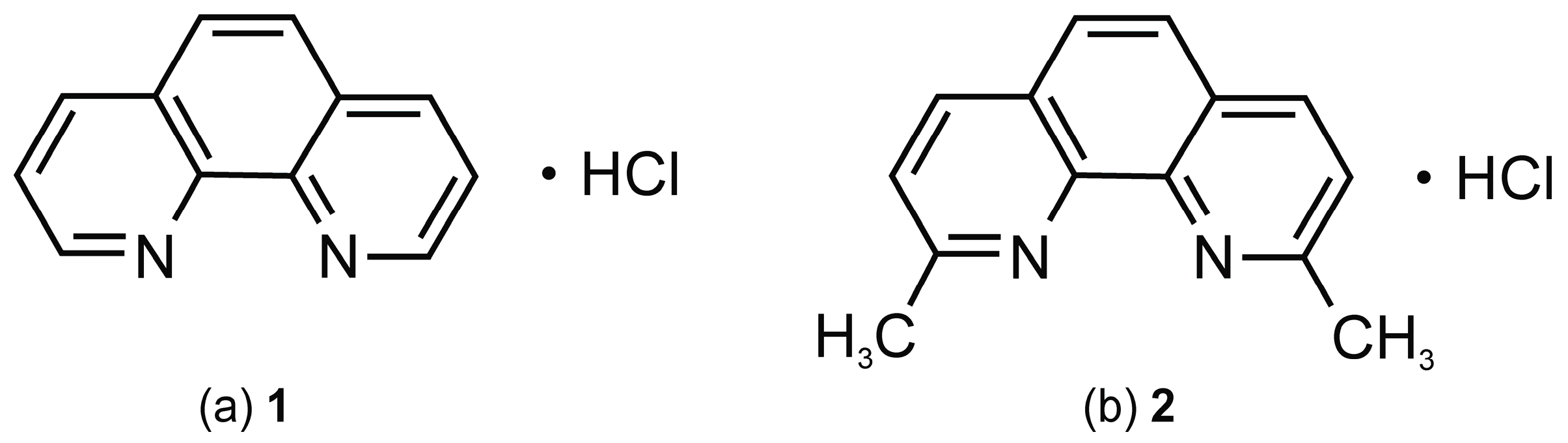
New Insights into Solid Form Stability and Hydrate Formation: o-Phenanthroline HCl and Neocuproine HCl

Abstract
1. Introduction
2. Results
2.1. Thermal Behavior of the Hydrates of o-Phenanthroline HCl and Neocuproine HCl
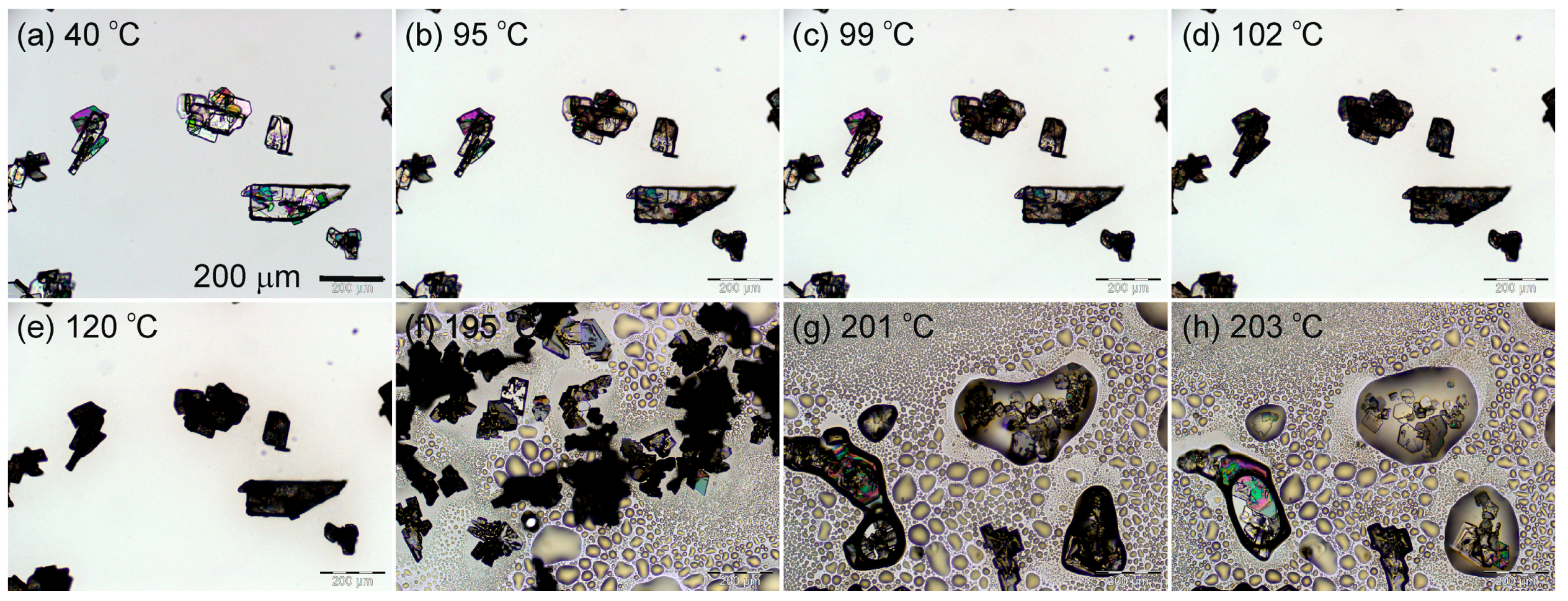
2.1.1. Hot-Stage Microscopy
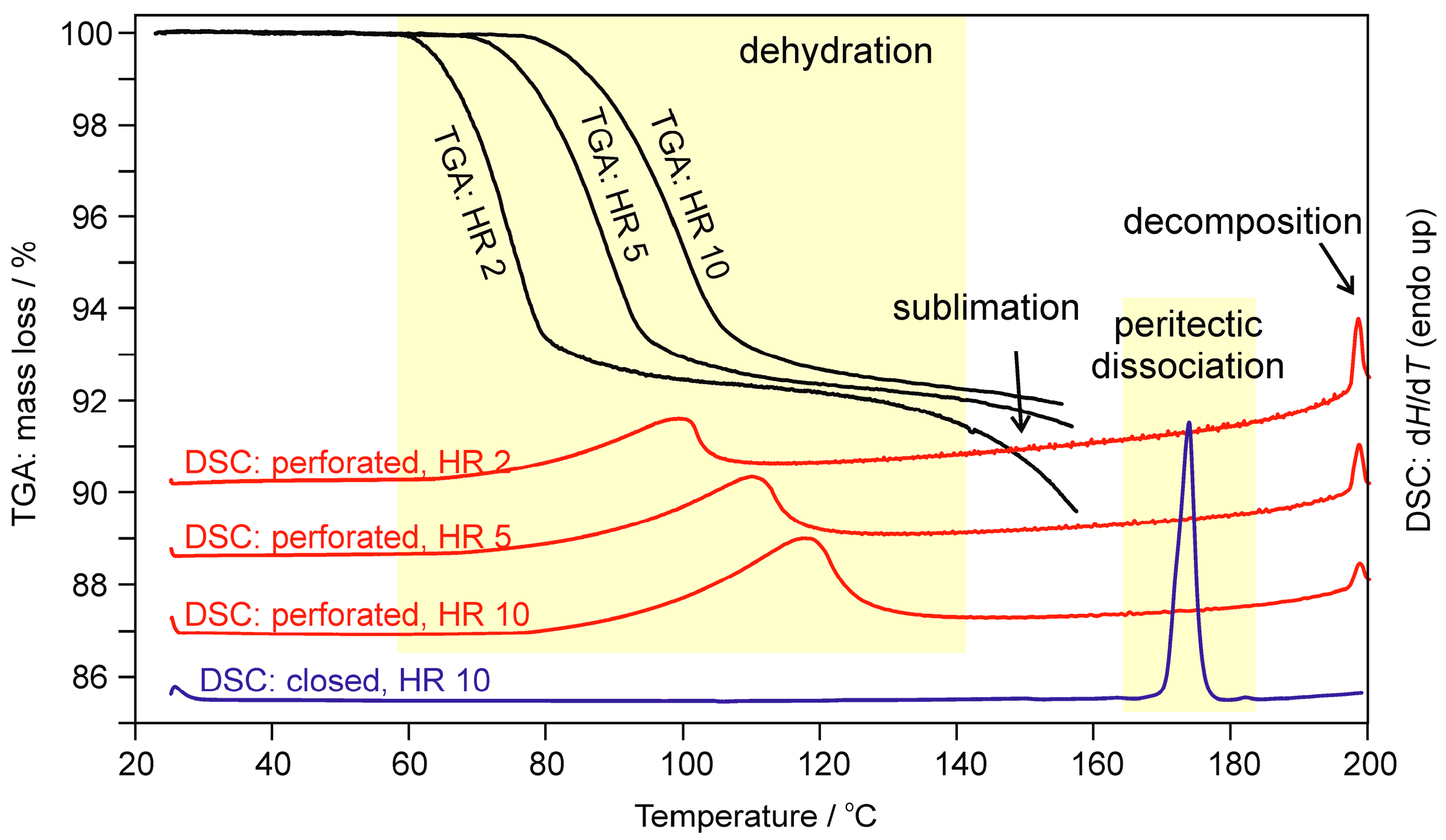
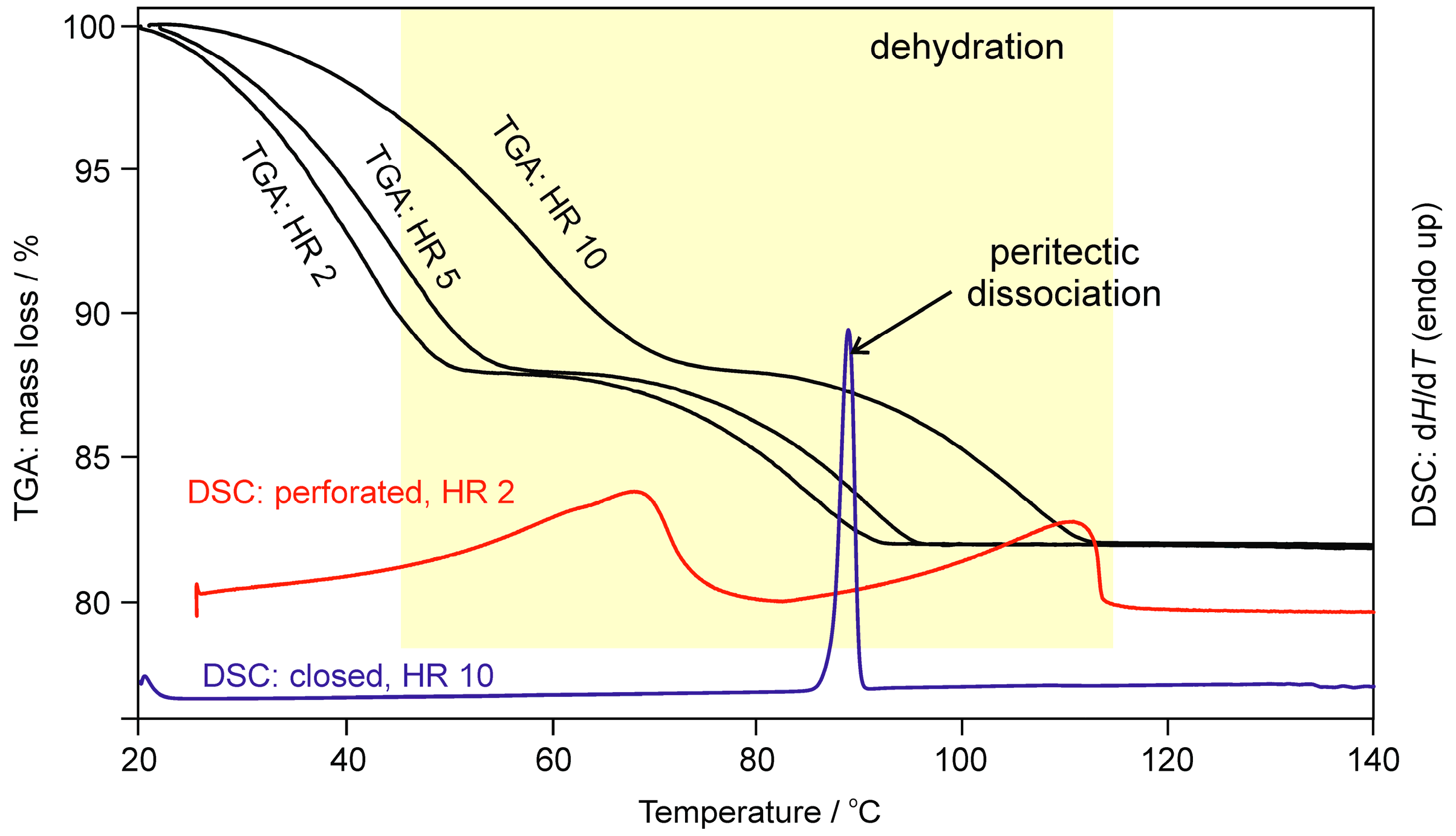
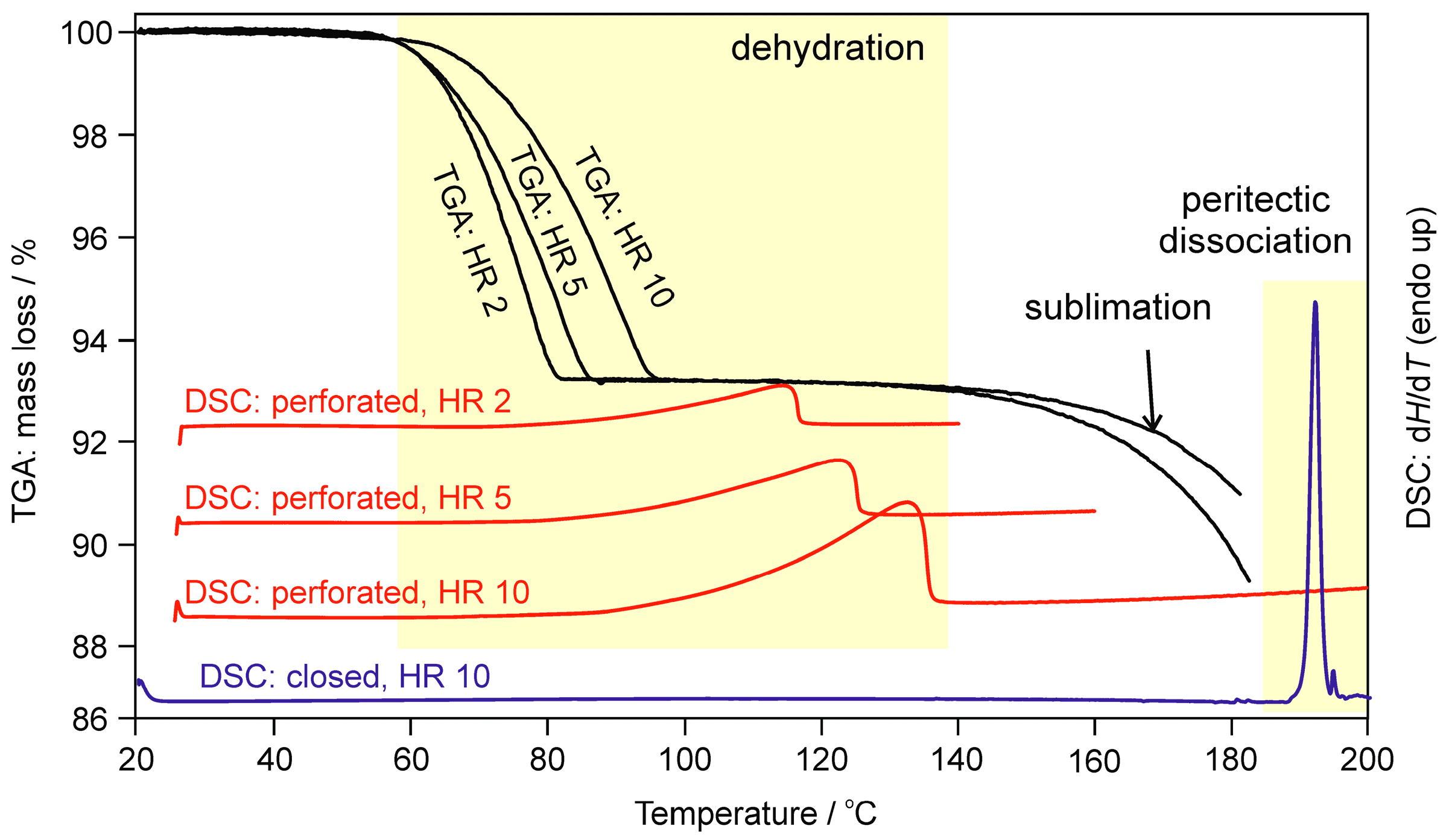
2.1.2. Differential Scanning Calorimetry and Thermogravimetric Analysis
2.2. Moisture-Dependent Phase Transformations
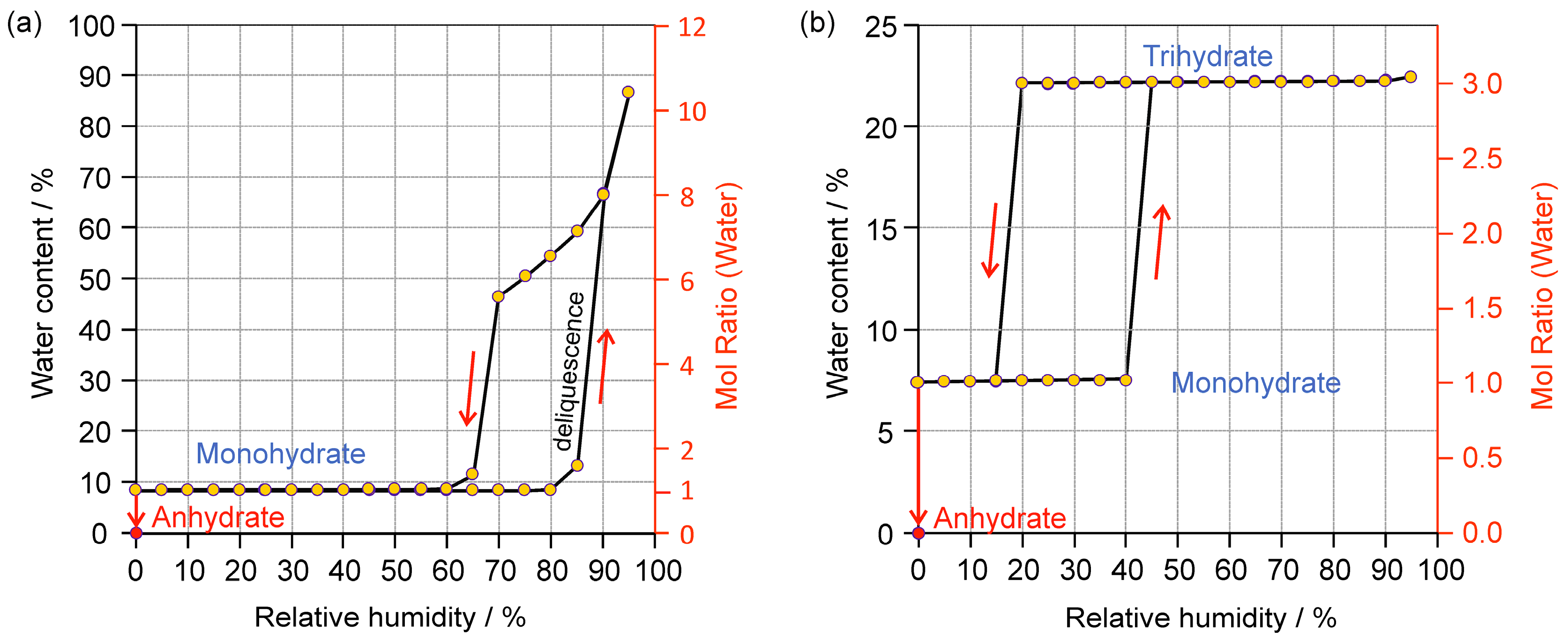
2.2.1. Gravimetric Moisture Sorption/Desorption Experiments
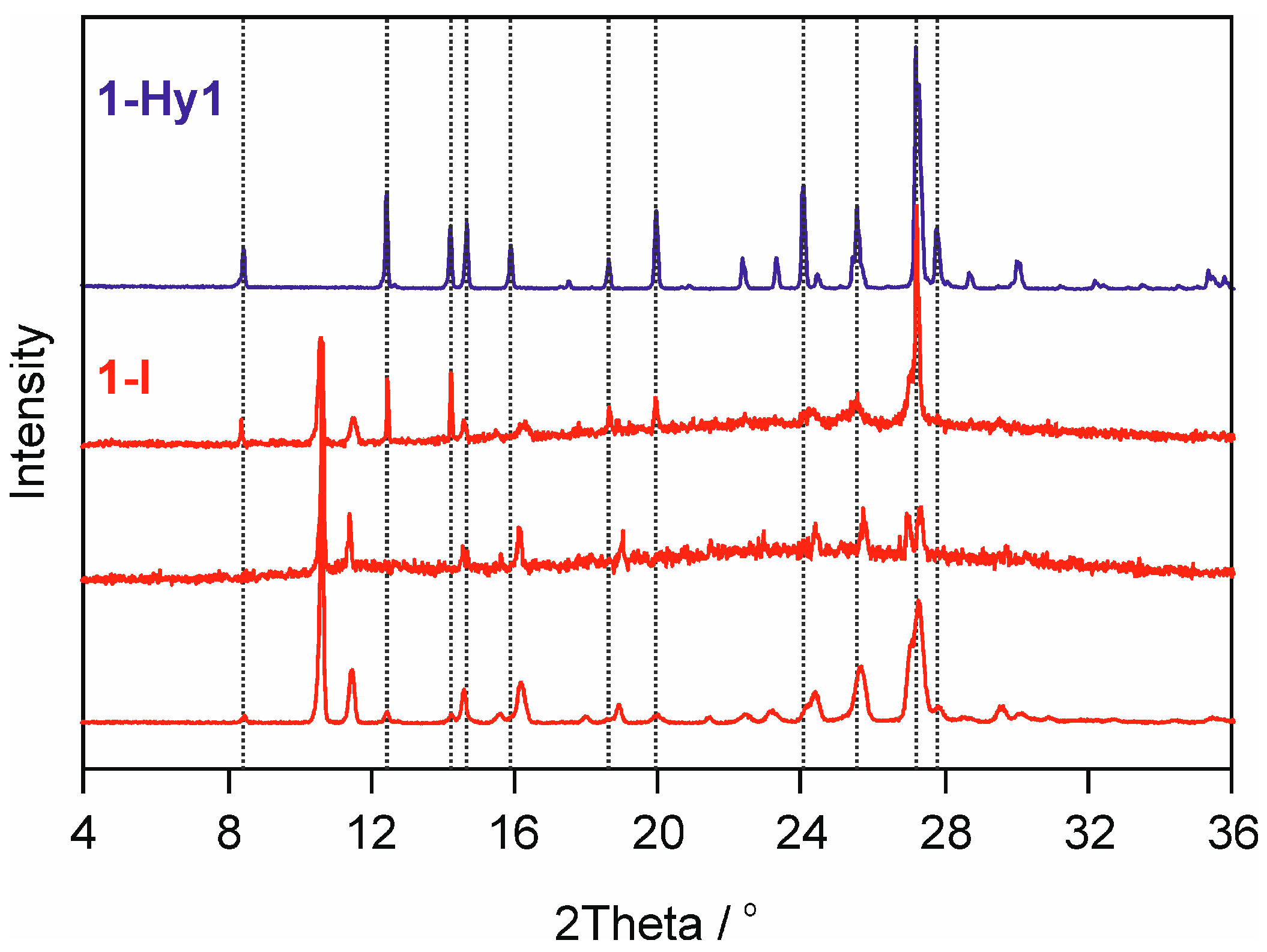
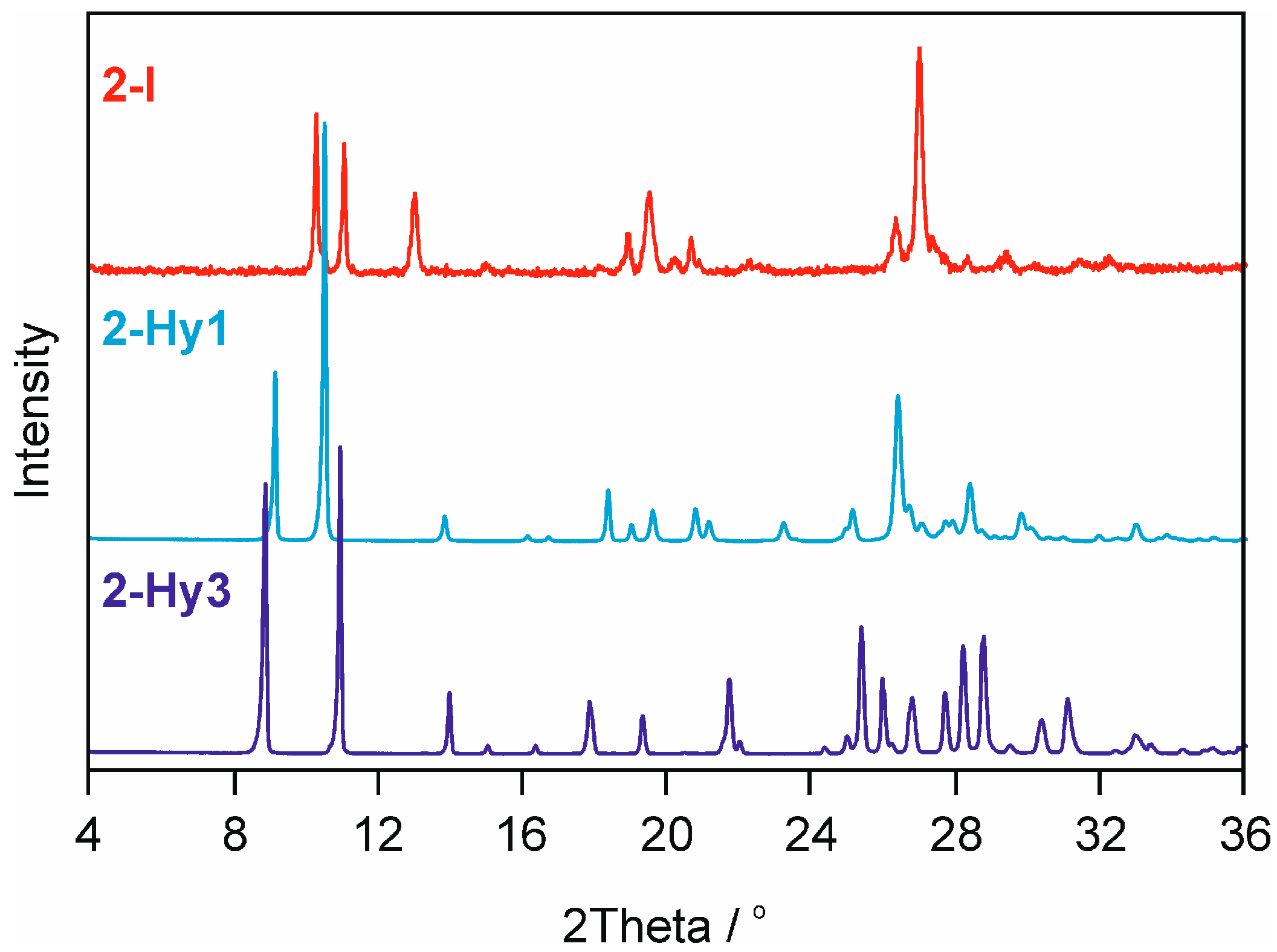
