
Intestinal Transport Characteristics and Metabolism of C-Glucosyl Dihydrochalcone, Aspalathin

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Physicochemical Characteristics of Aspalathin
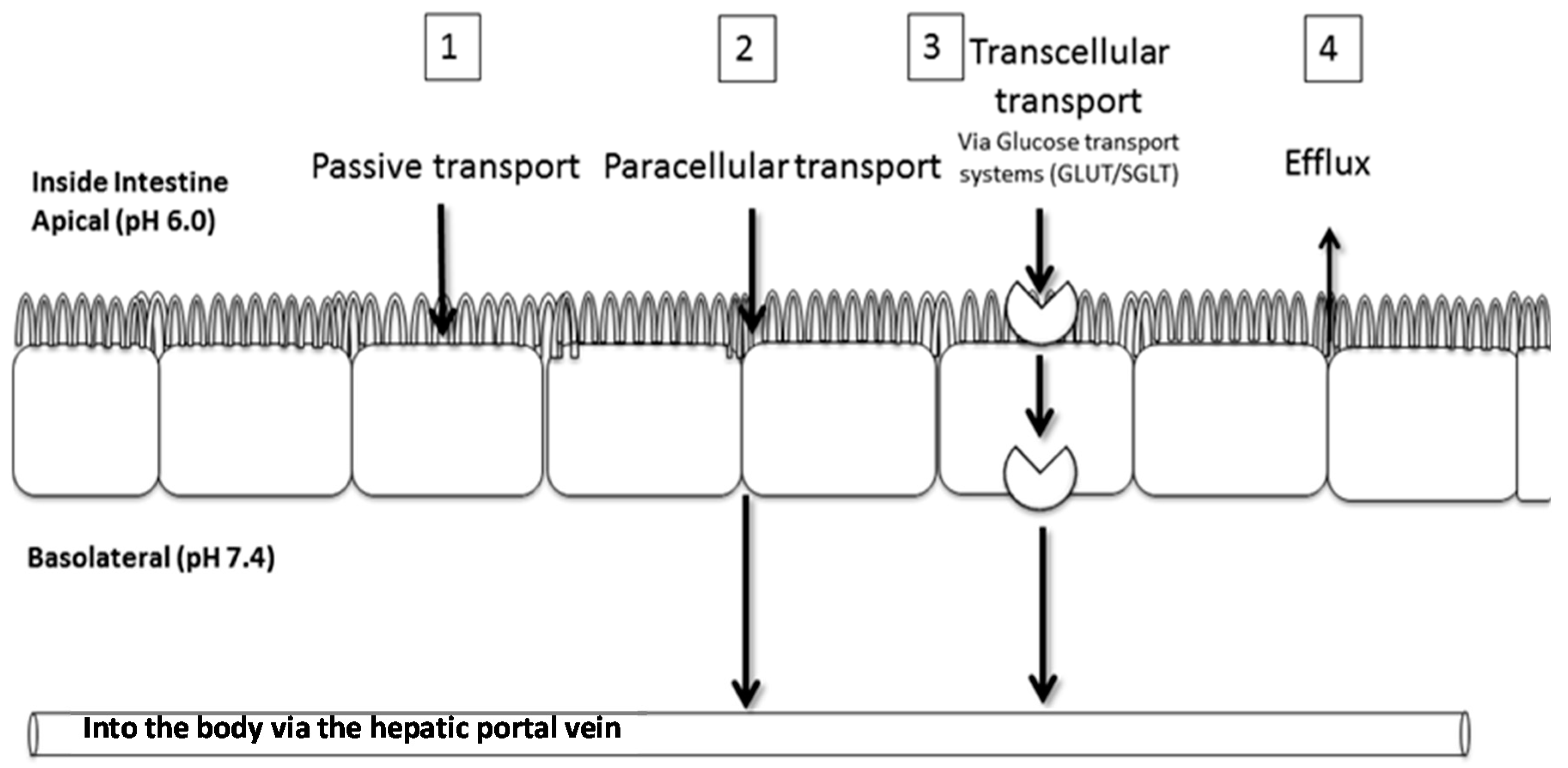
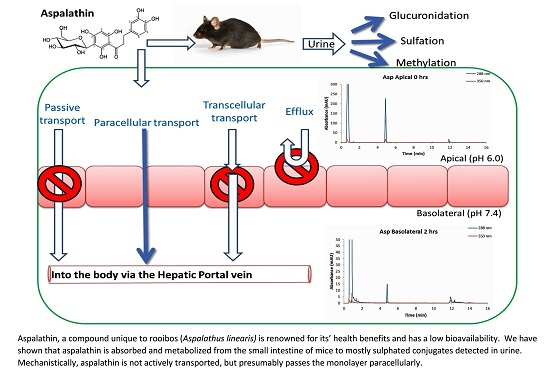
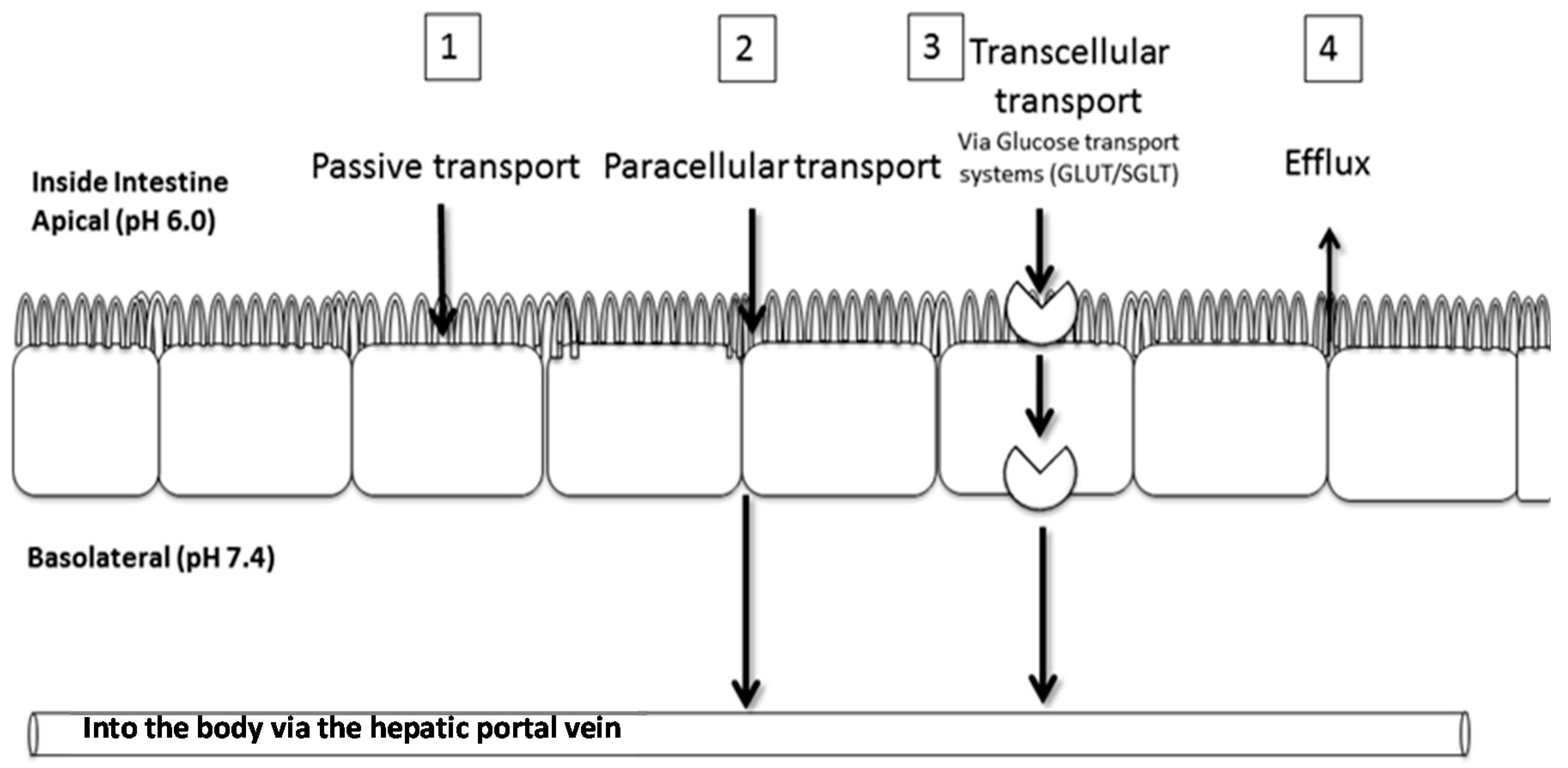
2.2. Transport of Aspalathin across a Caco-2 Monolayer
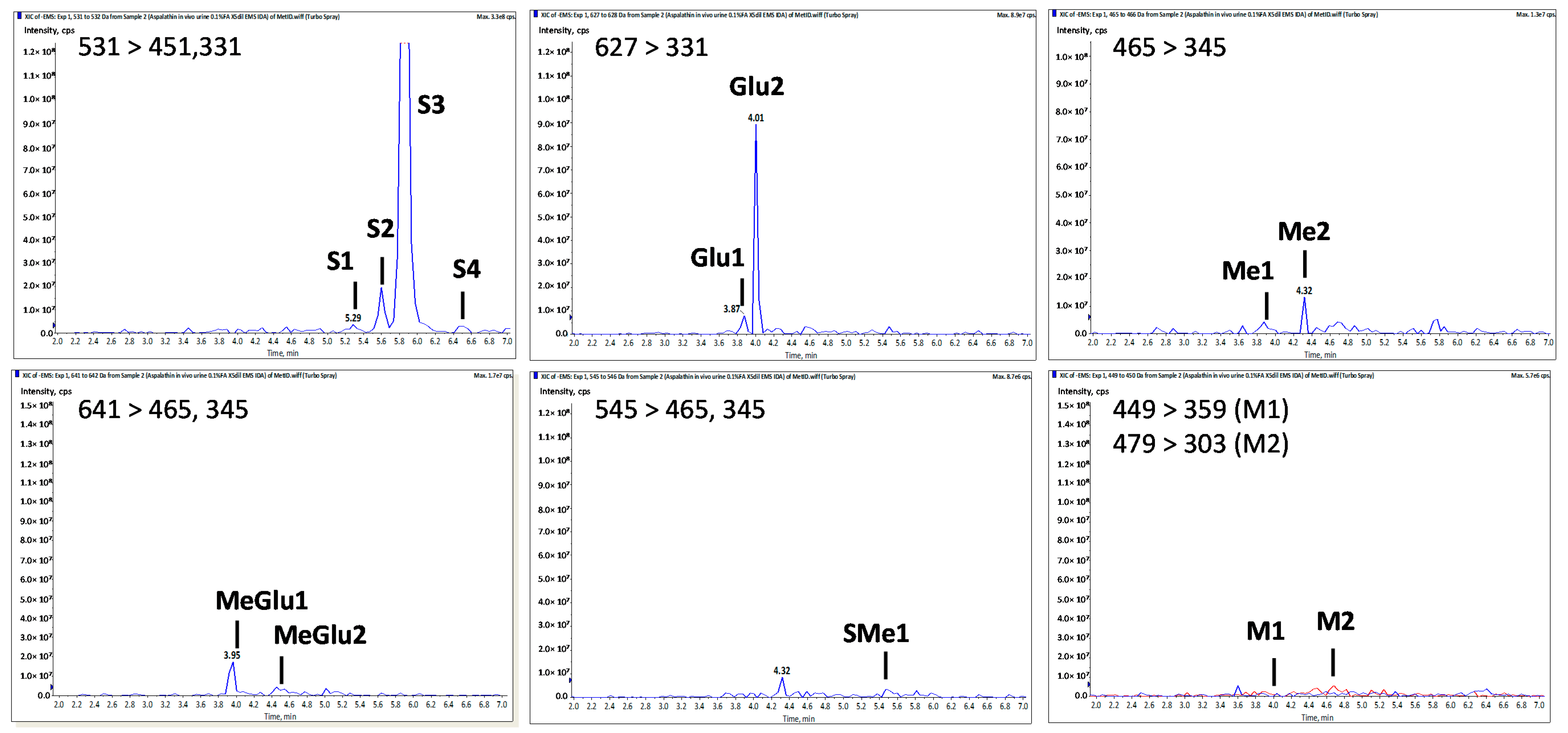
2.3. Aspalathin Metabolites in Mouse Urine
3. Discussion
4. Materials and Methods
4.1. Reagents and Extracts
4.2. Physicochemical Characterisation
4.3. Caco-2 Transport Experiments
4.4. In Vivo and In Vitro Metabolism of Aspalathin
4.4.1. Treatment and Sample Collection
4.4.2. Metabolite Identification by MS
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Joubert, E.; De Beer, D. Rooibos (Aspalathus linearis) beyond the farm gate: From herbal tea to potential phytopharmaceutical. S. Afr. J. Bot. 2011, 77, 869–886. [Google Scholar] [CrossRef]
- Muller, C.J.F.; Malherbe, C.J.; Chellan, N.; Yagasaki, K.; Miura, Y.; Joubert, E. Potential of rooibos, its major C-glucosyl flavonoids and Z-2-(β-d-glucopyranosyloxy)-3-phenylpropenoic acid in prevention of metabolic syndrome. Crit. Rev. Food Sci. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Courts, F.L.; Williamson, G. The C-glycosyl flavonoid, aspalathin, is absorbed, methylated and glucuronidated intact in humans. Mol. Nutr. Food Res. 2009, 53, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Stalmach, A.; Mullen, W.; Pecorari, M.; Serafini, M.; Crozier, A. Bioavailability of C-linked dihydrochalcone and flavanone glucosides in humans following ingestion of unfermented and fermented rooibos teas. J. Agric. Food Chem. 2009, 57, 7104–7111. [Google Scholar] [CrossRef] [PubMed]
