
Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy

Abstract
:1. Introduction
2. Identification and Molecular Characterization of the Plasma Membrane Citrate Transporter SLC13A5
3. Cellular and Subcellular Localization of NaCT Protein
4. Functional Differences between Human and Rodent NaCTs
5. Transcriptional Regulation of NaCT Expression
6. Biochemical and Metabolic Phenotype of Slc13a5-Knockout Mouse
7. Functional Loss of SLC13A5 as a Cause of Epilepsy in Humans
8. Molecular Mechanisms Underlying SLC13A5-Associated Epilepsy
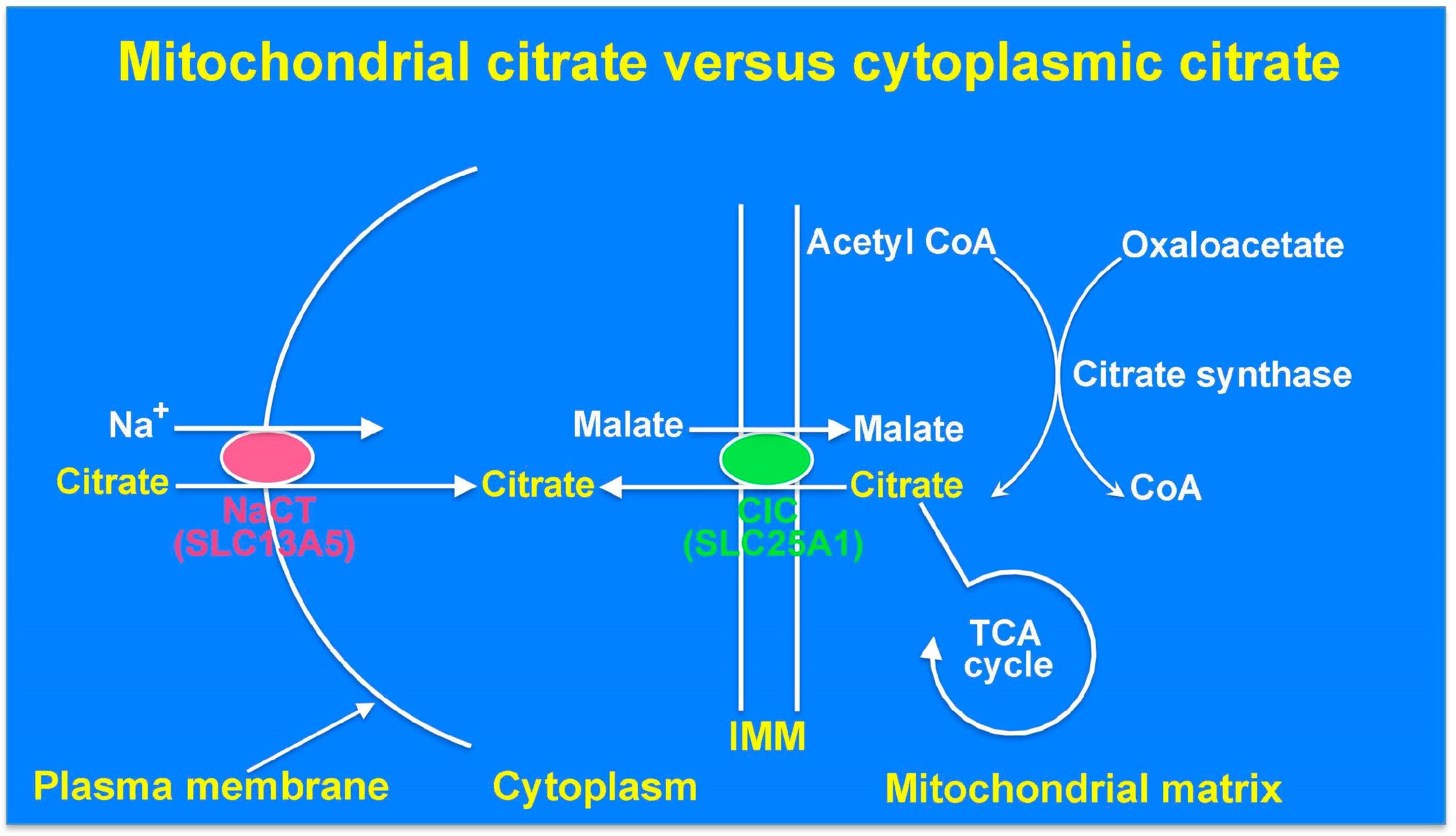
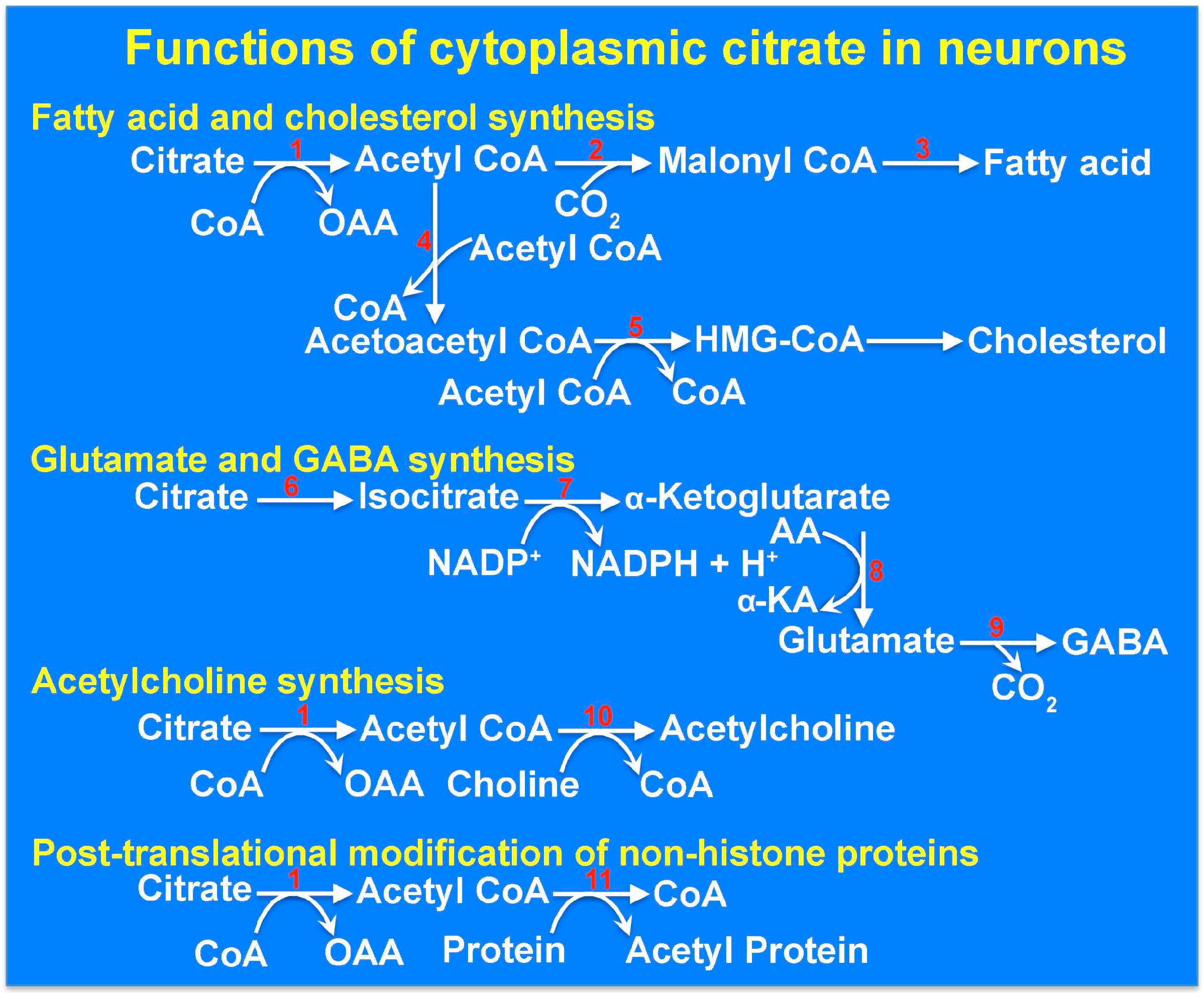
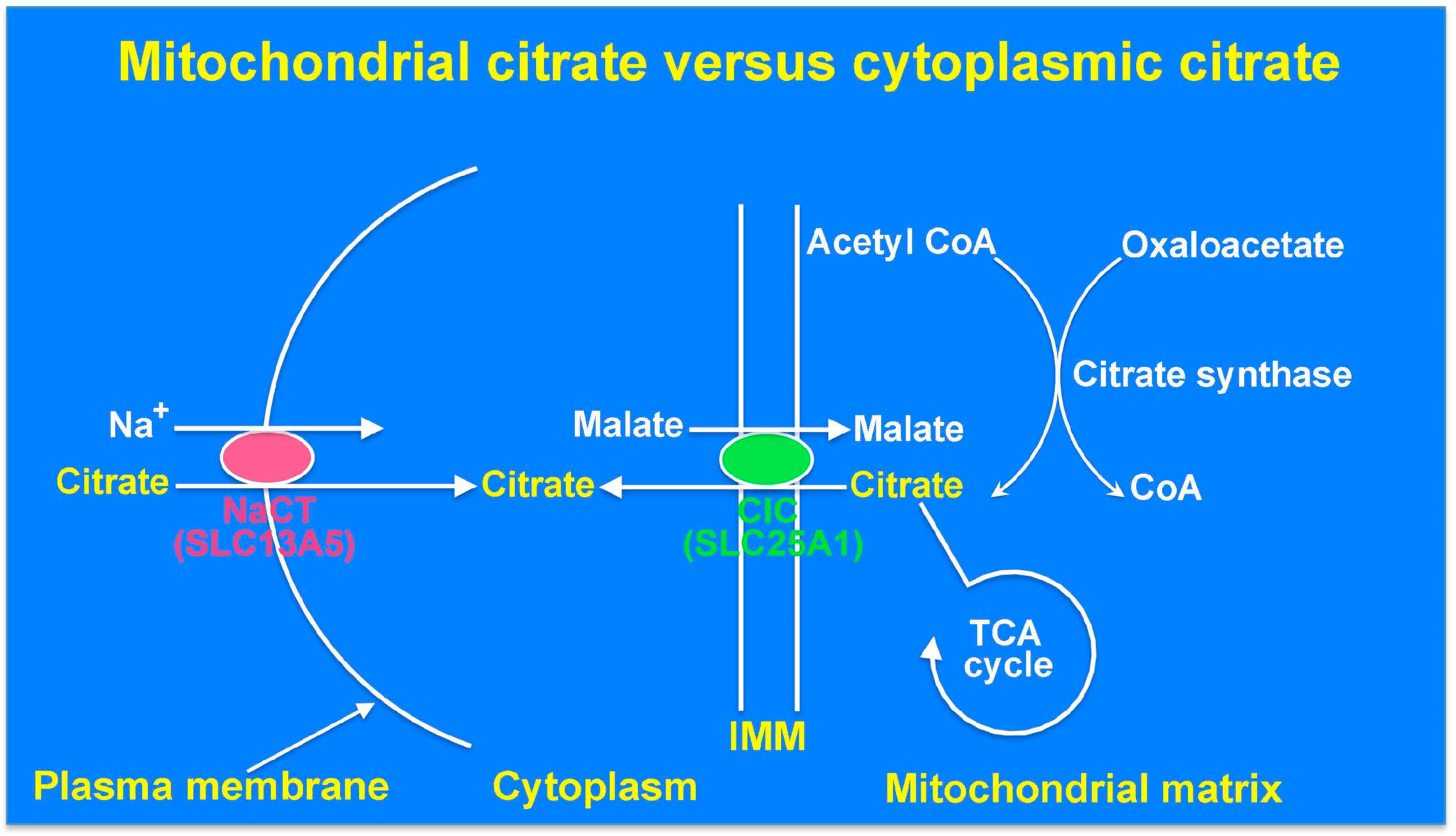
9. The Cytoplasmic Citrate Deficit Hypothesis: Mitochondrial Citrate versus Cytoplasmic Citrate in Relation to Epilepsy
10. The Interneuron Energy Hypothesis: A Mechanism Linking Low Cytoplasmic Citrate to Seizure Susceptibility
11. The Zinc Chelation Hypothesis: A Mechanism Linking High Extracellular Citrate to Seizure Susceptibility
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hodgkinson, A. The relation between citric acid and calcium metabolism with particular reference to primary hyper-parathyroidism and idiopathic hypercalcemia. Clin. Sci. 1963, 24, 167–178. [Google Scholar] [PubMed]
- Fraenkl, S.A.; Muser, J.; Groell, R.; Reinhard, G.; Orgul, S.; Flammer, J.; Goldblum, D. Plasma citrate levels as a potential biomarker for glaucoma. J. Ocul. Pharmacol. Ther. 2011, 27, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Gopal, E.; Martin, P.M.; Itagaki, S.; Miyauchi, S.; Prasad, P.D. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008, 10, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, M.J.; Clemencon, B.; Hediger, M.A.; Markovich, D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol. Asp. Med. 2013, 34, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Forster, M.J. Caloric restriction and the aging process: A critique. Free Radic. Biol. Med. 2014, 73, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Ruetenik, A.; Barrientos, A. Dietary restriction, mitochondrial function and aging: From yeast to humans. Biochim. Biophys. Acta 2015, 1847, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Willmes, D.M.; Birkenfeld, A.L. The role of INDY in metabolic regulation. Comput. Struct. Biotechnol. 2013, 6, e201303020. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.P.; Rogina, B. The role of INDY in metabolism, health and longevity. Front. Genet. 2015, 6, 204. [Google Scholar] [PubMed]
- Inoue, K.; Fei, Y.J.; Huang, W.; Zhuang, L.; Chen, Z.; Ganapathy, V. Functional identity of Drosophila melanogaster Indy as a cation-independent, electroneutral transporter for tricarboxylic acid-cycle intermediates. Biochem. J. 2002, 367, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Knauf, F.; Rogina, B.; Jiang, Z.; Aronson, P.S.; Helfand, S.L. Functional characterization and immunolocalization of the transporter encoded by the life-extending gene Indy. Proc. Natl. Acad. Sci. USA 2002, 99, 14315–14319. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+-coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Inoue, K.; Fei, Y.J.; Zhuang, L.; Gopal, E.; Miyauchi, S.; Ganapathy, V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem. J. 2004, 378, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Babu, E.; Ramachandran, S.; Bhutia, Y.D.; Prasad, P.D.; Ganapathy, V. Species-specific influence of lithium on the activity of SLC13A5 (NaCT): Lithium-induced activation is specific for the transporter in primates. J. Pharmacol. Exp. Ther. 2015, 353, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yodoya, E.; Wada, M.; Shimada, A.; Katsukawa, H.; Okada, N.; Yamamoto, A.; Ganapathy, V.; Fujita, T. Functional and molecular identification of sodium-coupled dicarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. J. Neurochem. 2006, 97, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem. J. 2003, 374, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Silverstone, T. Lithium and weight gain. Int. Clin. Psycopharmacol. 1990, 5, 217–225. [Google Scholar] [CrossRef]