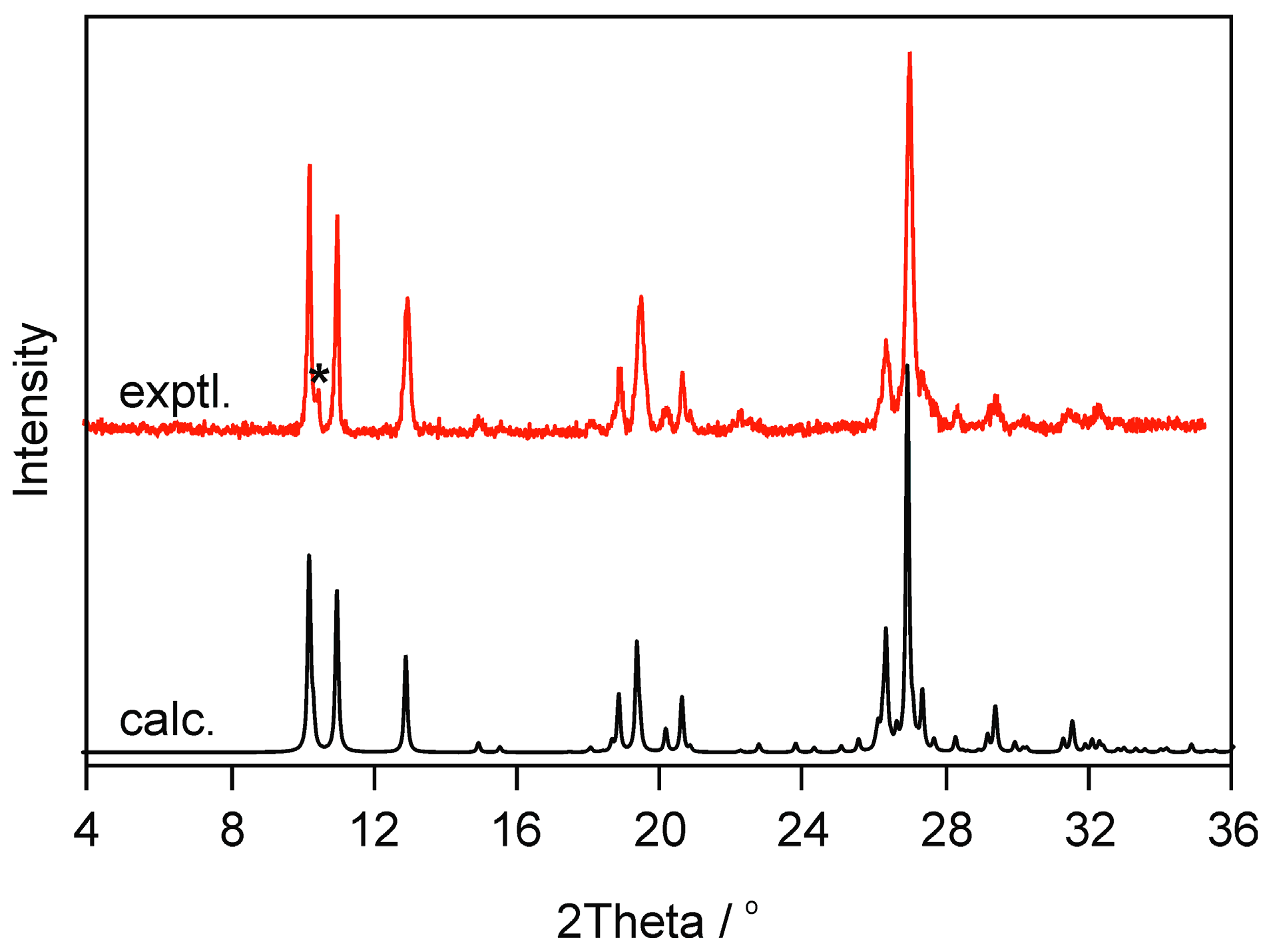
2.2.2. Moisture Controlled PXRD Experiments
2.3. Crystal Structures and Intermolecular Energy Calculations
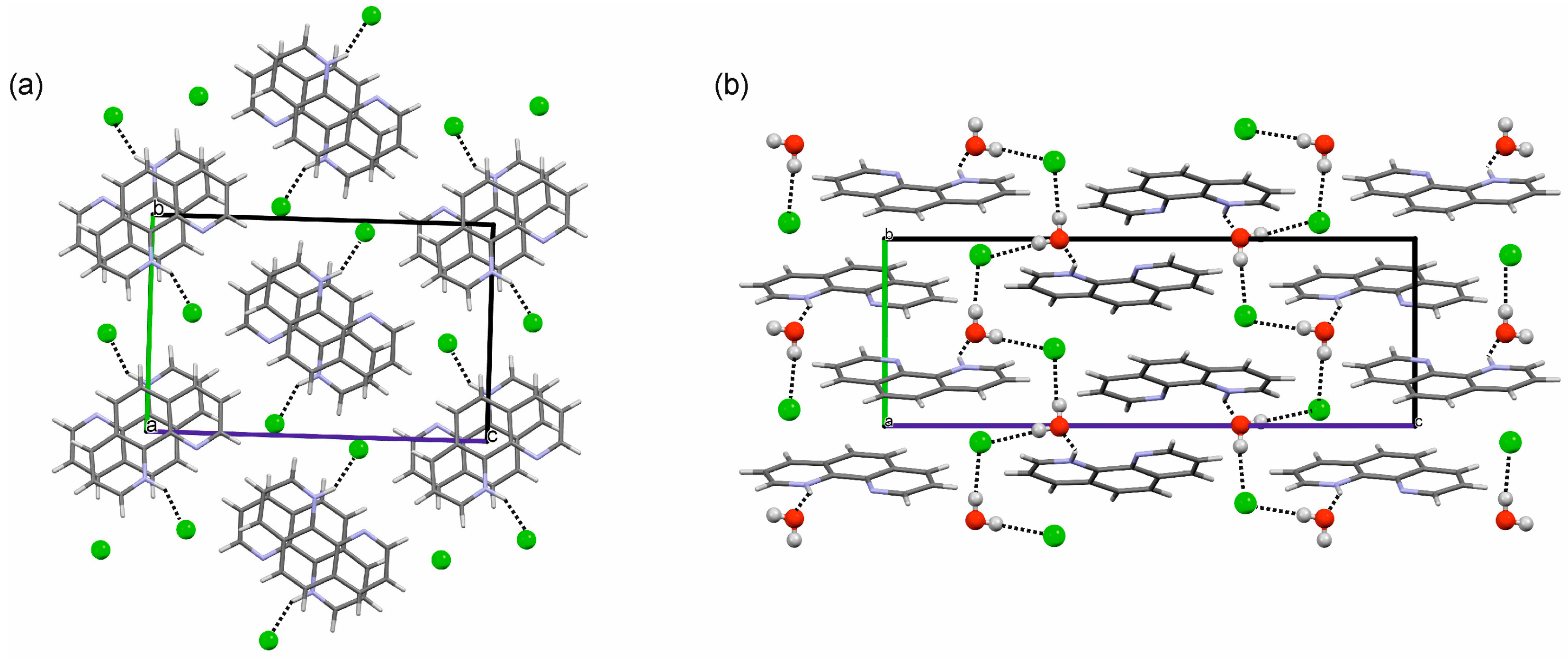
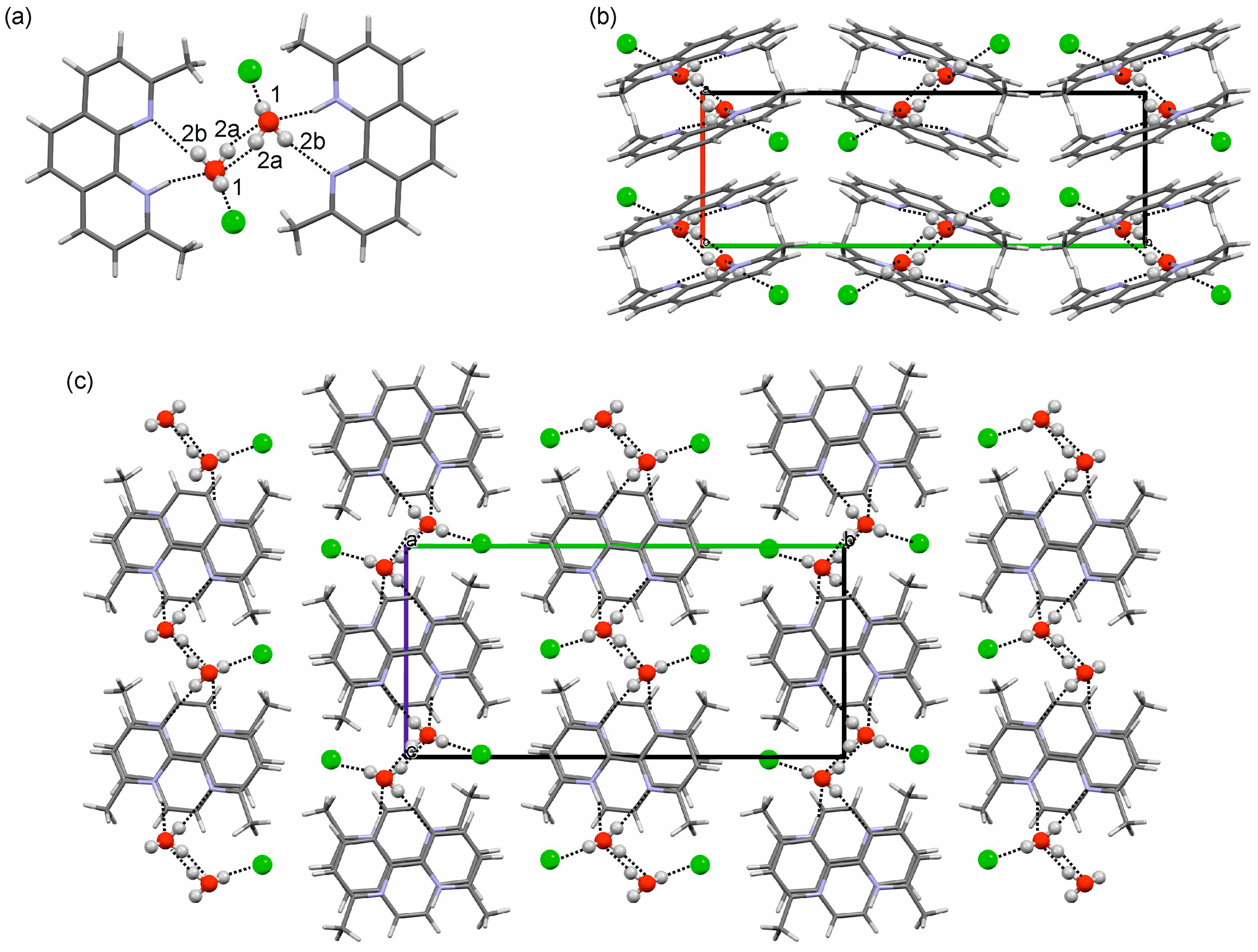
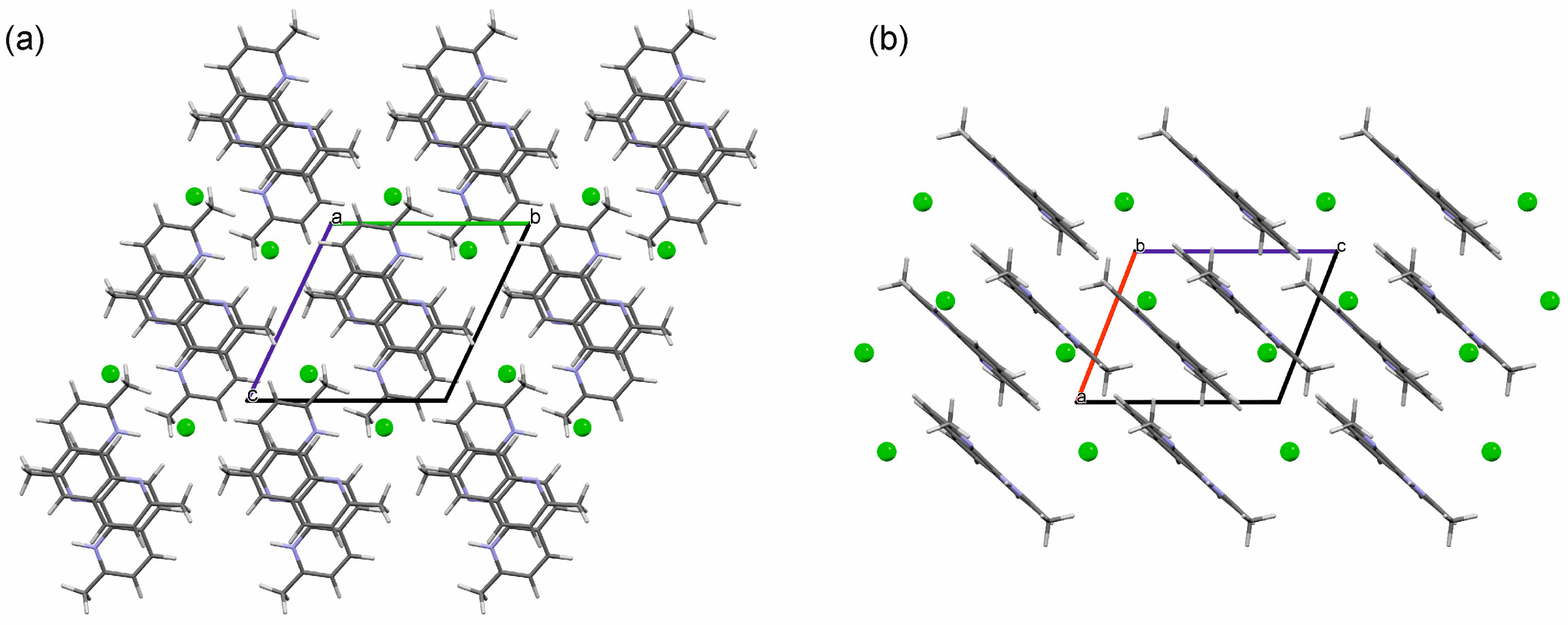
2.3.1. o-Phenanthroline HCl Crystal Structures
2.3.2. Neocuproine HCl Monohydrate Crystal Structure
2.3.3. Computationally Generated Neocuproine HCl Anhydrate Structures
2.4. Stability of Hydrates Estimated from Lattice Energy Calculations
3. Discussion
3.1. Hydrate Formation and Structural Role of Water in Phenanthrolinium Based HCl Salts
3.2. Stability and Handling of o-Phenanthroline HCl and Neocuproine HCl Solid-State Froms
3.3. Role of Computational Chemistry for Characterizing Organic Solid Forms