- Breiter, T.; Laue, C.; Kressel, G.; Gröll, S.; Engelhardt, U.H.; Hahn, A. Bioavailability and antioxidant potential of rooibos flavonoids in humans following the consumption of different rooibos formulations. Food Chem. 2011, 128, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, A.; Joost, H.G.; Schurmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. J. Parenter. Enter. Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Cañada, F.J.; Díaz, J.C.; Kroon, P.A.; Mclauchlan, R.; Faulds, C.B.; Plumb, G.W.; Morgan, M.R.; Williamson, G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2000, 468, 166–170. [Google Scholar] [CrossRef]
- Liu, W.; Wang, H.; Meng, F. In silico modeling of aspalathin and nothofagin against SGLT2. J. Theor. Comput. Chem. 2015, 14, 1550056. [Google Scholar] [CrossRef]
- White, J.R. Apple trees to sodium glucose co-transporter inhibitors: A review of SGLT2 inhibition. Clin. Diabetes 2010, 28, 5–10. [Google Scholar] [CrossRef]
- Courts, F.L.; Williamson, G. The occurrence, fate and biological activities of C-glycosyl flavonoids in the human diet. Crit. Rev. Food Sci. Nutr. 2013, 8398, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kawano, A.; Nakamura, H.; Hata, S.; Minakawa, M.; Miura, Y.; Yagasaki, K. Hypoglycemic effect of aspalathin, a rooibos tea component from Aspalathus linearis, in type 2 diabetic model db/db mice. Phytomedicine 2009, 16, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Minakawa, M.; Miura, Y.; Yagasaki, K. Aspalathin improves hyperglycemia and glucose intolerance in obese diabetic ob/ob mice. Eur. J. Nutr. 2013, 52, 1607–1619. [Google Scholar] [CrossRef]
