A Diastereoselective Synthesis of Dispiro[oxindole-cyclohexanone]pyrrolidines by 1,3-Dipolar Cycloaddition


Abstract
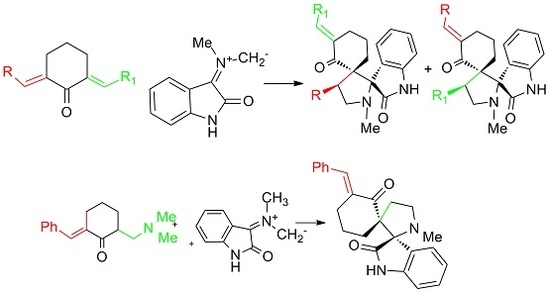
:
1. Introduction
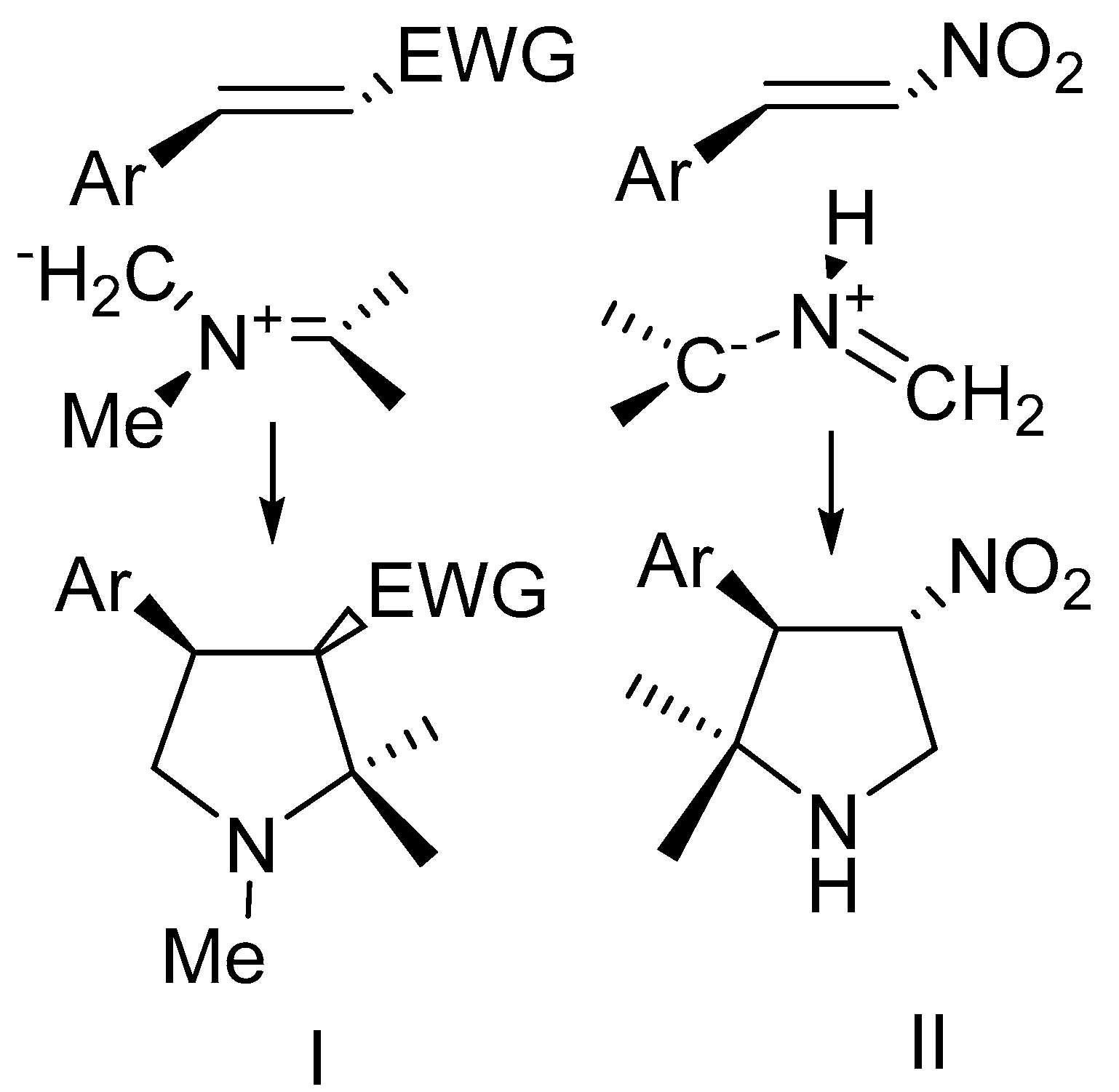