4. Materials and Methods
4.1. Materials and Preparation of Solid Forms
4.2. Thermal Analysis
4.3. Gravimetric Moisture Sorption/Desorption Experiments
4.4. Powder and Single-Crystal X-Ray Diffraction
4.5. Computational Generation of the Neocuproine HCl Anhydrate Crystal Energy Landscape
4.6. Modeling of the Pairwise Intermolecular Interactions
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| aw | water activity |
| CE-B3LYP | CrystalExplorer B3LYP calculations |
| CSD | Cambridge Structural Database |
| CSP | crystal structure prediction |
| Dehy | isostructural dehydrate |
| DFT-D (PBE-TS/D2) | dispersion corrected density functional calculations (Perdew-Burke-Ernzerhof, Tkatchenko and Scheffler/Grimme D2 dispersion correction) |
| DSC | differential scanning calorimetry |
| Elatt | lattice energy |
| HCl | hydrochloride |
| HSM | hot-stage microscopy |
| P2O5 | phosphorus pentoxide |
| PI | packing index |
| PXRD | powder X-ray diffraction |
| RH | relative humidity |
| RT | room temperature |
| SCXRD | single-crystal X-ray diffraction |
| TGA | thermogravimetric analysis |
| ∆U | potential energy difference |
| 1 | o-phenanthroline hydrochloride |
| 2 | neocuproine hydrochloride |
| I | anhydrate |
| Hy1 | monohydrate |
| Hy3 | trihydrate |
References and Note
- Bernstein, J. Polymorphism in Molecular Crystals; Clarendon Press: Oxford, UK, 2002. [Google Scholar] [CrossRef]
- Hilfiker, R. Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar] [CrossRef]