- Parrott, N.; Lukacova, V.; Fraczkiewicz, G.; Bolger, M.B. Predicting pharmacokinetics of drugs using physiologically based modeling—Application to food effects. AAPS J. 2009, 11, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kazuno, S.; Yanagida, M.; Shindo, N.; Murayama, K. Mass spectrometric identification and quantification of glycosyl flavonoids, including dihydrochalcones with neutral loss scan mode. Anal. Biochem. 2005, 347, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Joubert, E.; Gelderblom, W.C.A.; De Beer, D. Phenolic contribution of South African herbal teas to a healthy diet. Nat. Prod. Commun. 2009, 4, 701–718. [Google Scholar] [PubMed]
- Varma, M.V.; Steyn, S.J.; Allerton, C.; El-Kattan, A.F. Predicting clearance mechanism in drug discovery: Extended Clearance Classification System (ECCS). Pharm. Res. 2015, 32, 3785–3802. [Google Scholar] [CrossRef] [PubMed]
- Braune, A.; Blaut, M. Deglycosylation of puerarin and other aromatic C-glucosides by a newly isolated human intestinal bacterium. Environ. Microbiol. 2011, 13, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.J.F.; Joubert, E.; De Beer, D.; Sanderson, M.; Malherbe, C.J.; Fey, S.J.; Louw, J. Acute assessment of an aspalathin-enriched green rooibos (Aspalathus linearis) extract with hypoglycemic potential. Phytomedicine 2012, 20, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. 2010, 154, 103–116. [Google Scholar] [CrossRef]
- Kreuz, S.; Joubert, E.; Waldmann, K.H.; Ternes, W. Aspalathin, a flavonoid in Aspalathus linearis (rooibos), is absorbed by pig intestine as a C-glycoside. Nutr. Res. 2008, 28, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, J.D.; Joubert, E.; Manley, M.; De Beer, D.; Malherbe, C.J.; Gelderblom, W.C.A. In vitro hepatic biotransformation of