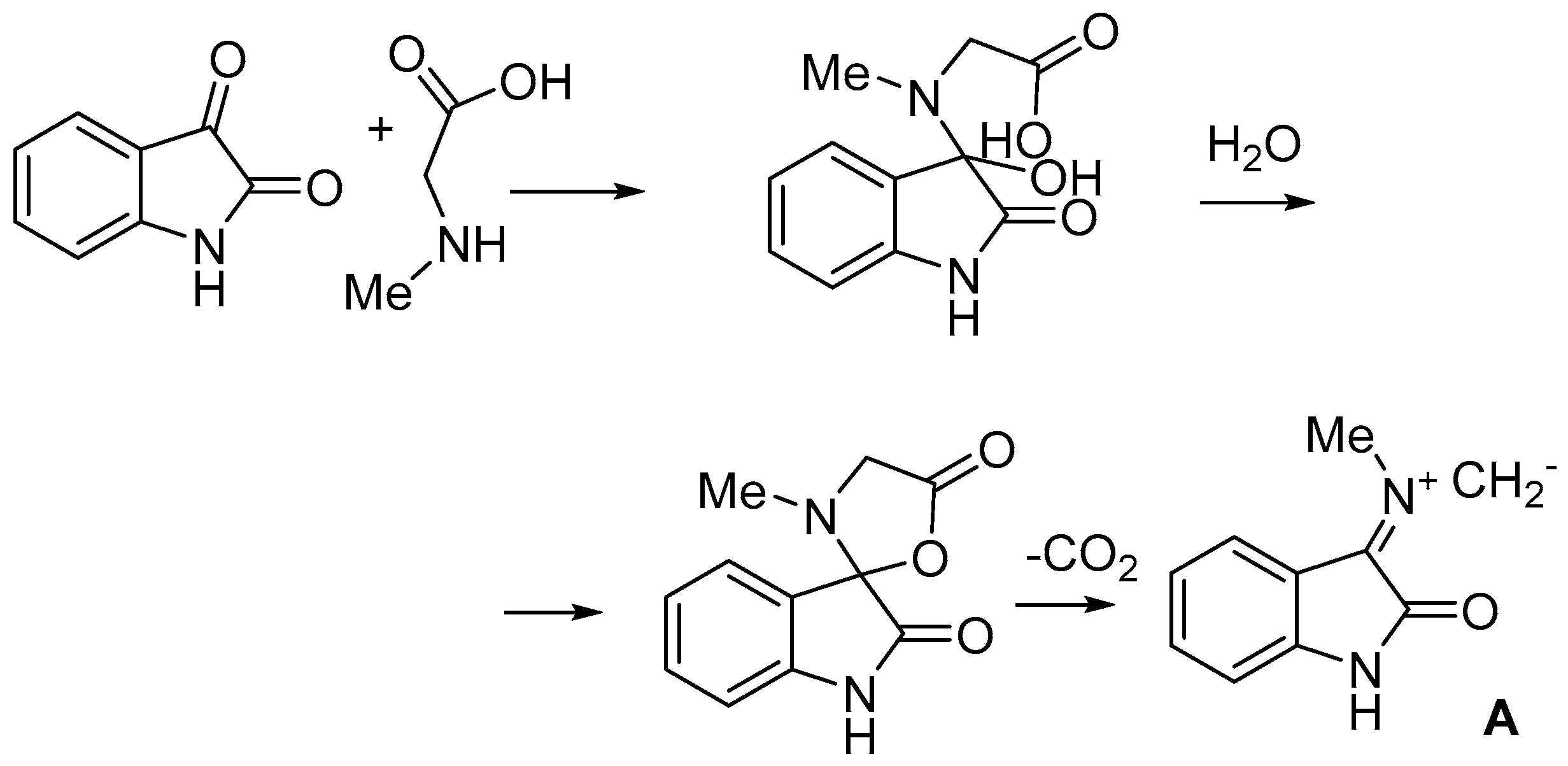
