
Protein Stability and Unfolding Following Glycine Radical Formation



Abstract
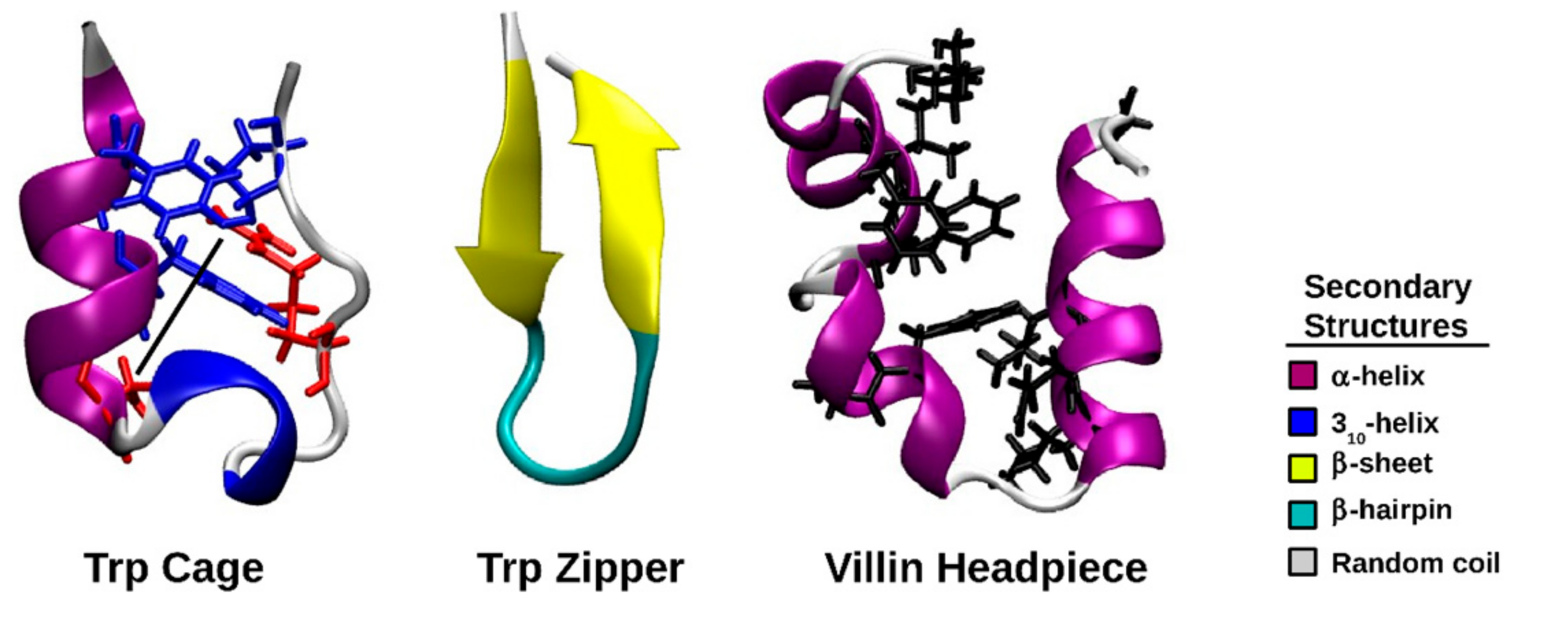
:1. Introduction
2. Results
2.1. Short-Term Analysis
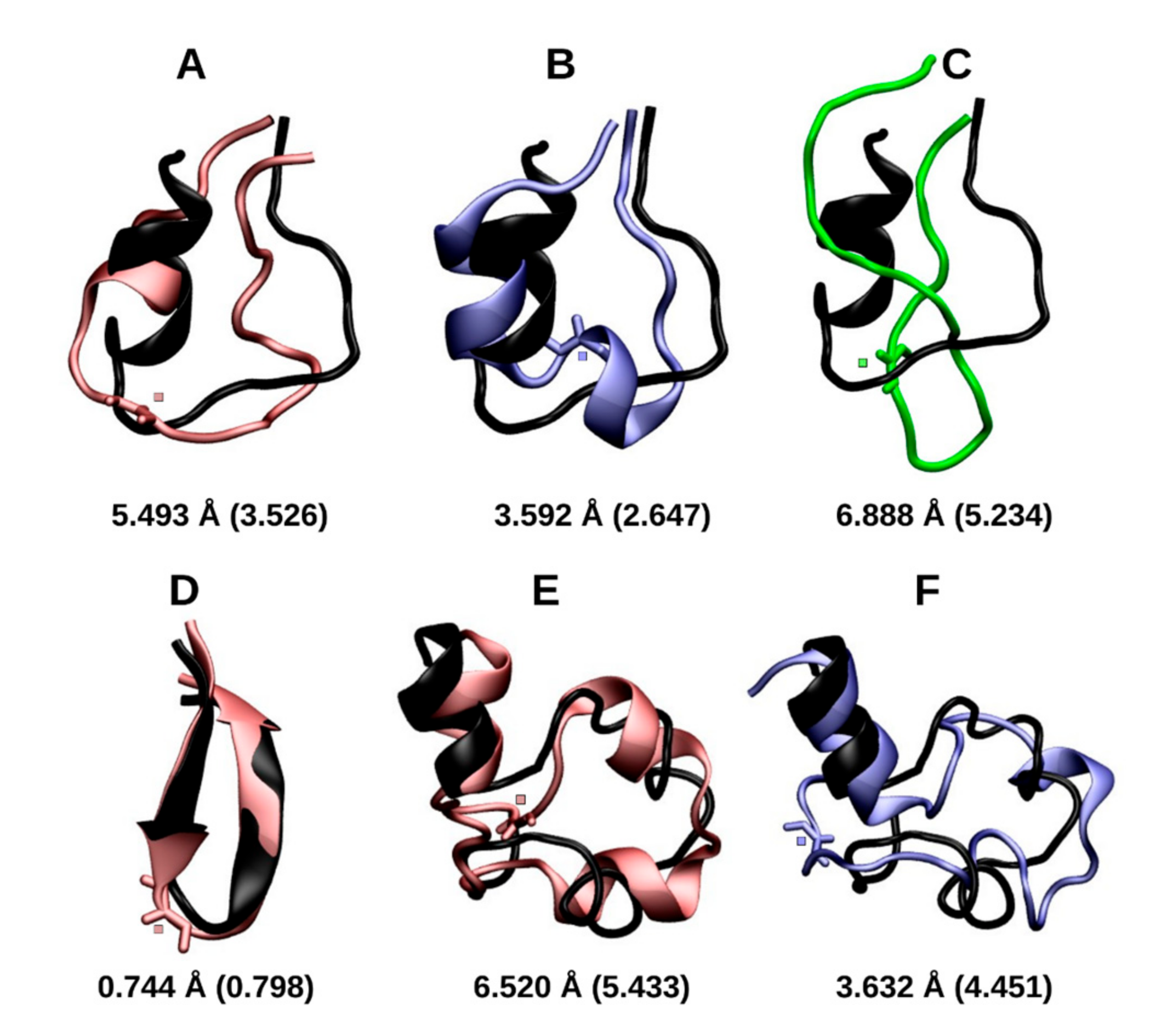
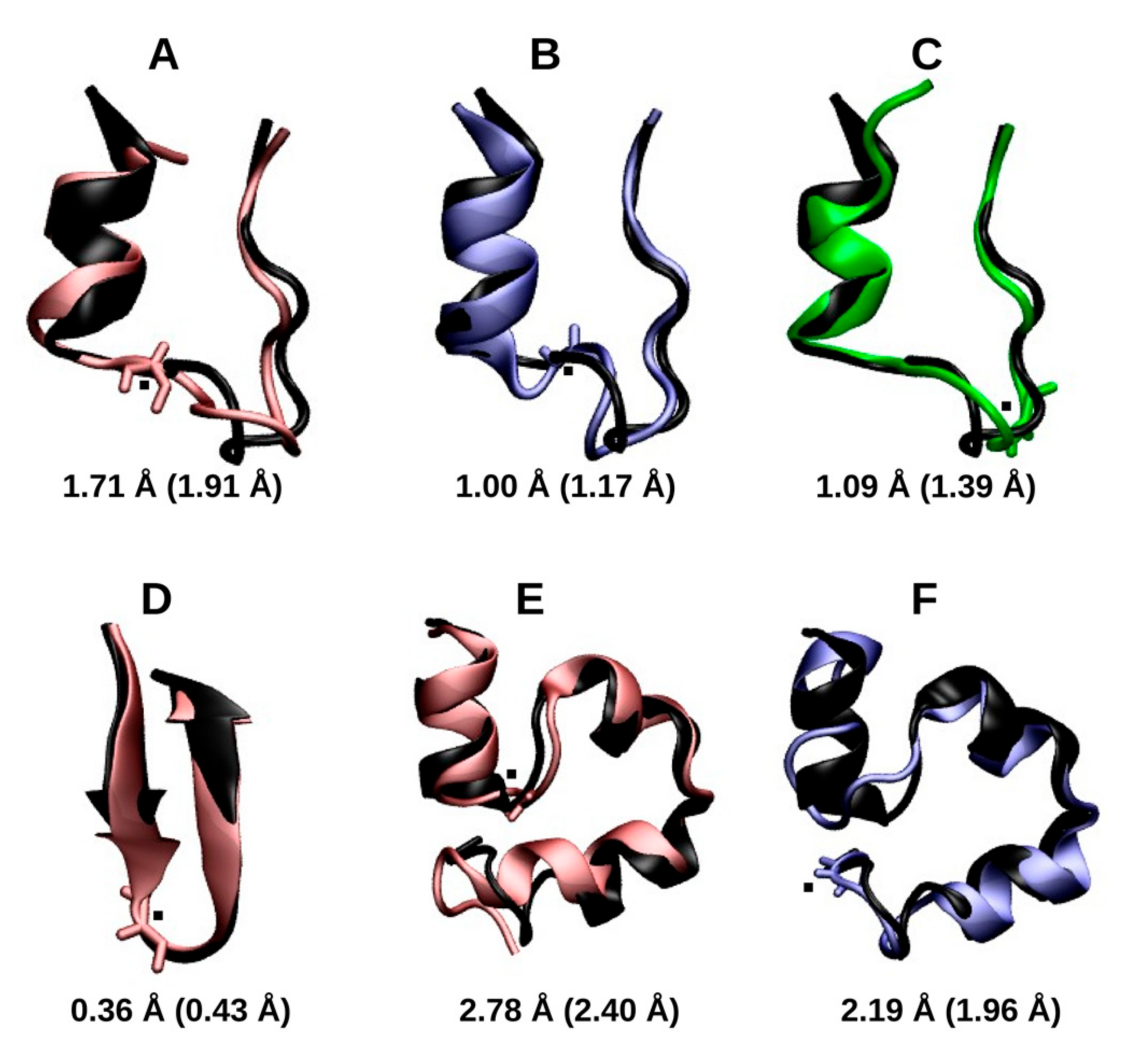
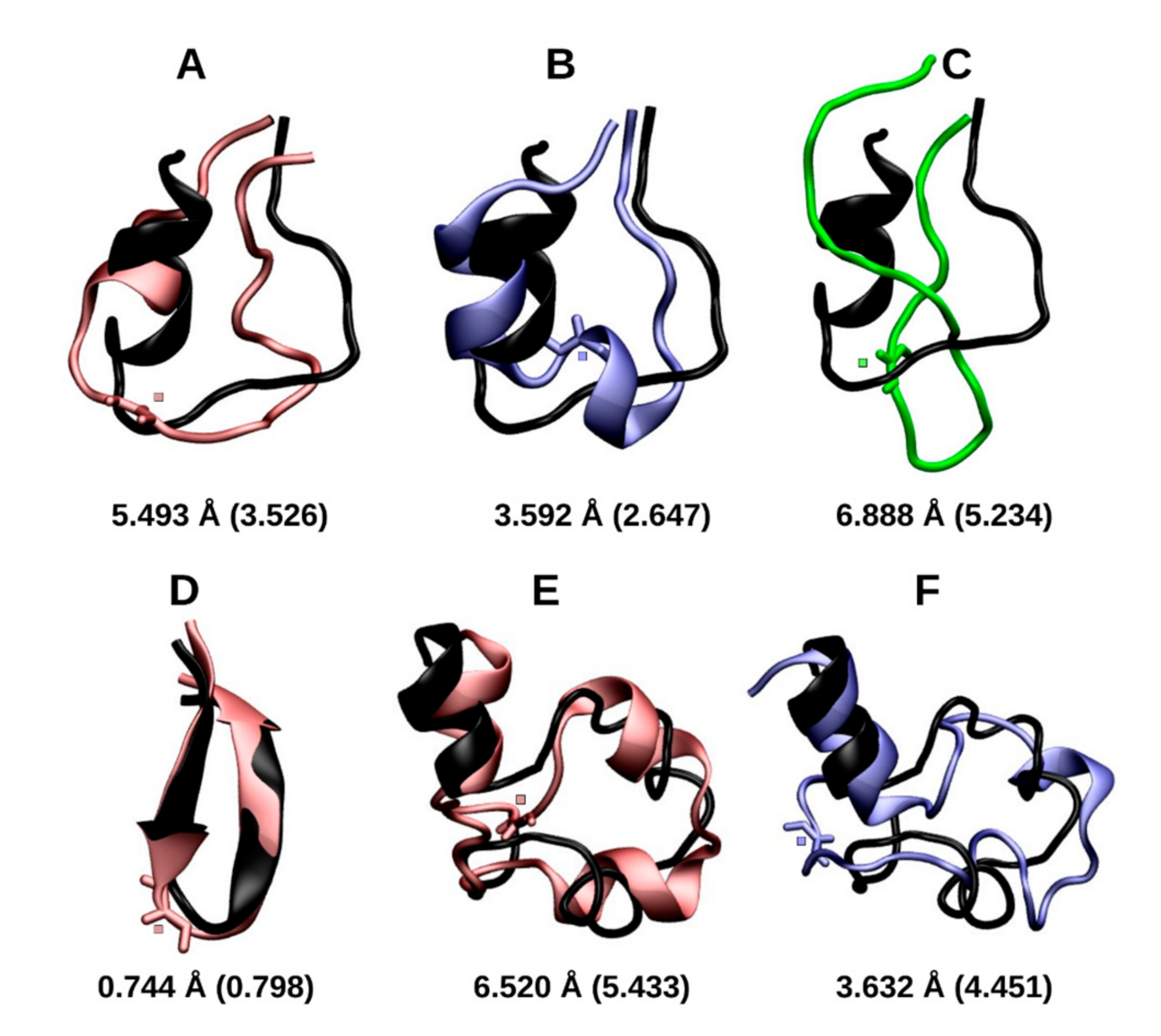
2.1.1. Cluster Analysis
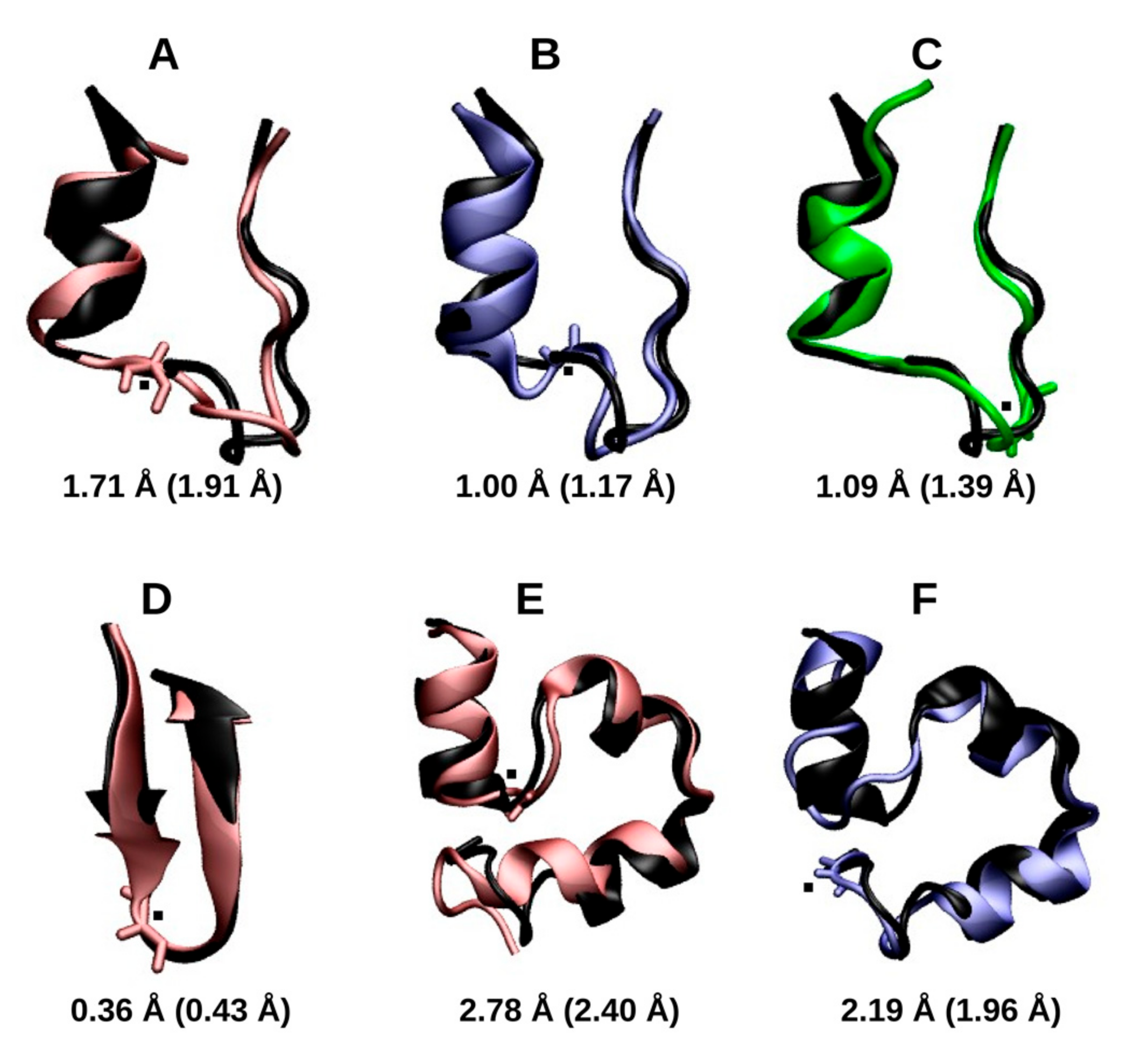
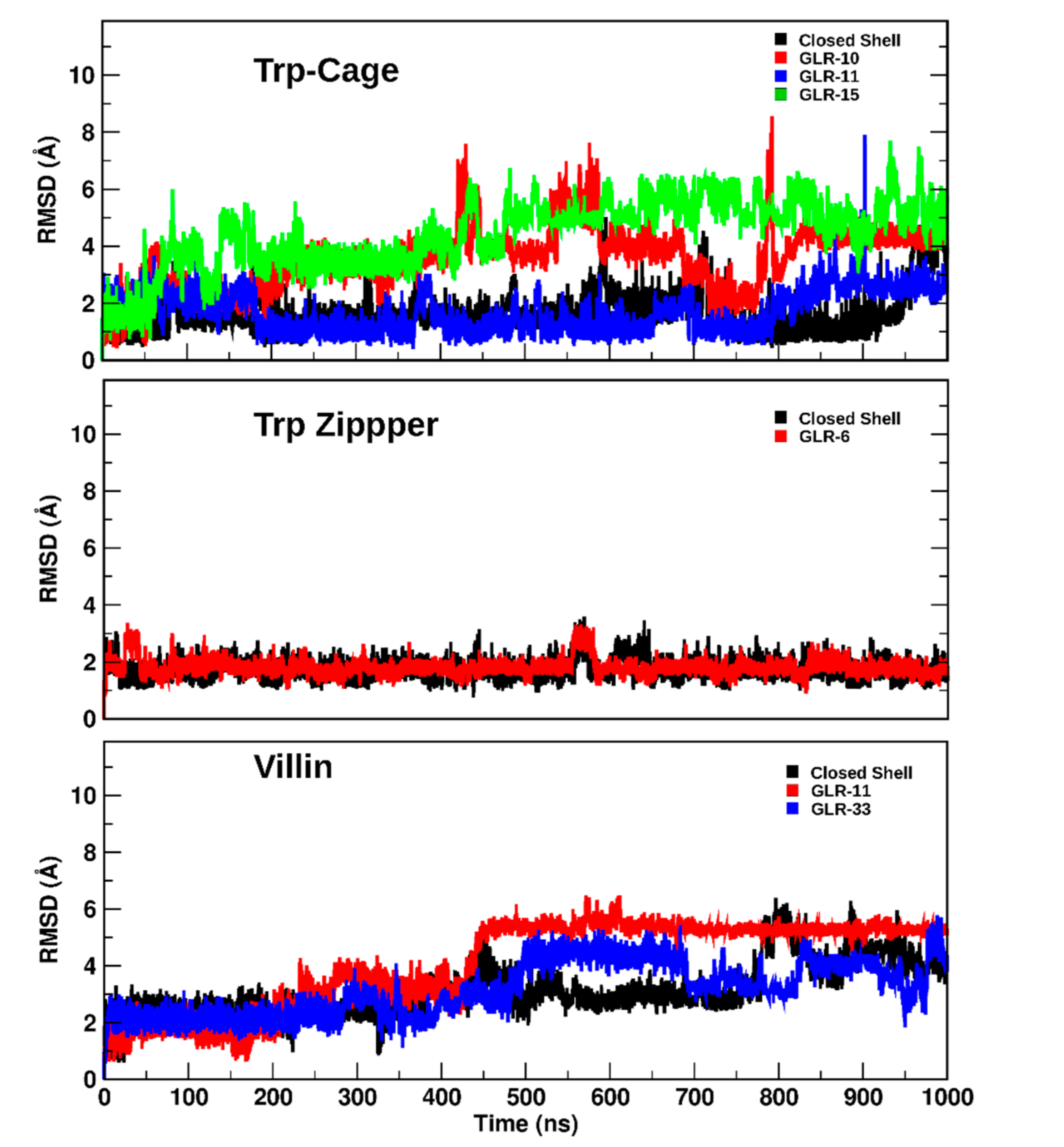
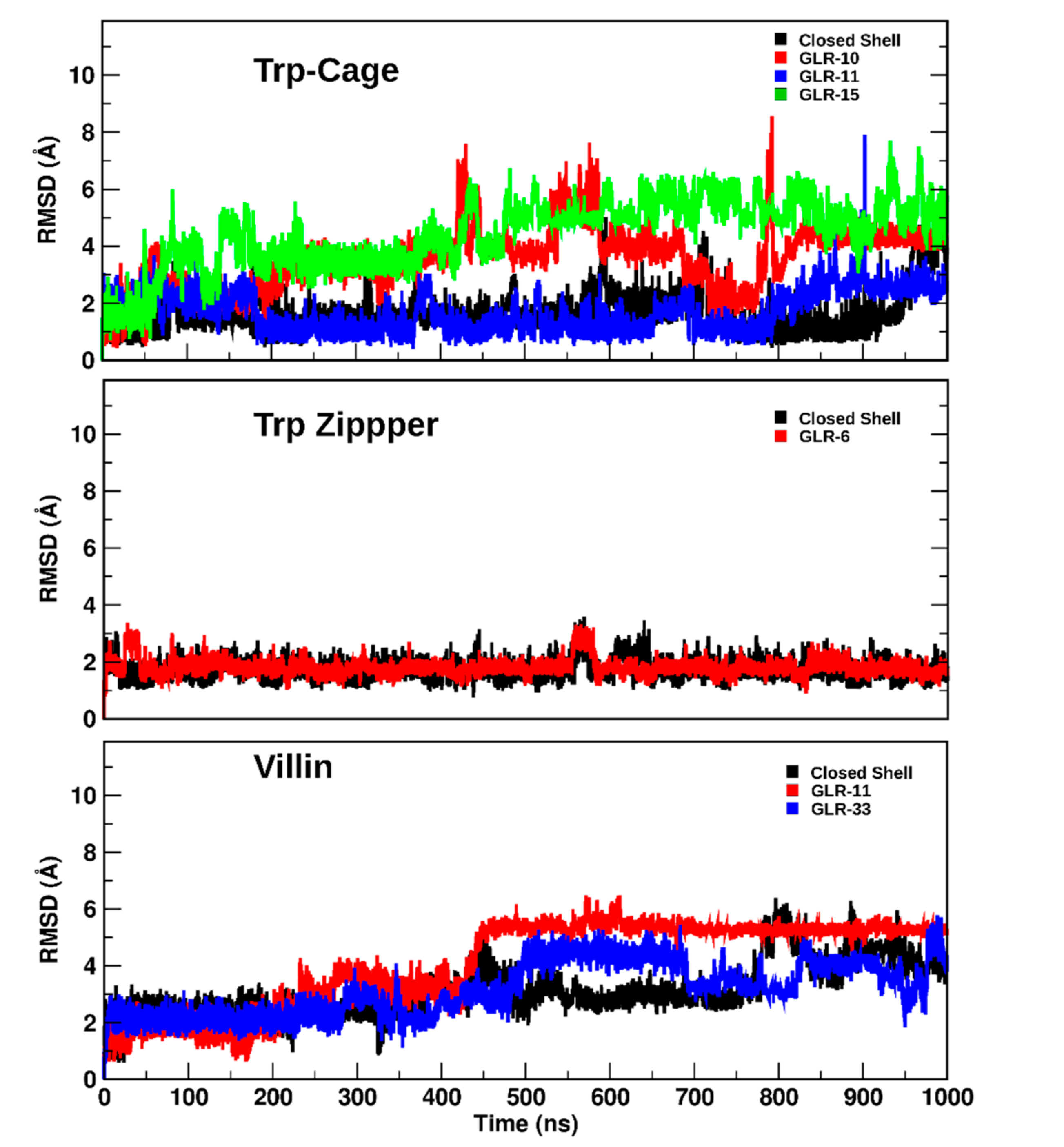
2.1.2. RMSD and Radius of Gyration
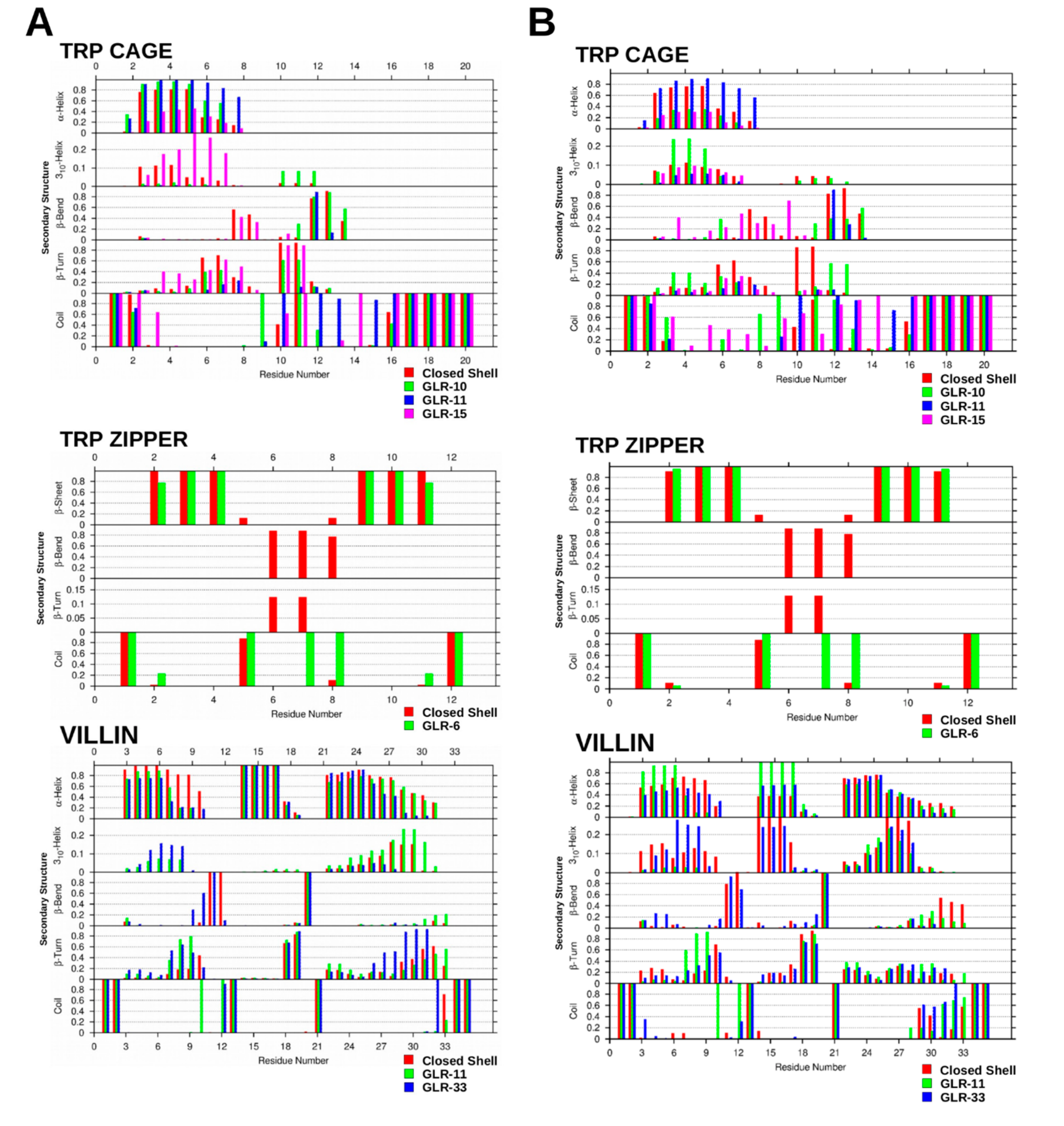
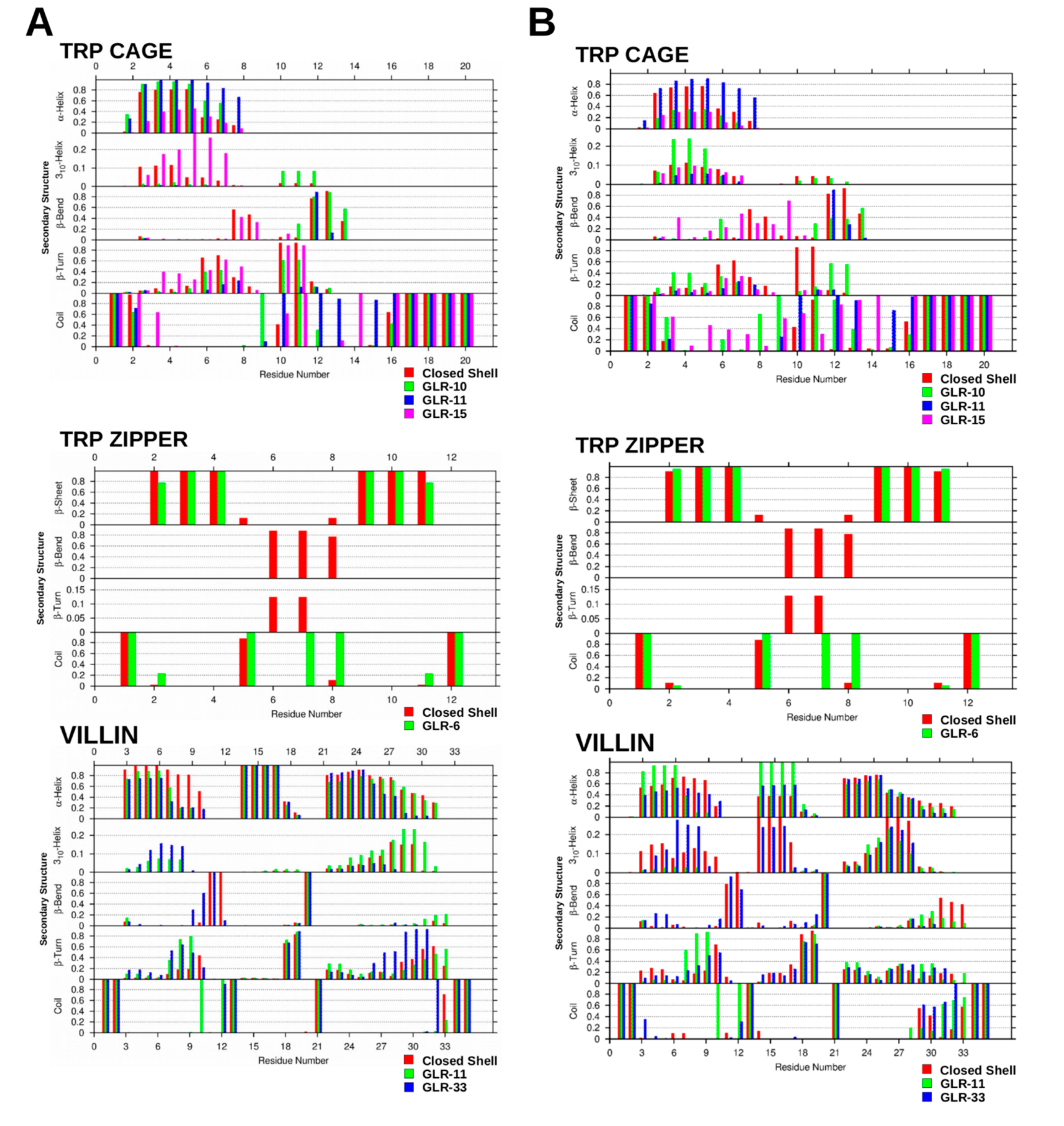
2.1.3. Secondary Structure Analysis
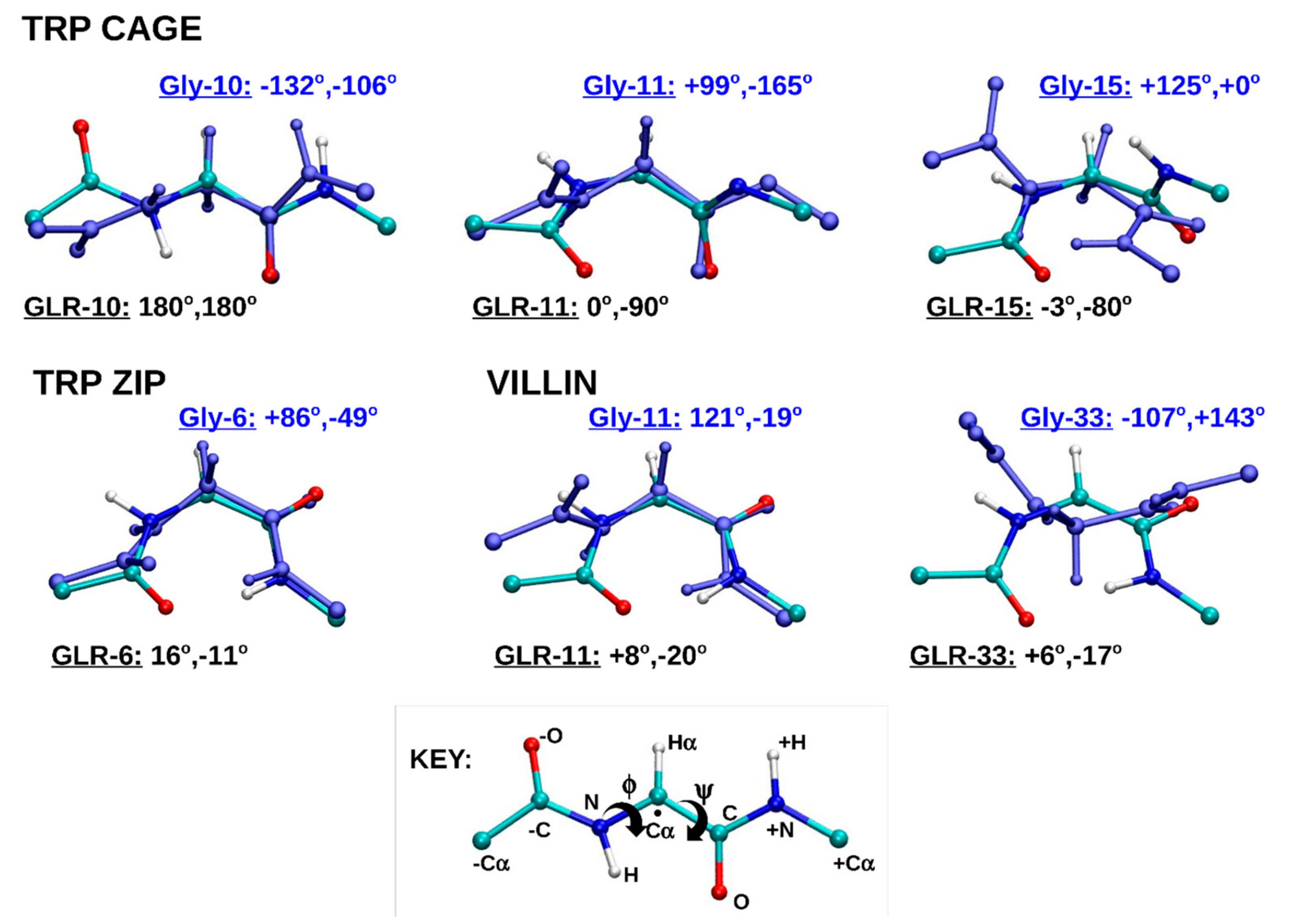
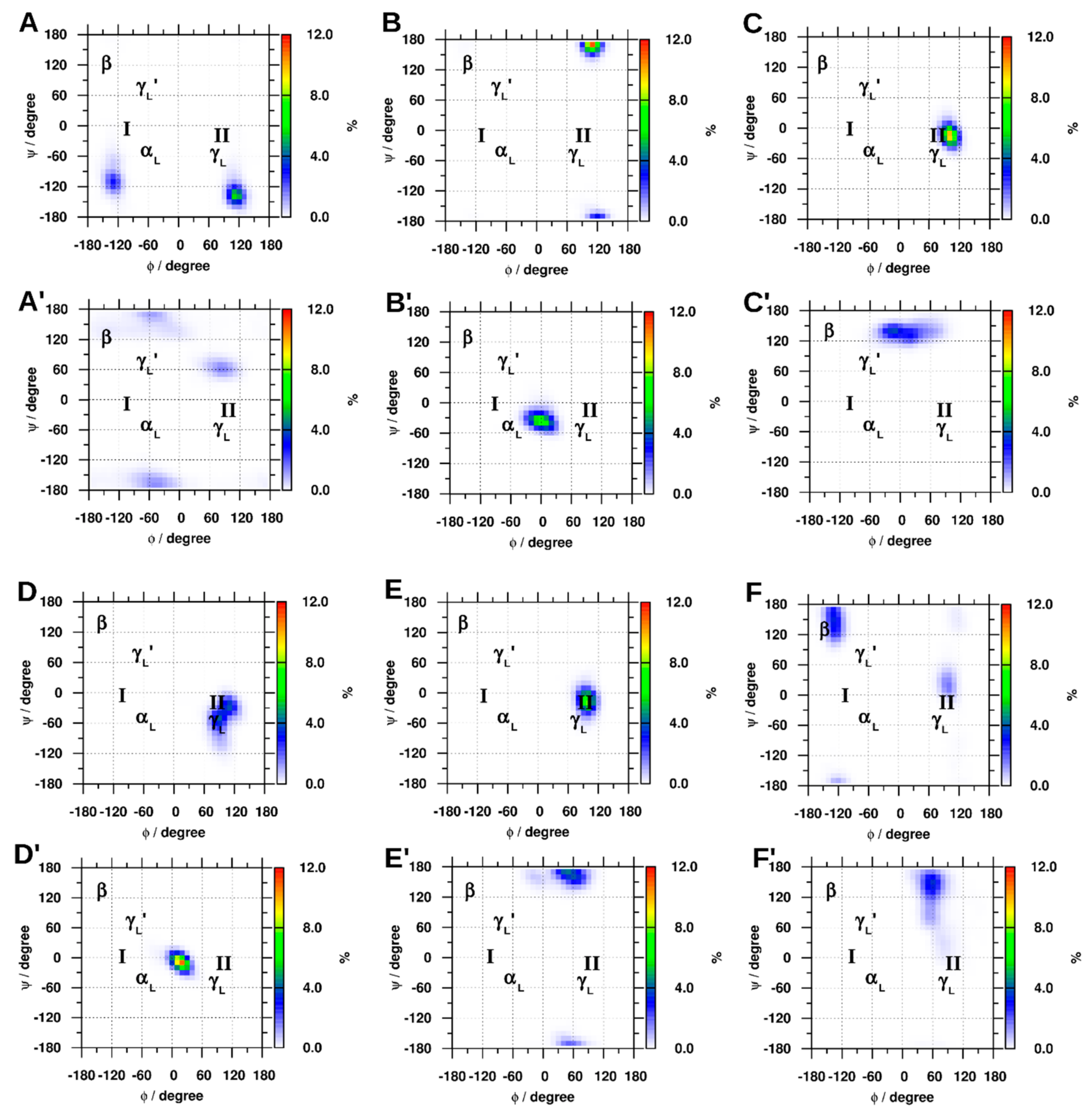
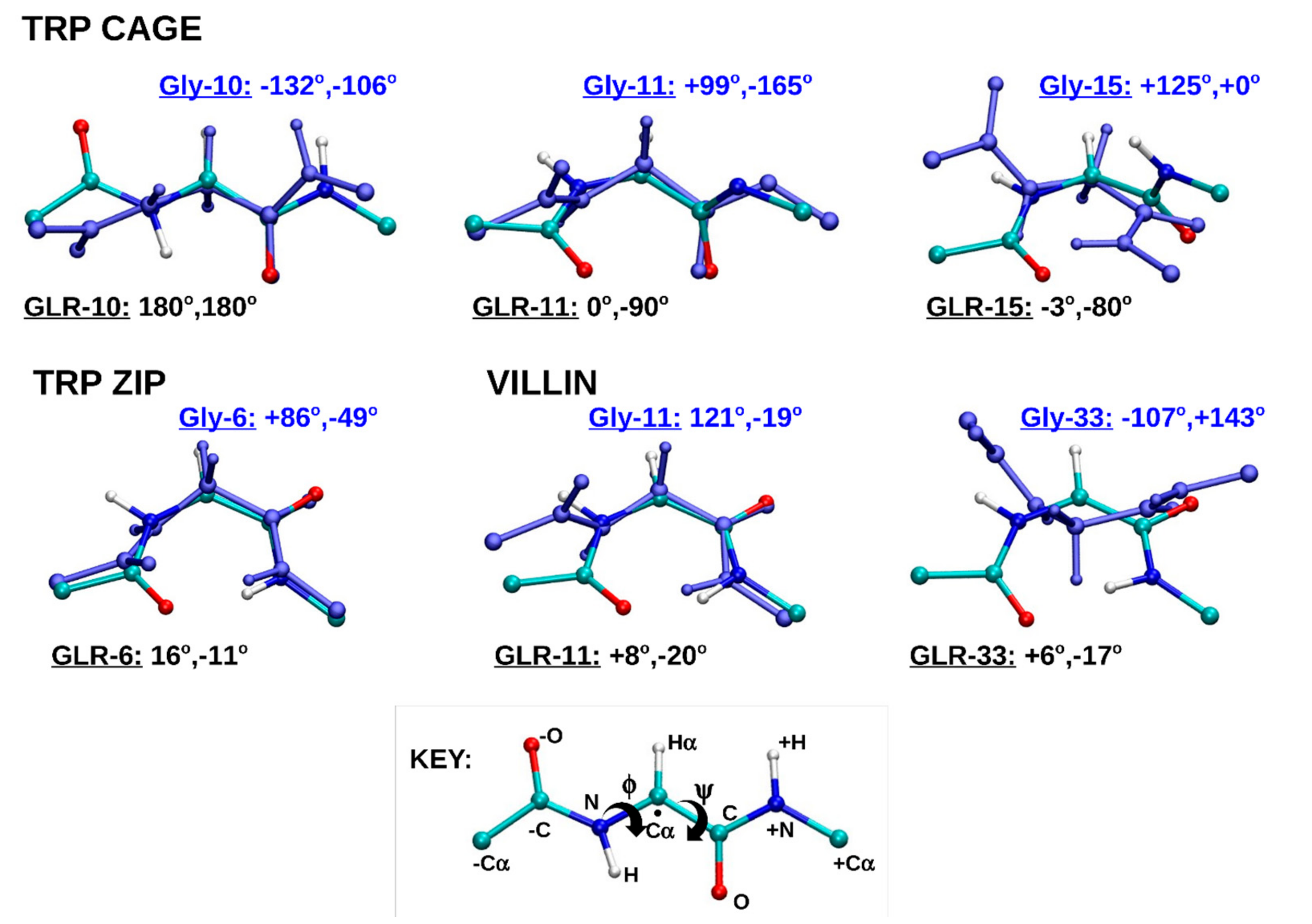
2.1.4. Dihedral Angles of Gly and GLR Residues
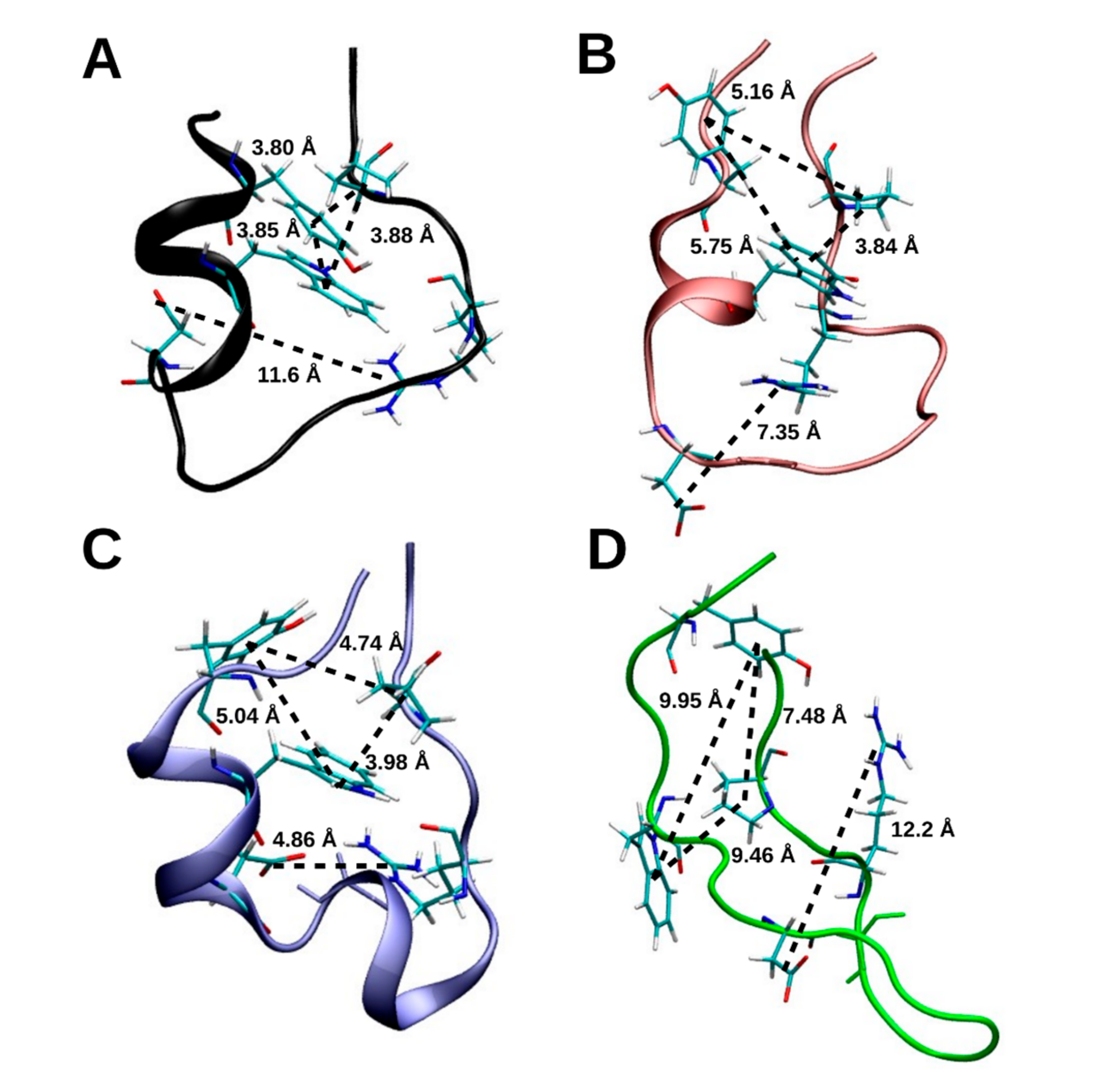
2.1.5. Hydrogen Bonding
2.2. Long-Term Effects
3. Discussion
3.1. Early Stage Radicalization
3.2. Prolonged Radical Exposure
3.3. General Effects of Gly Residue Radicalization on Stability
4. Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stadtman, E.R. Protein oxidation and aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Motherwell, M.S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 2013, 10, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Viehe, H.G.; Merényi, R.; Stella, L.; Janousek, Z. Capto-dative substituent effects in syntheses with radicals and radicophiles. Angew. Chem. Int. Ed. Engl. 1979, 18, 917–932. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. EPR studies on the selectivity of hydroxyl radical attack on amino acids and peptides. J. Chem. Soc. Perkin Trans. 1998, 2, 2617–2622. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Oliver, C. Metal-catalyzed oxidation of proteins. Physiological consequences. J. Biol. Chem. 1991, 266, 2005–2008. [Google Scholar] [PubMed]