- Baptista, T.; Teneud, L.; Contreras, Q.; Alastre, T.; Burguera, J.L.; de Burguera, M.; de Baptista, E.; Weiss, S.; Hernandez, L. Lithium and body weight gain. Pharmacopsychiatry 1995, 28, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H.; Gaszei, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Neuschafer-Rube, F.; Schraplau, A.; Schewe, B.; Lieske, S.; Krutzfeldt, J.M.; Ringel, S.; Henkel, J.; Birkenfeld, A.L.; Puschel, G.P. Arylhydrocarbon receptor-dependent mIndy (Slc13a5) induction as possible contributor to benzo[a]pyrene-induced lipid accumulation in hepatocytes. Toxicology 2015, 337, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Neuschafer-Rube, F.; Lieske, S.; Kuna, M.; Henkel, J.; Perry, R.J.; Erion, D.M.; Pesta, D.; Willmes, D.M.; Brachs, S.; von Loeffelholz, C.; et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes 2014, 63, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Von Loeffelholz, C.; Lieske, S.; Neuschafer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; Konig, J.; Fromm, M.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homology mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.H.; Perry, R.J.; Guebre-Egziabher, F.; Zhang, D.; Jurczak, M.; Fischer-Rosinsky, A.; Daniels, M.A.; Willmes, D.M.; Bhanot, S.; Bornstein, S.R.; et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging 2015, 7, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Brown, J.; Jones, J.C.; Cabral, S.; Futatsugi, K.; Gorgoglione, M.; Lanba, A.; Vera, N.B.; Zhu, Y.; Yan, Q.; et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 2015, 5, 17391. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Gosset, J.R.; Montgomery, J.I.; Gilbert, A.; Hayward, M.M.; Magee, T.V.; Cabral, S.; Uccello, D.P.; Bahnck, K.; Purkal, J.; et al. Optimization of a dicarboxylic series for in vivo inhibition of citrate transport by the solute carrier 13 (SLC13) family. J. Med. Chem. 2016, 59, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Krane, J.; Unsgard, G.; Petersen, S.B.; Schousboe, A. First direct demonstration of preferential release of citrate from astrocytes using [13C]NMR spectroscopy of cultured neurons and astrocytes. Neurosci. Lett. 1991, 128, 235–239. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Milenkovic, V.M.; Wetzel, C.H.; Rummele, P.; Geissler, E.K. Extracellular citrate in health and disease. Curr. Mol. Med. 2015, 15, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Juge, C.; Roubertie, A.; Heron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Klotz, J.; Porter, B.E.; Colas, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, F.; Eid, T.; Bergersen, L.H. Monocarboxylate transporters in temporal lobe epilepsy: Roles of lactate and ketogenic diet. Brain Struct. Funct. 2015, 220, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wolking, S.; Becker, F.; Bast, T.; Wiemer-Kruel, A.; Mayer, T.; Lerche, H.; Weber, Y.G. Focal epilepsy in glucose transporter type 1 (Glut1) defects: Case reports and a review of literature. J. Neurol. 2014, 261, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Muhlhausen, C.; Salomons, G.S.; Lukacs, Z.; Struys, E.A.; van der Knaap, M.S.; Ullrich, K.; Santer, R. Combined D2-/L2-hydroxyglutaric aciduria (SLC25A1 deficiency): Clinical course and effects of citrate treatment. J. Inherit. Metab. Dis. 2014, 37, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; MaBride, S.; Marcadier, J.L.; Michaud, J.; Al-Dirbashi, O.Y.; Schwartzentruber, J.; Beaulieu, C.L.; Katz, S.L.; Majewski, J.; FORGE Canada Consortium; et al. Severe neonatal presentation of mitochondrial citrate carrier (SLC25A1) deficiency. JIMD Rep. 2016, 30, 73–79. [Google Scholar] [PubMed]