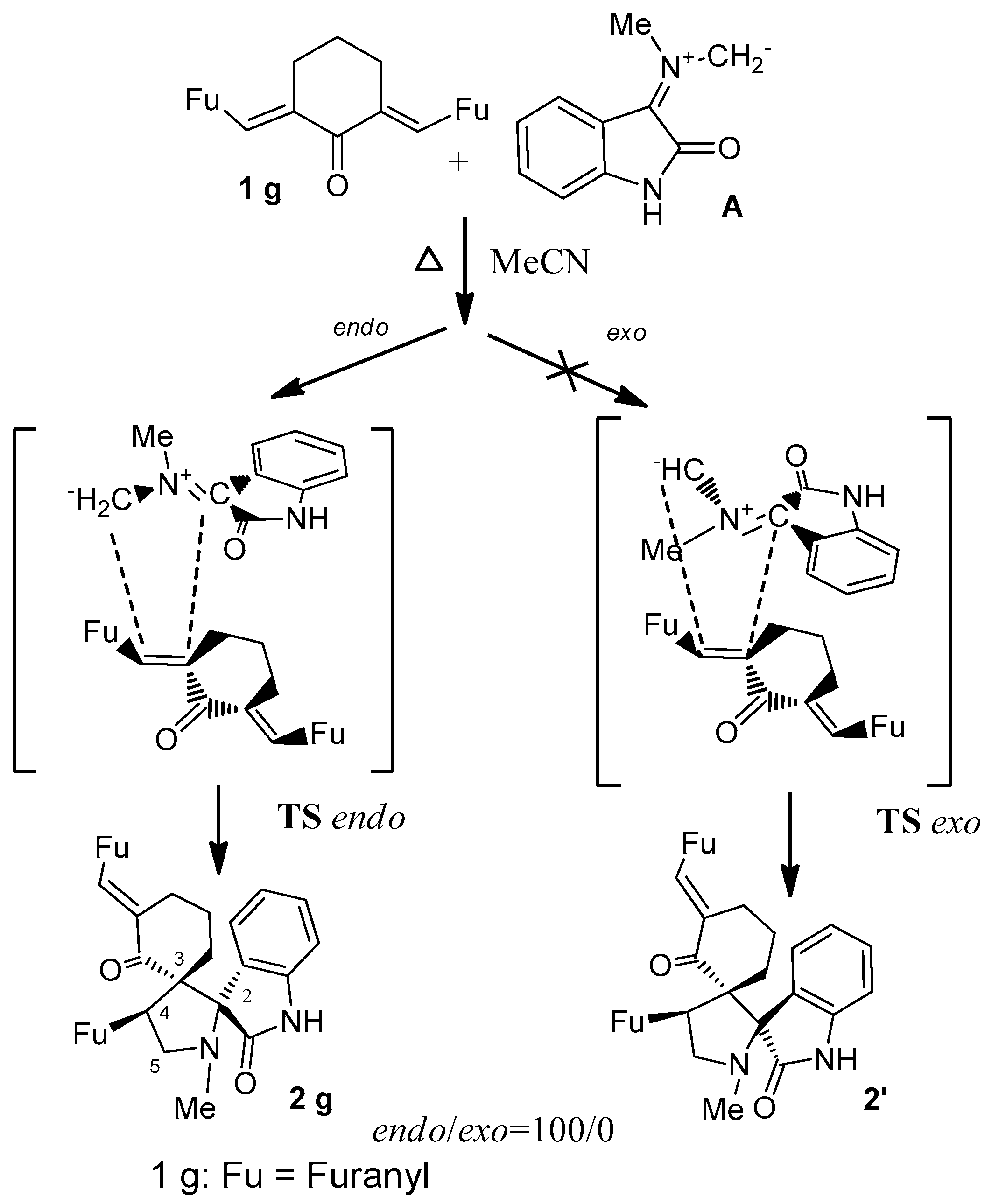
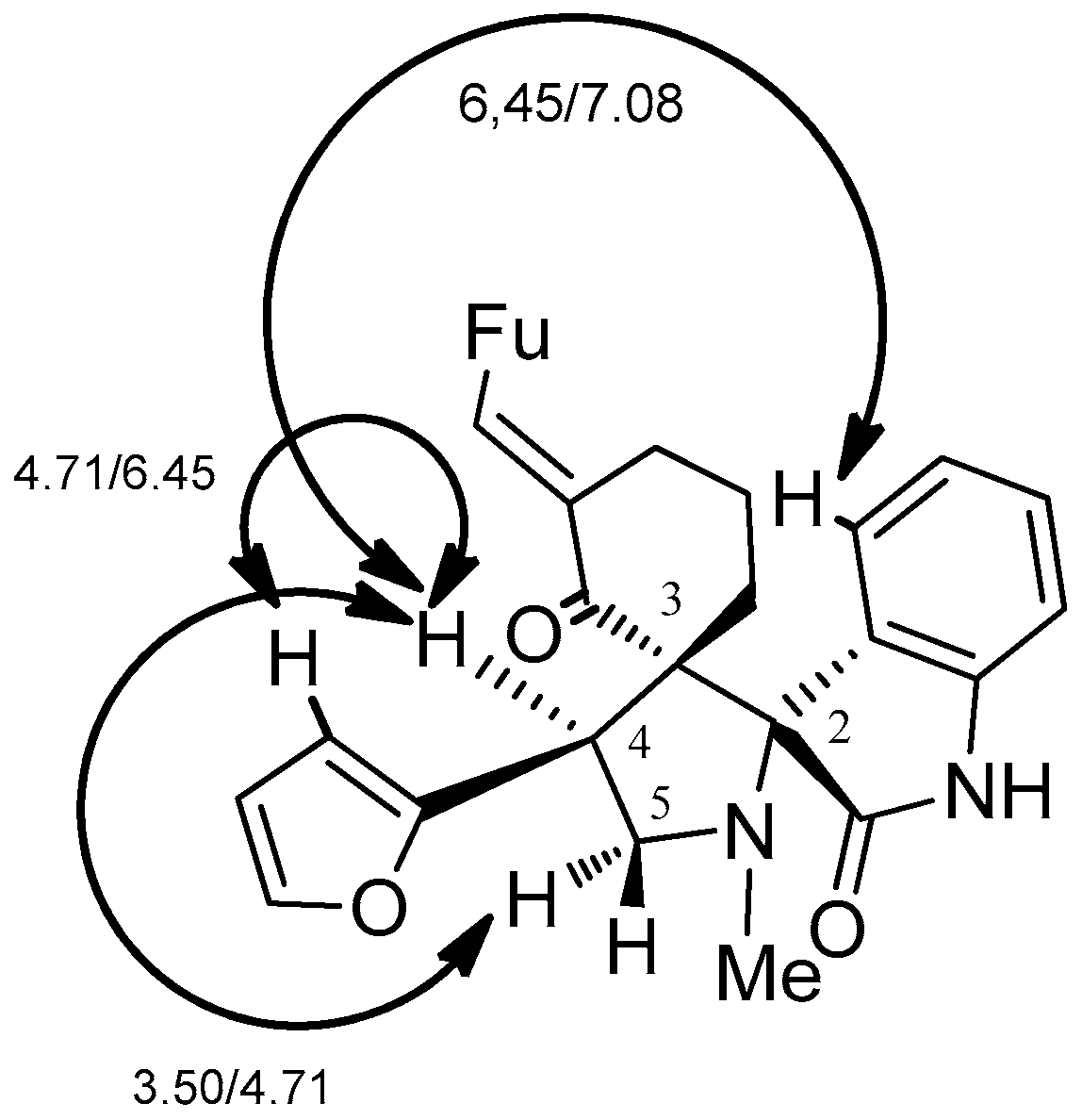