- Lu, H.-F.; Li, F.-Y.; Lin, S.-H. Site specificity of α-H abstraction reaction among secondary structure motif—An ab initio study. J. Comput. Chem. 2007, 28, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Farber, J.M.; Levine, R.L. Sequence of a peptide susceptible to mixed-function oxidation. Probable cation binding site in glutamine synthetase. J. Biol. Chem. 1986, 261, 4574–4578. [Google Scholar] [PubMed]
- Galano, A.; Alvarez-Idaboy, J.R.; Montero, L.A.; Vivier-Bunge, A. OH hydrogen abstraction reactions from alanine and glycine: A quantum mechanical approach. J. Comput. Chem. 2001, 22, 1138–1153. [Google Scholar] [CrossRef]
- Easton, C.J.; Hay, M.P. Preferential reactivity of glycine residues in free radical reactions of amino acid derivatives. J. Chem. Soc. Chem. Commun. 1986, 1, 55–57. [Google Scholar] [CrossRef]
- Garrison, W.M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chem. Rev. 1987, 87, 381–396. [Google Scholar] [CrossRef]
- Sharma, V.K. Oxidation of Amino Acids, Peptides and Proteins: Kinetics and Mechanism; Wiley and Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 162–204. [Google Scholar]
- Owen, M.C.; Viskolcz, B.; Csizmadia, I.G. Quantum chemical analysis of the unfolding of a penta-alanyl 310-Helix Initiated by HO•, HO2• and O2–•. J. Phys. Chem. B 2011, 115, 8014–8023. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Fritz-Wolf, K.; Kabsch, W.; Knappe, J.; Schultz, S.; Wagner, A.F.V. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Biol. 1999, 6, 969–975. [Google Scholar] [PubMed]