- Tian, F.; Qu, H.; Zimmermann, A.; Munk, T.; Joergensen, A.C.; Rantanen, J. Factors affecting crystallization of hydrates. J. Pharm. Pharmacol. 2010, 62, 1534–1546. [Google Scholar] [CrossRef] [PubMed]
- Rager, T.; Geoffroy, A.; Hilfiker, R.; Storey, J.M.D. The crystalline state of methylene blue: A zoo of hydrates. Phys. Chem. Chem. Phys. 2012, 14, 8074–8082. [Google Scholar] [CrossRef] [PubMed]
- Gal, S. Phenomenological study of water vapor sorption isotherms of solid sorbents. Chimia 1968, 22, 409–425. [Google Scholar]
- Griesser, U.J. The importance of solvates. In Polymorphism: In the Pharmaceutical Industry; Hilfiker, R., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 211–233. [Google Scholar]
- Proell, F. Thermomikroskopische und Differentialthermoanalytische Untersuchungen von Hydraten. Ph.D. Thesis, University of Innsbruck, Innsbruck, Austria, 1978. [Google Scholar]
- O’Neil, M.J. (Ed.) The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 14th ed.; Merck and Co., Inc.: Whitehouse Station, NJ, USA, 2006. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Hensen, K.; Spangenberg, B.; Bolte, M. Two phenanthroline hydrochlorides. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2000, C56, 208–210. [Google Scholar] [CrossRef]
- Thevenet, G.; Toffoli, P.; Rodier, N.; Ceolin, R. Crystal structure of o-phenanthroline hydrochloride and hydrobromide, C12H8N2.HX.H2O (X = Cl or Br). Acta Crystallogr. Sect. B 1977, B33, 2526–2529. [Google Scholar] [CrossRef]
- Fronczek, F.R. 1,10-Phenanthrolin-1-ium Chloride Monohydrate. Personal Communication, 2002.