- Li, Z.; Erion, D.M.; Maurer, T.S. Model-based assessment of plasma citrate flux into the liver: Implications for NaCT as a therapeutic target. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Ramautar, R.; Somsen, G.W.; de Jong, G.J. Direct sample injection for capillary electrophoretic determination of organic acids in cerebrospinal fluid. Anal. Bioanal. Chem. 2007, 387, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L. Brain energetics (thought needs food). Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 2016, 138, 14–52. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Kash, S.F.; Johnson, R.S.; Tecott, L.H.; Noebels, J.L.; Mayfield, R.D.; Hanahan, D.; Baekkeskov, S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. USA 1997, 94, 14060–14065. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hornfeldt, C.S.; Larson, A.A. Seizures induced by fluoroacetic acid and fluorocitric acid may involve chelation of divalent cations in the spinal cord. Eur. J. Pharmacol. 1990, 179, 307–313. [Google Scholar] [CrossRef]
- Field, T.; Coburn, J.; McCourt, J.; McBryde, W.A.E. Composition and stability of some metal citrate and diglycolate complexes in aqueous solution. Anal. Chim. Acta 1975, 74, 101–106. [Google Scholar] [CrossRef]
- Glusker, J. Citrate conformation and chelation: Enzymic implications. Acc. Chem. Res. 1980, 13, 345–352. [Google Scholar] [CrossRef]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1997, 17, 5711–5725. [Google Scholar] [PubMed]
- Amico-Ruvio, S.; Murthy, S.; Smith, T.; Popescu, G. Zinc effects on NMDA receptor gating kinetics. Biophys. J. 2011, 100, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Romero-Hernandez, A.; Simorowski, N.; Karakas, E.; Furukawa, H. Molecular basis for subtype specificity and high-affinity zinc inhibition in the GluN1-GluN2A NMDA receptor amino-terminal domain. Neuron 2016, 92, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, N.; Banke, T.; Wahl, P.; Sonnewald, U.; Schousboe, A. Citrate modulates the regulation by Zn2+ of N-methyl-d-aspartate receptor-mediated channel current and neurotransmitter release. Proc. Natl. Acad. Sci. USA 1995, 92, 3367–3370. [Google Scholar] [CrossRef] [PubMed]