aspalathin and nothofagin, dihydrochalcones of rooibos (Aspalathus linearis), and assessment of metabolite antioxidant activity. J. Agric. Food Chem. 2010, 58, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Elsevier Science: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Volpe, D.A. Application of method suitability for drug permeability classification. AAPS J. 2010, 12, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, H.J.R.; Hamman, J.H. Paracellular drug absorption enhancement through tight junction modulation. Expert Opin. Drug Deliv. 2013, 10, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, G.; Folkers, G.; Van de Waterbeemd, H. Shapes of membrane permeability–lipophilicity curves: Extension of theoretical models with an aqueous pore pathway. Eur. J. Pharm. Sci. 1998, 6, 321–329. [Google Scholar] [CrossRef]
- Thomson, A.B.; Keelan, M.; Thiesen, A.; Clandinin, M.T.; Ropeleski, M.J.; Wild, G. Small bowel review: Part II. Can. J. Gastroenterol. 2001, 15, 446–466. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Matthias, T. Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 2015, 14, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Furuse, M.; Fujimoto, K.; Tsukita, S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. USA 1999, 96, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Mandell, K.J.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J. Biol. Chem. 2005, 280, 11665–11674. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Tang, D.W.; Hsieh, H.Y.; Wu, W.S.; Lin, B.X.; Chuang, E.Y.; Sung, H.W.; Mi, F.L. Nanoparticle-induced tight-junction opening for the transport of an anti-angiogenic sulfated polysaccharide across Caco-2 cell monolayers. Acta Biomater. 2013, 9, 7449–7459. [Google Scholar] [CrossRef] [PubMed]