- Logan, D.T.; Andersson, J.; Sjöberg, B.-M.; Nordlund, P. A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science 1999, 283, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.J.; Roseboom, W.; Albracht, S.P.J.; Spormann, A.M. A stable organic free radical in anaerobic benzylsuccinate synthase of Azoarcus sp. strain T. J. Biol. Chem. 2001, 276, 12924–12927. [Google Scholar] [CrossRef] [PubMed]
- Prigge, S.T.; Kolhekar, A.S.; Eipper, B.A.; Mains, R.E.; Amzel, L.M. Substrate-mediated electron transfer in peptidylglycine alpha-hydroxylating monooxygenase. Nat. Struct. Biol. 1999, 6, 976–983. [Google Scholar] [PubMed]
- Rauk, A.; Armstrong, D.A.; Fairlie, D.P. Is oxidative damage by β-amyloid and prion peptides mediated by hydrogen atom transfer from glycine α-carbon to methionine sulfur within β-sheets? J. Am. Chem. Soc. 2000, 122, 9761–9767. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zagorski, M.G.; Surewicz, W.K.; Krafft, G.A.; Perry, G. Mechanisms of neurotoxicity associated with amyloid β deposition and the role of free radicals in the pathogenesis of Alzheimer’s disease: A critical appraisal. Chem. Res. Toxicol. 1997, 10, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Brunk, U.T.; Rodgers, K.J. Oxidized proteins: Mechanisms of removal and consequences of accumulation. IUBMB Life 2009, 61, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: Insights into mechanism of neurodegeneration from redox proteomics. Antioxid. Redox Signal. 2006, 8, 2021–2037. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.J.; Hume, P.M.; Morris, J.G.; Dean, R.T. Evidence for l-dopa incorporation into cell proteins in patients treated with levodopa. J. Neurochem. 