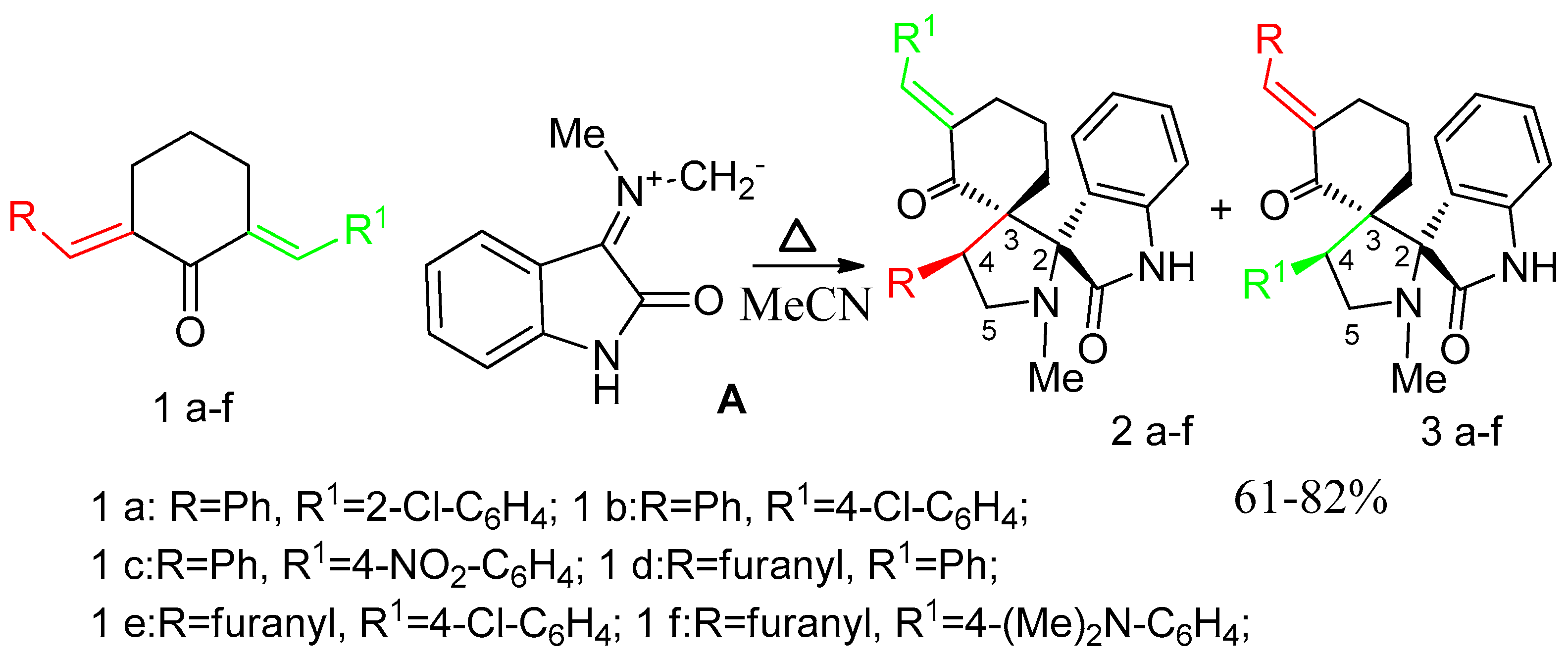
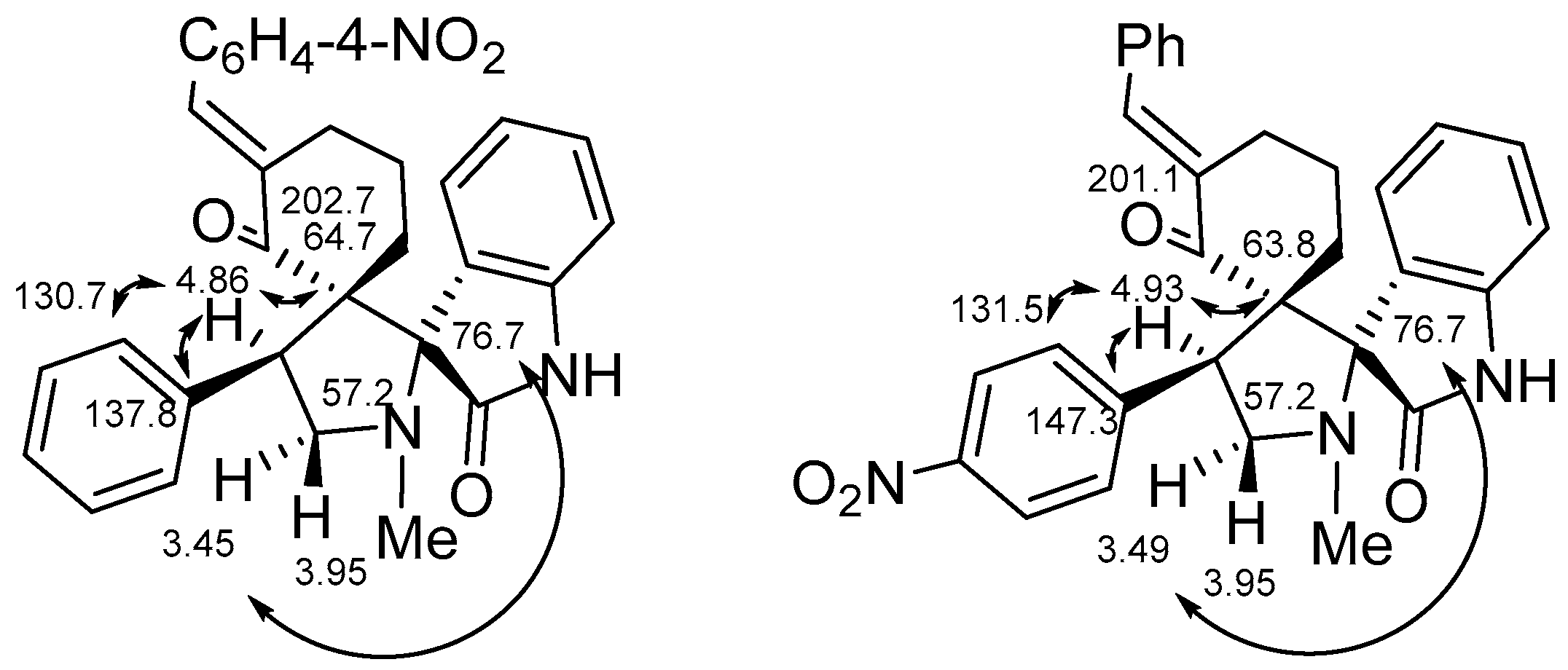