- Jayalakshmi, K.; Ghoshal, U.C.; Kumar, S.; Misra, A.; Roy, R.; Khetrapal, C.L. Assessment of small intestinal permeability using 1H-NMR spectroscopy. J. Gastrointest. Liver Dis. 2009, 18, 27–32. [Google Scholar]
- Huang, M.; Du Plessis, J.; Du Preez, J.; Hamman, J.; Viljoen, A. Transport of aspalathin, a rooibos tea flavonoid, across the skin and intestinal epithelium. Phyther. Res. 2008, 22, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhu, L.; Li, Y.; Zheng, H.; Yu, J.; Lu, L.; Liu, Z. In vitro study of UGT metabolism and permeability of orientin and isoorientin, two active flavonoid C-glycosides. Drug Metab. Lett. 2016, 10, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Bae, J.S. Anti-inflammatory effects of aspalathin and nothofagin from rooibos (Aspalathus linearis) in vitro and in vivo. Inflammation 2015, 38, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Achilonu, M.C.; Kendrekar, P.S.; Joubert, E.; Ferreira, D.; Bonnet, S.L.; Van der Westhuizen, J.H. Concise and scalable synthesis of aspalathin, a powerful plasma sugar-lowering natural product. J. Nat. Prod. 2013, 77, 583–588. [Google Scholar] [CrossRef] [PubMed]
- De Beer, D.; Malherbe, C.J.; Beelders, T.; Willenburg, E.L.; Brand, D.J.; Joubert, E. Isolation of aspalathin and nothofagin from rooibos (Aspalathus linearis) using high-performance countercurrent chromatography: Sample loading and compound stability considerations. J. Chromatogr. A 2015, 1381, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.P.; Young, R.J. Getting physical in drug discovery: A contemporary perspective on solubility and hydrophobicity. Drug Discov. Today 2010, 15, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Alelyunas, Y.W.; Pelosi-kilby, L.; Turcotte, P.; Kary, M.; Spreen, R.C. A high throughput dried DMSO Log D lipophilicity measurement based on 96-well shake-flask and atmospheric pressure photoionization mass spectrometry detection. J. Chromatogr. A 2010, 1217, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Smetanova, L.; Stetinova, V.; Kholova, D.; Kvetina, J.; Smetana, J.; Svoboda, Z. Caco-2 cells and Biopharmaceutics Classification System (BCS) for prediction of transepithelial transport of xenobiotics (model drug: Caffeine). Neuro Endocrinol. Lett. 2009, 30 (Suppl. 1), 101–105. [Google Scholar] [PubMed]
- Johnston, K.; Sharp, P.; Clifford, M.N.; Morgan, L. Dietary polyphenols decrease glucose uptake by human intestinal Caco-2 cells. FEBS Lett. 2005, 579, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Caudill, M.A. Biochemical, Physiological, and Molecular Aspects of Human Nutrition; Elsevier Health Sciences: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Ingels, F.; Beck, B.; Oth, M.; Augustijns, P. Effect of simulated intestinal fluid on drug permeability estimation across Caco-2 monolayers. Int. J. Pharm. 2004, 274, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Aspalathin is available from commercial sources. |

{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | Values |
|---|---|
| Molecular weight (g/mol) | 452.1 |
| Kinetic solubility (µM) at pH 2, pH 6.5 and in FaSSIF at pH 6.5 | 153, 123, 119 |
| Log D at pH 7.4 | 0.13 |
| Treatment | Papp a-b (cm/s) c | Papp b-a (cm/s) c | % Passage |
|---|---|---|---|
| Caffeine (n = 9) a | 6.50 ± 0.99 × 10−5 | 7.23 ± 0.23 × 10−5 | 79.50 ± 1.23 |
| Aspalathin (n = 18) a | 1.73 ± 0.97 × 10−6 | 2.15 ± 0.23 × 10—6; efflux ratio 1.1 | 4.95 ± 2.11 d |