2006, 98, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.T.; Dunlop, R.; Hume, P.; Rodgers, K.J. Proteolytic ‘defences’ and the accumulation of oxidized peptides in cataractogenesis and atherogenesis. Biochem. Soc. Symp. 2003, 70, 135–146. [Google Scholar] [CrossRef]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-J.; Jang, S.; Wu, C.-C.; Liu, Y.-L.; Li, F.-Y. Site specificity of OH α-H abstraction reaction for a β-hairpin peptide: An ab initio study. J. Comput. Chem. 2011, 32, 3409–3422. [Google Scholar] [CrossRef] [PubMed]
- Nauser, T.; Casi, G.; Koppenol, W.H.; Schöneich, C. Reversible intramolecular hydrogen transfer between cysteine thiyl radicals and glycine and alanine in model peptides: Absolute rate constants derived from pulse radiolysis and laser flash photolysis. J. Phys. Chem. B 2008, 112, 15034–15044. [Google Scholar] [CrossRef] [PubMed]
- Tarabek, P.; Bonifacic, M.; Beckert, D. Oxidation of cyclic dipeptides by photoinduced H-atom abstraction. a laser flash FT EPR and Optical Spectroscopy Study. J. Phys. Chem. A 2007, 111, 4958–4964. [Google Scholar] [CrossRef] [PubMed]
- Croft, A.; Easton, C.J.; Radom, L. Design of radical-resistant amino acid residues: A combined theoretical and experimental investigation. J. Am. Chem. Soc. 2003, 125, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Sperling, J. Elad, D. Photoalkylation of proteins. J. Am. Chem. Soc. 1971, 93, 3839–3840. [Google Scholar]