- Braga, D.; Grepioni, F.; Maini, L.; Prosperi, S.; Gobetto, R.; Chierotti, M.R. From unexpected reactions to a new family of ionic co-crystals: The case of barbituric acid with alkali bromides and cesium iodide. Chem. Commun. 2010, 46, 7715–7717. [Google Scholar] [CrossRef] [PubMed]
- Hensen, K.; Pullmann, P.; Bats, J.W. Crystal and Molecular-Structures of Chlorotrispyridinium-Bis-(Tetrachloroaluminate(Iii)) and A New Modification of Pyridinium Chloride. Z. Anorg. Allg. Chem. 1988, 556, 62–69. [Google Scholar] [CrossRef]
- Buttery, J.H.; Junk, P.C.; Mutrofin, S.; Skelton, B.W.; Whitaker, C.R.; White, A.H. The structural systematics of protonation of some important nitrogen-base ligands, IV. Some ethane-1,2-diaminium univalent anion salt/1,10-phenanthroline (hydrate) arrays. Z. Anorg. Allg. Chem. 2006, 632, 1326–1339. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Griesser, U.J. Insights into Hydrate Formation and Stability of Morphinanes from a Combination of Experimental and Computational Approaches. Mol. Pharm. 2014, 11, 3145–3163. [Google Scholar] [CrossRef] [PubMed]
- Brittain, H.G.; Morris, K.R.; Boerrigter, S.X.M. Polymorphism in Pharmaceutical Solids; Brittain, H.G., Ed.; Informa Healthcare: New York, NY, USA; London, UK, 2009; Volume 192, pp. 233–281. [Google Scholar]
- Price, S.L. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2003. [Google Scholar]
- Chisholm, J.A.; Motherwell, S. COMPACK: A program for identifying crystal structure similarity using distances. J. Appl. Crystallogr. 2005, 38, 228–231. [Google Scholar] [CrossRef]
- Whalley, E. The difference in the intermolecular forces on H2O and D2O. Trans. Faraday Soc. 1957, 53, 1578–1585. [Google Scholar] [CrossRef]
- Whalley, E. The hydrogen bond in ice. Hydrog. Bond 1976, 3, 1425–1470. [Google Scholar]
- Thierfelder, C.; Hermann, A.; Schwerdtfeger, P.; Schmidt, W.G. Strongly bonded water monomers on the ice Ih basal plane: Density-functional calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74, 045422. [Google Scholar] [CrossRef]
- Beran, G.J.O.; Nanda, K. Predicting organic crystal lattice energies with chemical accuracy. J. Phys. Chem. Lett. 2010, 1, 3480–3487. [Google Scholar] [CrossRef]
- Braun, D.E.; Oberacher, H.; Arnhard, K.; Orlova, M.; Griesser, U.J. 4-Aminoquinaldine monohydrate polymorphism: Prediction and impurity aided discovery of a difficult to access stable form. CrystEngComm 2016, 18, 4053–4067. [Google Scholar] [CrossRef] [PubMed]
- Infantes, L.; Fabian, L.; Motherwell, W.D.S. Organic crystal hydrates: What are the important factors for formation. CrystEngComm 2007, 9, 65–71. [Google Scholar] [CrossRef]
- Thevenet, G.; Rodier, N. Crystal structure of o-phenanthrolinium nitrate monohydrate-o-phenanthroline (C12H9N2)NO3.H2O.C12H8N2. Bull. Soc. Chim. Fr. 1981, 11–12, 437–440. [Google Scholar]
- Seo, D.M.; Boyle, P.D.; Allen, J.L.; Han, S.-D.; Jonsson, E.; Johansson, P.; Henderson, W.A. Solvate Structures and Computational/Spectroscopic Characterization of LiBF4 Electrolytes. J. Phys. Chem. C 2014, 118, 18377–18386. [Google Scholar] [CrossRef]
- Braun, D.E. Crystal Polymorphism of Drug Compounds: Statistical Aspects, Analytical Strategies and Case Studies. Ph.D. Thesis, University of Innsbruck, Innsbruck, Austria, 2008. [Google Scholar]
- Aaltonen, J.; Alleso, M.; Mirza, S.; Koradia, V.; Gordon, K.C.; Rantanen, J. Solid form screening—A review. Eur. J. Pharm. Biopharm. 2009, 71, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.E.; Koztecki, L.H.; McMahon, J.A.; Price, S.L.; Reutzel-Edens, S.M. Navigating the Waters of Unconventional Crystalline Hydrates. Mol. Pharm. 2015, 12, 3069–3088. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.R.; Griesser, U.J.; Eckhardt, C.J.; Stowell, J.G. Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Adv. Drug Deliv. Rev. 2001, 48, 91–114. [Google Scholar] [CrossRef]
- Petrova, R.I.; Peresypkin, A.; Mortko, C.J.; McKeown, A.E.; Lee, J.; Williams, J.M. Rapid conversion of API hydrates to anhydrous forms in aqueous media. J. Pharm. Sci. 2009, 98, 4111–4118. [Google Scholar] [CrossRef] [PubMed]
- Baias, M.; Dumez, J.N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [PubMed]
- Selent, M.; Nyman, J.; Roukala, J.; Ilczyszyn, M.; Oilunkaniemi, R.; Bygrave, P.J.; Laitinen, R.; Jokisaari, J.; Day, G.M.; Lantto, P. Clathrate Structure Determination by Combining Crystal Structure Prediction with Computational and Experimental 129Xe NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5386. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Hejczyk, K.E.; Bithell, E.G.; Day, G.M.; Jones, W. Polymorph Identification and Crystal Structure Determination by a Combined Crystal Structure Prediction and Transmission Electron Microscopy Approach. Chem. Eur. J. 2013, 19, 7874–7882. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L.; Braun, D.E.; Reutzel-Edens, S.M. Can computed crystal energy landscapes help understand pharmaceutical solids? Chem. Commun. 2016, 52, 7065–7077. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Day, G.M. Static and lattice vibrational energy differences between polymorphs. CrystEngComm 2015, 17, 5154–5165. [Google Scholar] [CrossRef]
- Markvardsen, A.J.; David, W.I.F.; Johnson, J.C.; Shankland, K. A probabilistic approach to space-group determination from powder diffraction data. Acta Crystallogr. Sect. A Found. Crystallogr. 2001, A57, 47–54. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-Cell Refinement from Powder Diffraction Scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Coelho, A.A. Topas Academic V5; Coelho Software: Brisbane, Australia, 2012. [Google Scholar]
- Oxford Diffraction Ltd. CrysAlis CCD and CrysAlis RED; Version 1.171.31.5; Oxford Diffraction Ltd.: Abingdon, UK, 2006. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, J.M.A.; Cheeseman, R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Gaussian Inc.: Wallingford, CT, USA, 2009.