| Aspalathin with high glucose (n = 8) b | 2.90 ± 0.75 × 10−7 | N/A | 2.34 ± 2.35 d |
| Treatment | Inhibited Protein | Papp a-b b − Inhibitor | Papp a-b b + Inhibitor | Papp b-a b − Inhibitor | Papp b-a b + Inhibitor | Efflux Ratio − Inhibitor | Efflux Ratio + Inhibitor |
|---|---|---|---|---|---|---|---|
| Phloridzin | SGLT1 | 1.73 ± 0.97 | 1.47 ± 1.10 c | 2.09 ± 0.23 | 1.47 ± 0.9 c | 1.21 | 0.99 c |
| Phloretin | GLUT2 | 1.73 ± 0.97 | 1.67 ± 0.43 c | 2.09 ± 0.23 | 1.95 ± 1.1 c | 1.21 | 1.33 c |
| Verapamil | PgP | 1.73 ± 0.97 | 1.84 ± 0.20 c | 2.09 ± 0.23 | 1.94 ± 0.68 c | 1.21 | 1.1 c |
| Treatment | Concentration | Papp (cm/s) a-b × 10−6 |
|---|---|---|
| Caffeine | 260 µM | 67.88 ± 0.99 |
| Aspalathin | 1 µM | 2.28 ± 0.09 |
| 150 µM | 1.73 ± 0.97 | |
| SB1 Aspalathin | 0.38 mg/mL a | 2.00 ± 1.10 |
| SB1 Nothafagin | 1.92 ± 1.10 | |
| SB1 Isoorientin | 1.81 ± 1.10 | |
| SB1 Orientin | 1.99 ± 1.30 | |
| PEF1 Aspalathin | 0.15 mg/mL b | 2.11 ± 0.20 |
| PEF1 Nothofagin | 2.22 ± 0.30 | |
| PEF1 Isoorientin | 1.69 ± 0.20 | |
| PEF1 Orientin | 1.92 ± 0.20 |
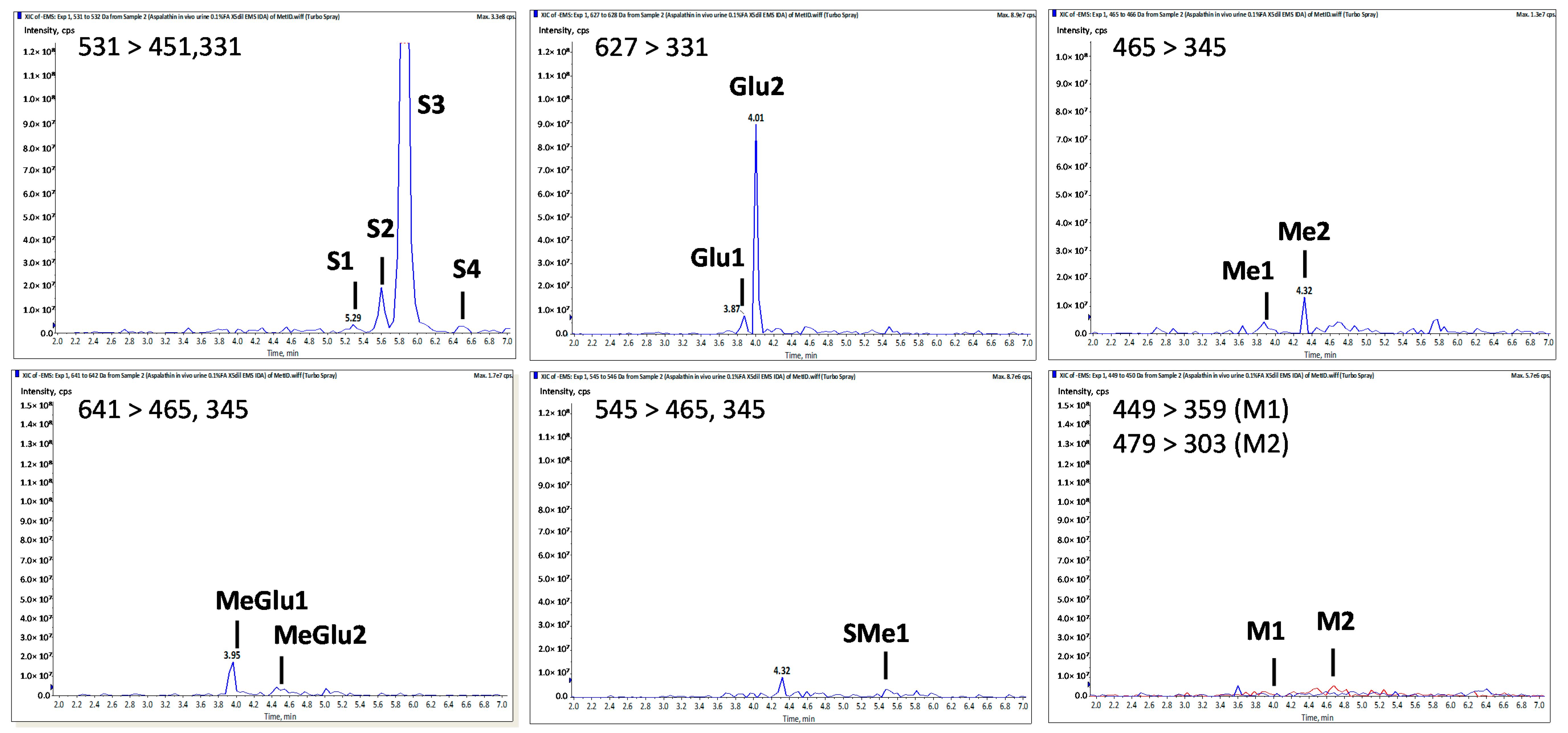
| Peak No. | tR a | [M − H]− (m/z) | MS2 Fragment ioNs (m/z) | Tentative Identity | |||||
|---|---|---|---|---|---|---|---|---|---|
| [M − H − 176]− | [M − H − 80]− | Sugar Moiety Cleavage (0,3X0−, 0,2X0−) | α-β Cleavage (X0α,β A−) | Z0−, Z0α,β, A− | Other Fragments | ||||
| Asp | 4.20 | 451 | 361, 331 | 209, 179 | 289, 167 | 239, 125 | Aspalathin b | ||
| S1 | 5.30 | 531 | 451 | 361, 331 | 289 | Aspalathin-O-SO3H | |||
| S2 | 5.60 | 531 | 451 | 361, 331 | 209, 179 | 289, 167 | 239, 125 | Aspalathin-O-SO3H | |
| S3 | 5.85 | 531 | 451 | 361, 331 | 209, 179 | 289, 167 | 239, 125 | Aspalathin-O-SO3H | |