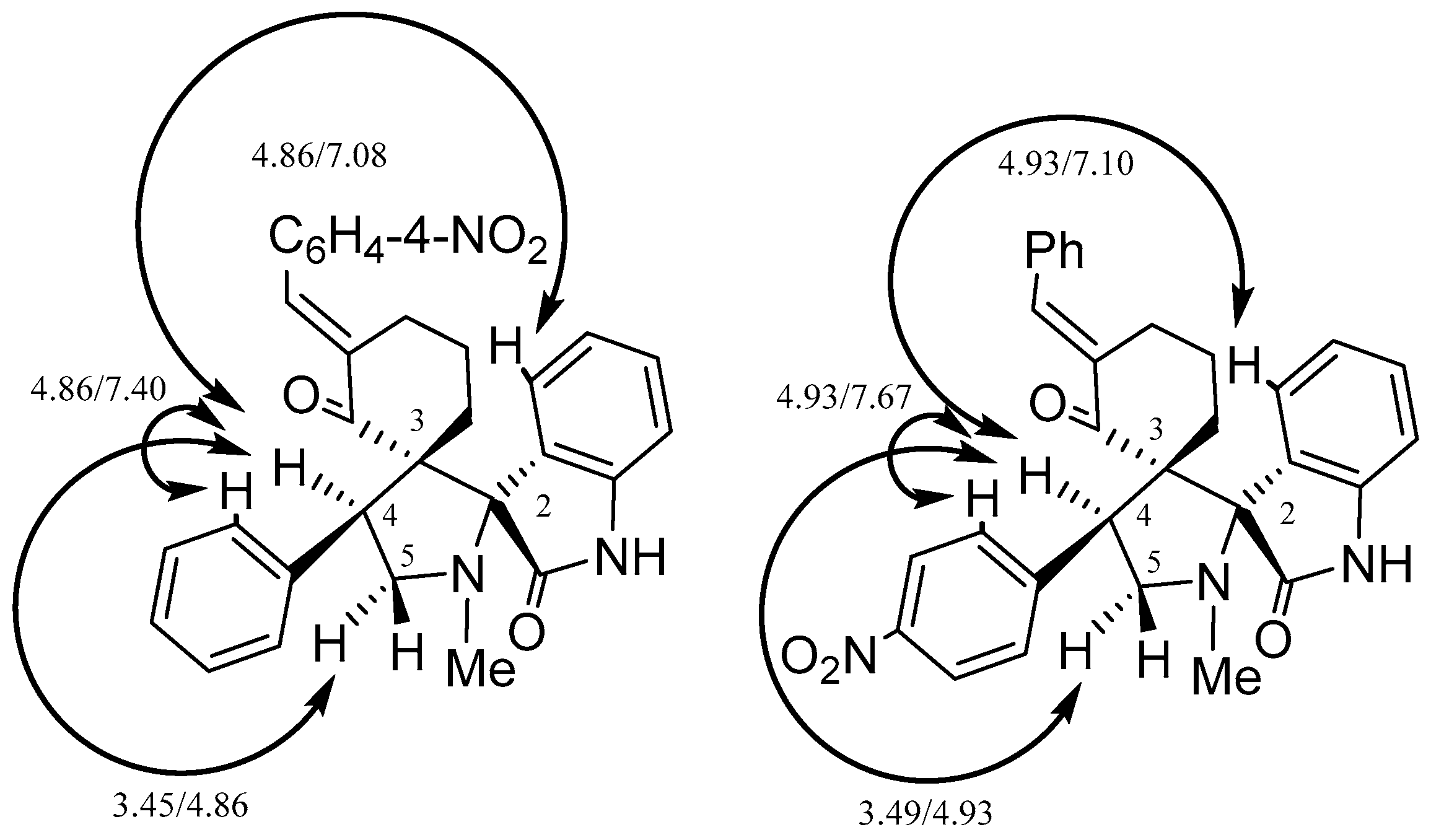