- Vergnano, A.M.; Rebola, N.; Savtchenko, L.P.; Pinheiro, P.S.; Casado, M.; Kieffer, B.L.; Rusakov, D.A.; Mulle, C.; Paoletti, P. Zinc dynamics and action at excitatory synapses. Neuron 2014, 82, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K. Zinc in neurotransmission. Nutrition 2011, 31, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Serraz, B.; Grand, T.; Paoletti, P. Altered zinc sensitivity of NMDA receptors harboring clinically-relevant mutations. Neuropharmacology 2016, 109, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.; Hildebrand, M.; Mullen, S.; Hildebrand, J.; Berkovic, S.; Petrou, S. Synaptic Zn2+ and febrile seizure susceptibility. Br. J. Pharmacol. 2017, 174, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Goldschen-Ohm, M.; Haroldson, A.; Jones, M.; Pearce, R. A nonequilibrium binary elements-based kinetic model for benzodiazepine regulation of GABAA receptors. J. Gen. Physiol. 2014, 144, 27–39. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Intermediates | Concentration |
|---|---|
| Tricarboxylates | |
| Citrate (10 ATP/mole) | ~160 μM |
| Isocitrate (10 ATP/mole) | <10 μM |
| Dicarboxylates | |
| α-Ketoglutarate (7.5 ATP/mole) | ~10 μM |
| Succinate (4 ATP/mole) | ~40 μM |
| Fumarate (2.5 ATP/mole) | <10 μM |
| Malate (2.5 ATP/mole) | ~35 μM |
| Oxaloacetate | <10 μM |
| Monocarboxylates | |
| Lactate (15 ATP/mole) | ~1 mM |
| Pyruvate (12.5 ATP/mole) | ~70 μM |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. https://doi.org/10.3390/molecules22030378
Bhutia YD, Kopel JJ, Lawrence JJ, Neugebauer V, Ganapathy V. Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules. 2017; 22(3):378. https://doi.org/10.3390/molecules22030378
Chicago/Turabian StyleBhutia, Yangzom D., Jonathan J. Kopel, John J. Lawrence, Volker Neugebauer, and Vadivel Ganapathy. 2017. "Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy" Molecules 22, no. 3: 378. https://doi.org/10.3390/molecules22030378