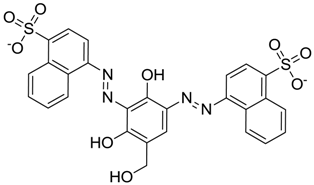 | Dye (analogue chocolate brown) |
| ZINC03830434 | 0.017 | 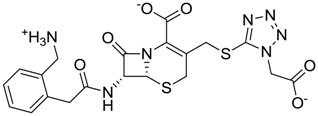 | Antibiotic (ceforanide) |
| ZINC03831159 | 0.154 | 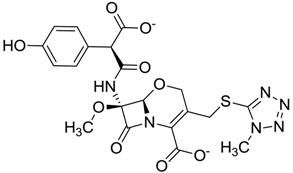 | Antibiotic (analogue moxalactam disodium) * |
| ZINC00538465 | 0.235 | 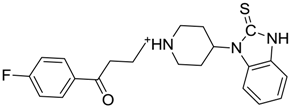 | NAI |
| ZINC11616761 | 0.276 | 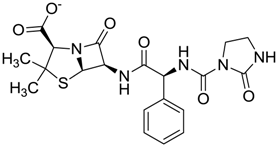 | Antibiotic (analogue ampicillin) |
| ZINC03831436 | 0.278 | 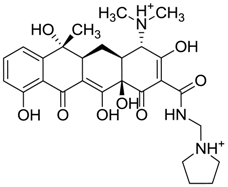 | Antibiotic (rolitetracycline) |
| ZINC01532344 | 0.287 |  | Antibiotic (flucloxacillin sodium) |
| ZINC03831437 | 0.328 | 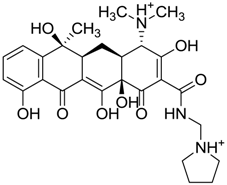 | Antibiotic (rolitetracycline) |
| ZINC11616328 | 0.436 |  | Antibiotic (rolitetracycline) |
| ZINC03830394 | 0.441 |  | Antibiotic (cefamandole) |
| ZINC01530562 | 0.455 | 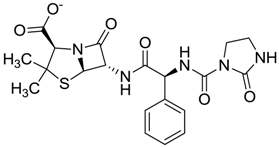 | Antibiotic (analogue mezlocilline) |
| ZINC04534031 | 0.466 | 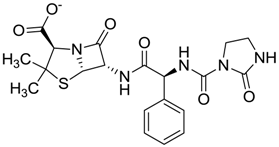 | Antibiotic (analogue mezlocilline) |
| ZINC03097990 | 0.544 | 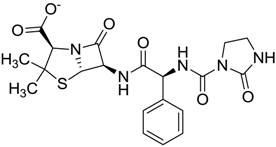 | Antibiotic (analogue mezlocilline) |
| ZINC04097293 | 0.610 | 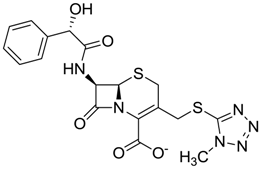 | Antibiotic (cefamandole) |
| ZINC03830551 | 1.080 |  | Antibiotic (analogue tetracycline) |
| Name | Clinical Use | % Lysis on INC-5 at 50 µg/mL | LC50 (µg/mL) on INC-5 | % Lysis on NINOA at 50 µg/mL | LC50 (µg/mL) on NINOA |
|---|---|---|---|---|---|
| Etofyllin clofibrate | Antilipemic | 21 | ND | 60 | 18.4 ± 0.9 |
| Flucloxacillin sodium | Antibiotic | 61 | 26.1 ± 1.4 | 81 | 23.2 ± 1.4 |
| Piperacillin sodium | Antibiotic | 65 | 15.8 ± 1.4 | 43 | ND |
| Cefoperazone sodium | Antibiotic | 71 | 23 ± 1.8 | 64 | 25.8 ± 0.7 |
| Benznidazole | Antichagasic | 56 | 40.6 ± 2.4 | 69 | 46.6 ± 1.9 |
| Nifurtimox | Antichagasic | 51 | 46.7 ± 5.2 | 63 | 33.1 ± 1.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palos, I.; Lara-Ramirez, E.E.; Lopez-Cedillo, J.C.; Garcia-Perez, C.; Kashif, M.; Bocanegra-Garcia, V.; Nogueda-Torres, B.; Rivera, G. Repositioning FDA Drugs as Potential Cruzain Inhibitors from Trypanosoma cruzi: Virtual Screening, In Vitro and In Vivo Studies. Molecules 2017, 22, 1015. https://doi.org/10.3390/molecules22061015
Palos I, Lara-Ramirez EE, Lopez-Cedillo JC, Garcia-Perez C, Kashif M, Bocanegra-Garcia V, Nogueda-Torres B, Rivera G. Repositioning FDA Drugs as Potential Cruzain Inhibitors from Trypanosoma cruzi: Virtual Screening, In Vitro and In Vivo Studies. Molecules. 2017; 22(6):1015. https://doi.org/10.3390/molecules22061015
Chicago/Turabian StylePalos, Isidro, Edgar E. Lara-Ramirez, Julio Cesar Lopez-Cedillo, Carlos Garcia-Perez, Muhammad Kashif, Virgilio Bocanegra-Garcia, Benjamin Nogueda-Torres, and Gildardo Rivera. 2017. "Repositioning FDA Drugs as Potential Cruzain Inhibitors from Trypanosoma cruzi: Virtual Screening, In Vitro and In Vivo Studies" Molecules 22, no. 6: 1015. https://doi.org/10.3390/molecules22061015