| S4 | 6.50 | 531 | 361, 331 | Aspalathin-O-SO3H | |||||
| Me1 | 3.90 | 465 | 345 | 209 | 289 | 251 | Me-O-Aspalathin | ||
| Me2 | 4.30 | 465 | 375, 345 | 209, 179 | 303, 167 | 447, 125 | Me-O-Aspalathin | ||
| SMe1 | 5.50 | 545 | 465 | 375, 345 | 303 | Me-O-Aspalathin-O-SO3H | |||
| Glu1 | 3.90 | 627 | 507 | 331 | Aspalathin-O-Gluc | ||||
| Glu2 | 4.00 | 627 | 331, 269 | Aspalathin-O-Gluc | |||||
| MeGlu1 | 3.95 | 641 | 465 | 375, 345 | 209 | 303, 167 | 447, 259, 125 | Me-O-Aspalathin-O-Gluc | |
| MeGlu2 | 4.50 | 641 | 465 | 375, 345 | 209 | 303, 167 | 447, 125 | Me-O-Aspalathin-O-Gluc | |
| M1 | 4.05 | 449 | 359, 329 | 223, 193 | Eriodictyol-C-glucoside | ||||
| M2 | 4.80 | 479 | 303 | 167 | Me-O-tetrahydroxy-dihydrochalcone-O-Gluc | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowles, S.; Joubert, E.; De Beer, D.; Louw, J.; Brunschwig, C.; Njoroge, M.; Lawrence, N.; Wiesner, L.; Chibale, K.; Muller, C. Intestinal Transport Characteristics and Metabolism of C-Glucosyl Dihydrochalcone, Aspalathin. Molecules 2017, 22, 554. https://doi.org/10.3390/molecules22040554
Bowles S, Joubert E, De Beer D, Louw J, Brunschwig C, Njoroge M, Lawrence N, Wiesner L, Chibale K, Muller C. Intestinal Transport Characteristics and Metabolism of C-Glucosyl Dihydrochalcone, Aspalathin. Molecules. 2017; 22(4):554. https://doi.org/10.3390/molecules22040554
Chicago/Turabian StyleBowles, Sandra, Elizabeth Joubert, Dalene De Beer, Johan Louw, Christel Brunschwig, Mathew Njoroge, Nina Lawrence, Lubbe Wiesner, Kelly Chibale, and Christo Muller. 2017. "Intestinal Transport Characteristics and Metabolism of C-Glucosyl Dihydrochalcone, Aspalathin" Molecules 22, no. 4: 554. https://doi.org/10.3390/molecules22040554