- Schwarzberg, M.; Sperling, J.; Elad, D. Photoalkylation of peptides. Visible light-induced conversion of glycine residues into branched alpha-amino acids. J. Am. Chem. Soc. 1973, 95, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Knappe, J.; Wagner, A.F.V. Glycyl free radical in pyruvate formate-lyase: Synthesis, structure characteristics, and involvement in catalysis. Methods Enzymol. 1995, 258, 343–362. [Google Scholar] [PubMed]
- Rabus, R.; Wilkes, H.; Behrends, A.; Armstroff, A.; Fischer, T.; Pierik, A.J.; Widdel, F. Anaerobic initial reaction of n-alkanes in a denitrifying bacterium: Evidence for (1-methylpentyl)succinate as initial product and for involvement of an organic radical in n-hexane metabolism. J. Bacteriol. 2001, 183, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Freddolino, P.L.; Harrison, C.B.; Liu, L.; Schulten, K. Challenges in protein-folding simulations. Nature Phys. 2010, 6, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Szori, M.; Csizmadia, I.G.; Viskolcz, B. Conformation-dependent ·OH/H2O2 hydrogen abstraction reaction cycles of Gly and Ala residues: A comparative theoretical study. J. Phys. Chem. B 2012, 116, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Toth, L.; Jojart, B.; Komaromi, I.; Csizmadia, I.G.; Viskolcz, B. The effect of newly developed OPLS-AA alanyl radical parameters on peptide secondary structure. J. Chem. Theory Comput. 2012, 8, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Strodel, B.; Csizmadia, I.G.; Viskolcz, B. Radical formation initiates solvent-dependent unfolding and β-sheet formation in a model helical peptide. J. Phys. Chem. B 2016, 120, 4878–4889. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Pabit, S.A.; Roitberg, A.E.; Hagen, S.J. Smaller and faster: The 20-residue Trp-cage protein folds in 4 μs. J. Am. Chem. Soc. 2002, 124, 12952–12953. [Google Scholar] [CrossRef] [PubMed]