- Turner, M.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Karamertzanis, P.G.; Pantelides, C.C. Ab initio crystal structure prediction—I. Rigid molecules. J. Comput. Chem. 2005, 26, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Karamertzanis, P.G.; Pantelides, C.C. Ab initio crystal structure prediction. II. Flexible molecules. Mol. Phys. 2007, 105, 273–291. [Google Scholar] [CrossRef]
- Habgood, M.; Sugden, I.J.; Kazantsev, A.V.; Adjiman, C.S.; Pantelides, C.C. Efficient Handling of Molecular Flexibility in Ab Initio Generation of Crystal Structures. J. Chem. Theory Comput. 2015, 11, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Coombes, D.S.; Price, S.L.; Willock, D.J.; Leslie, M. Role of Electrostatic Interactions in Determining the Crystal Structures of Polar Organic Molecules. A Distributed Multipole Study. J. Phys. Chem. 1996, 100, 7352–7360. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-Centered Monopoles From Molecular Electrostatic Potentials—The Need For High Sampling Density in Formamide Conformational-Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Price, S.L.; Leslie, M.; Welch, G.W.A.; Habgood, M.; Price, L.S.; Karamertzanis, P.G.; Day, G.M. Modelling organic crystal structures using distributed multipole and polarizability-based model intermolecular potentials. Phys. Chem. Chem. Phys. 2010, 12, 8478–8490. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.J. Distributed multipole analysis: Stability for large basis sets. J. Chem. Theory Comput. 2005, 1, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.J. GDMA: A Program for Performing Distributed Multipole Analysis of Wave Functions Calculated Using the Gaussian Program System; University of Cambridge: Cambridge, UK, 2010. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID a | Space Group | a (Å) | b (Å) | c (Å) | 𝛂 (°) | 𝛃 (°) | 𝛄 (°) | Elatt (kJ mol−1) | ∆Elatt (kJ mol−1) | PI b (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1_745 | P | 7.119 | 9.504 | 9.870 | 113.68 | 108.59 | 92.14 | −538.7 | 0.00 | 76.5 |
| 2_106 | P21/n | 9.474 | 7.345 | 16.370 | 90 | 92.53 | 90 | −537.1 | 1.63 | 76.4 |
| 3_3345 | P | 7.009 | 9.543 | 10.217 | 65.64 | 75.28 | 68.69 | −536.9 | 1.81 | 75.4 |
| 4_3308 | P21/n | 10.750 | 6.671 | 16.143 | 90 | 94.42 | 90 | −535.3 | 3.44 | 75.3 |
| 5_4999 | P | 6.945 | 9.461 | 10.265 | 114.74 | 103.22 | 98.47 | −534.7 | 4.04 | 75.8 |
| 6_4078 | C2/c | 18.783 | 9.468 | 13.392 | 90 | 104.40 | 90 | −533.3 | 5.39 | 75.4 |
| 7_343 | C2/c | 16.778 | 10.571 | 13.379 | 90 | 102.80 | 90 | −533.2 | 5.50 | 75.3 |
| 8_399 | P | 7.185 | 9.520 | 10.059 | 113.62 | 92.29 | 110.70 | −532.8 | 5.89 | 75.3 |
| 9_5489 | P21/n | 6.570 | 18.550 | 9.580 | 90 | 97.76 | 90 | −532.7 | 6.02 | 75.4 |
| 10_268 | P21/n | 6.813 | 10.484 | 16.581 | 90 | 100.95 | 90 | −532.6 | 6.17 | 74.7 |
| 11_395 | I2/a | 13.158 | 10.074 | 17.514 | 90 | 94.58 | 90 | −532.3 | 6.44 | 75.3 |
| 12_790 | P212121 | 6.637 | 10.073 | 17.329 | 90 | 90.00 | 90 | −531.9 | 6.83 | 75.1 |
| 13_212 | C2/c | 16.705 | 10.680 | 13.247 | 90 | 101.63 | 90 | −531.8 | 6.90 | 75.3 |
| 14_139 | P21/n | 7.256 | 16.884 | 9.469 | 90 | 94.08 | 90 | −531.7 | 7.00 | 75.2 |
| 15_136 | P21/n | 10.027 | 6.656 | 17.567 | 90 | 95.77 | 90 | −531.3 | 7.46 | 74.3 |
| 16_2716 | Pna21 | 6.627 | 16.767 | 10.463 | 90 | 90 | 90 | −531.2 | 7.54 | 74.8 |
| Structure | Elatt/kJ mol−1 | ∆Utrs (Hy1→I) a/kJ mol−1 | ∆Utrs (Hy1→dehy) a/kJ mol−1 | ∆Utrs (dehy→1) b/kJ mol−1 | ||||
|---|---|---|---|---|---|---|---|---|
| PBE-TS c | PBE-D2 d | PBE-TS c | PBE-D2 d | PBE-TS c | PBE-D2 d | PBE-TS c | PBE-D2 d | |
| 1-I | −509.67 | −480.91 | −29.28 | −29.05 | − | − | 16.47 | 14.56 |
| 1-dehy | −493.20 | −466.35 | − | − | −45.75 | −43.61 | − | − |
| 1-Hy1 | −597.95 | −568.96 | − | − | − | − | − | − |
| 2-I | −538.72 | −529.93 | −19.98 | −20.79 | − | − | 14.06 | 12.56 |
| 2-dehy | −524.66 | −517.37 | − | − | −45.75 | −43.61 | − | − |
| 2-Hy1 | −617.70 | −609.72 | − | − | − | − | − | − |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, D.E.; Raabe, K.; Schneeberger, A.; Kahlenberg, V.; Griesser, U.J. New Insights into Solid Form Stability and Hydrate Formation: o-Phenanthroline HCl and Neocuproine HCl. Molecules 2017, 22, 2238. https://doi.org/10.3390/molecules22122238
Braun DE, Raabe K, Schneeberger A, Kahlenberg V, Griesser UJ. New Insights into Solid Form Stability and Hydrate Formation: o-Phenanthroline HCl and Neocuproine HCl. Molecules. 2017; 22(12):2238. https://doi.org/10.3390/molecules22122238
Chicago/Turabian StyleBraun, Doris E., Katharina Raabe, Anna Schneeberger, Volker Kahlenberg, and Ulrich J. Griesser. 2017. "New Insights into Solid Form Stability and Hydrate Formation: o-Phenanthroline HCl and Neocuproine HCl" Molecules 22, no. 12: 2238. https://doi.org/10.3390/molecules22122238
APA StyleBraun, D. E., Raabe, K., Schneeberger, A., Kahlenberg, V., & Griesser, U. J. (2017). New Insights into Solid Form Stability and Hydrate Formation: o-Phenanthroline HCl and Neocuproine HCl. Molecules, 22(12), 2238. https://doi.org/10.3390/molecules22122238