- Neidigh, J.W.; Fesinmeyer, R.M.; Andersen, N.H. Designing a 20-residue protein. Nat. Struct. Biol. 2002, 9, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Skelton, N.J.; Starovasnik, M.A. Tryptophan zippers: Stable, monomeric β-hairpins. Proc. Natl. Acad. Sci. USA 2001, 98, 5578–5583. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Hofrichter, J.; Eaton, W.A. The protein folding ‘speed limit’. Curr. Opin. Struct. Biol. 2004, 14, 76–88. [Google Scholar] [CrossRef] [PubMed]
- McNight, C.J.; Matsudaira, P.T.; Kim, P.S. NMR structure of the 35-residue villin headpiece subdomain. Nat. Struct. Biol. 1997, 4, 180–184. [Google Scholar] [CrossRef]
- Komáromi, I.; Owen, M.C.; Murphy, R.; Lovas, S. Development of glycyl radical parameters for the OPLS-AA/L force field. J. Comput. Chem. 2008, 29, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Lifson, S.; Warshel, A. Consistent force field for calculations of conformations, vibrational spectra, and enthalpies of cycloalkane and n-alkane molecules. J. Chem. Phys. 1968, 49, 5116–5129. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Cruz-Torres, A.; Ruiz-Santoyo, E. Kinetics and mechanism of the gas-phase OH hydrogen abstraction reaction from methionine: A quantum mechanical approach. Int. J. Chem. Kinet. 2003, 35, 212–221. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Cruz-Torres, A.; Ruiz-Santoyo, E. Rate coefficients and mechanism of the gas phase OH hydrogen abstraction reaction from serine: A quantum mechanical approach. J. Mol. Struct. (Theochem) 2003, 629, 165–174. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Bravo-Pérez, G.; Ruiz-Santoyo, E. Mechanism and rate coefficients of the gas phase OH hydrogen abstraction reaction from asparagine: A quantum mechanical approach. J. Mol. Struct. (Theochem) 2002, 617, 77–86. [Google Scholar] [CrossRef]
- Beke, T.; Czajlik, A.; Csizmadia, I.G.; Perczel, A. Determining suitable lego-structures to estimate stability of larger nanostructures using computational methods. Phys. Biol. 2006, 3, S26–S39. [Google Scholar] [CrossRef] [PubMed]
- Streicher, W.W.; Makhatadze, G.I. Unfolding thermodynamics of Trp-cage, a 20 residue miniprotein, Studied by differential scanning calorimetry and circular dichroism spectroscopy. Biochemistry 2007, 46, 2876–2880. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Eaton, W.A.; Hofrichter, J. Experimental tests of villin subdomain folding simulations. J. Mol. Biol. 2003, 329, 625–630. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. Protein folding kinetics and thermodynamics from atomistic simulation. Proc. Natl. Acad. Sci. USA 2012, 109, 17845–17850. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Zacharias, M. Role of tryptophan side chain dynamics on the Trp-cage mini-protein folding studied by molecular dynamics simulations. PLoS ONE 2014, 9, e88383. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J.; Ortner, M.; Jaritz, M.; Lackner, P.; Flöckner, H. Helmholtz free energies of atom pair interactions in proteins. Fold Des. 1996, 1, 289–298. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comp. Chem. 2005, 26, 1701–1719. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesener, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D. Temperature and size dependence for Monte Carlo simulations of TIP4P water. Mol. Phys. 1985, 56, 1381–1392. [Google Scholar] [CrossRef]
- Berendsen, H.J. C.; Postma, J.P. M. van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Juan, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Number of Clusters | Proportion of Structures in Most Populated Cluster/% | Frame of Middle Structure of Largest Cluster/ns |
|---|---|---|---|
| CS Trp cage | 77 | 56.6 | 44.5 |
| Trp cage(GLR10) | 81 | 24.4 | 89.1 |
| Trp cage(GLR11) | 51 | 43.3 | 4.0 |
| Trp cage(GLR15) | 170 | 20.7 | 46.8 |
| CS Trp zipper | 6 | 97.8 | 63.7 |
| Trp zipper(GLR6) | 7 | 92.6 | 84.5 |
| CS Villin | 71 | 63.3 | 44.7 |
| Villin(GLR11) | 218 | 18.3 | 28.6 |
| Villin(GLR33) | 70 | 68.4 | 66.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owen,, M.C.; Csizmadia, I.G.; Viskolcz, B.; Strodel, B. Protein Stability and Unfolding Following Glycine Radical Formation. Molecules 2017, 22, 655. https://doi.org/10.3390/molecules22040655
Owen, MC, Csizmadia IG, Viskolcz B, Strodel B. Protein Stability and Unfolding Following Glycine Radical Formation. Molecules. 2017; 22(4):655. https://doi.org/10.3390/molecules22040655
Chicago/Turabian StyleOwen,, Michael C., Imre G. Csizmadia, Béla Viskolcz, and Birgit Strodel. 2017. "Protein Stability and Unfolding Following Glycine Radical Formation" Molecules 22, no. 4: 655. https://doi.org/10.3390/molecules22040655
APA StyleOwen,, M. C., Csizmadia, I. G., Viskolcz, B., & Strodel, B. (2017). Protein Stability and Unfolding Following Glycine Radical Formation. Molecules, 22(4), 655. https://doi.org/10.3390/molecules22040655