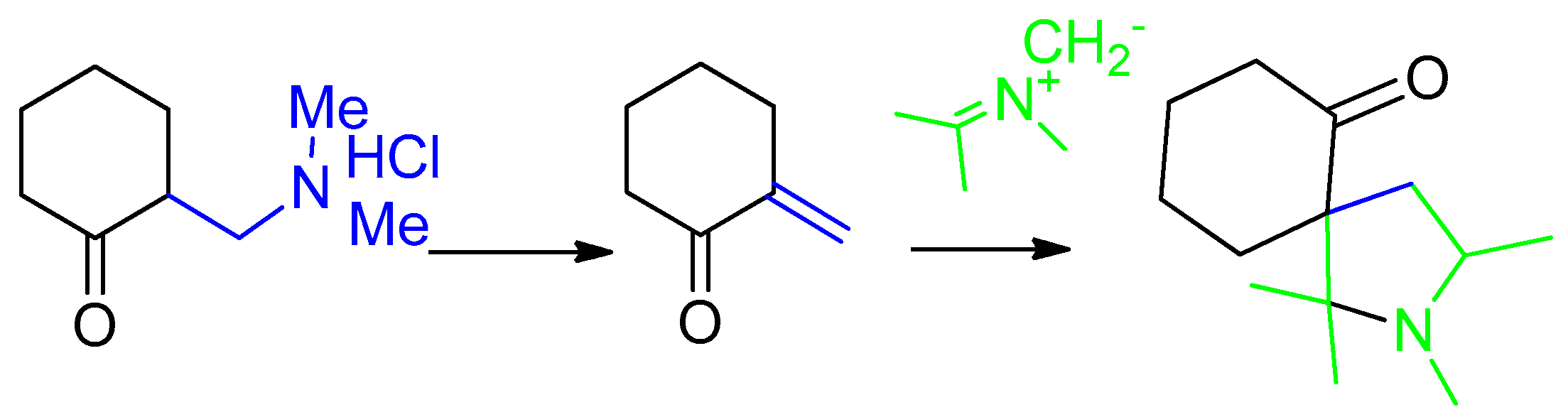
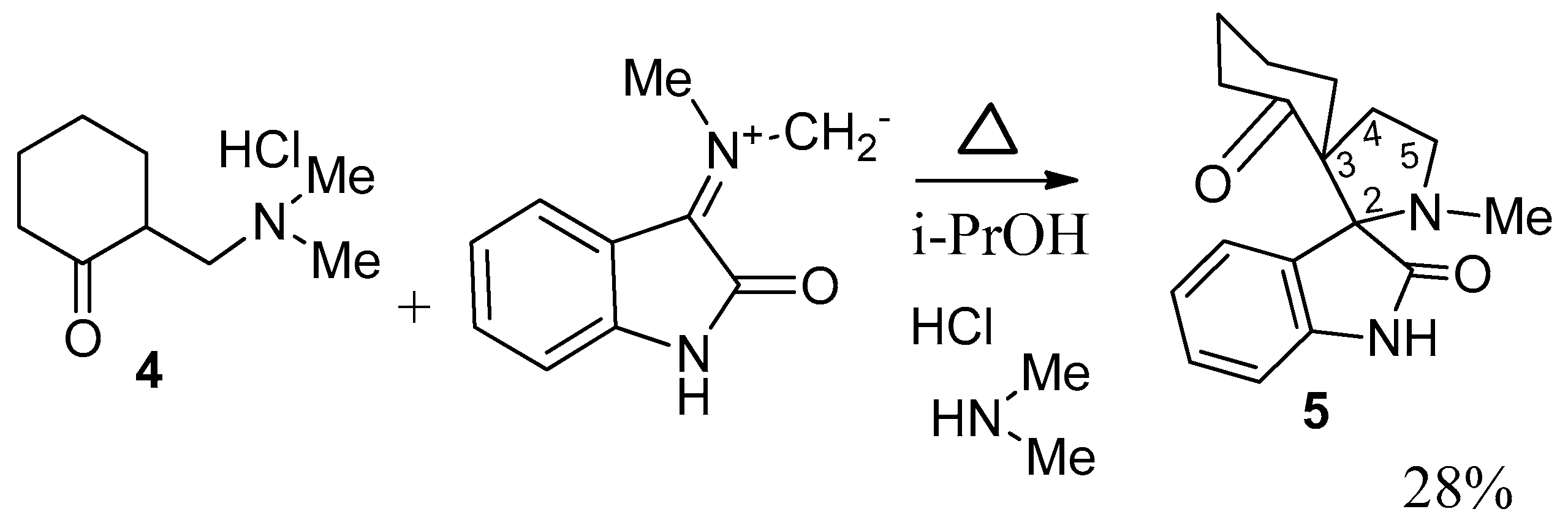
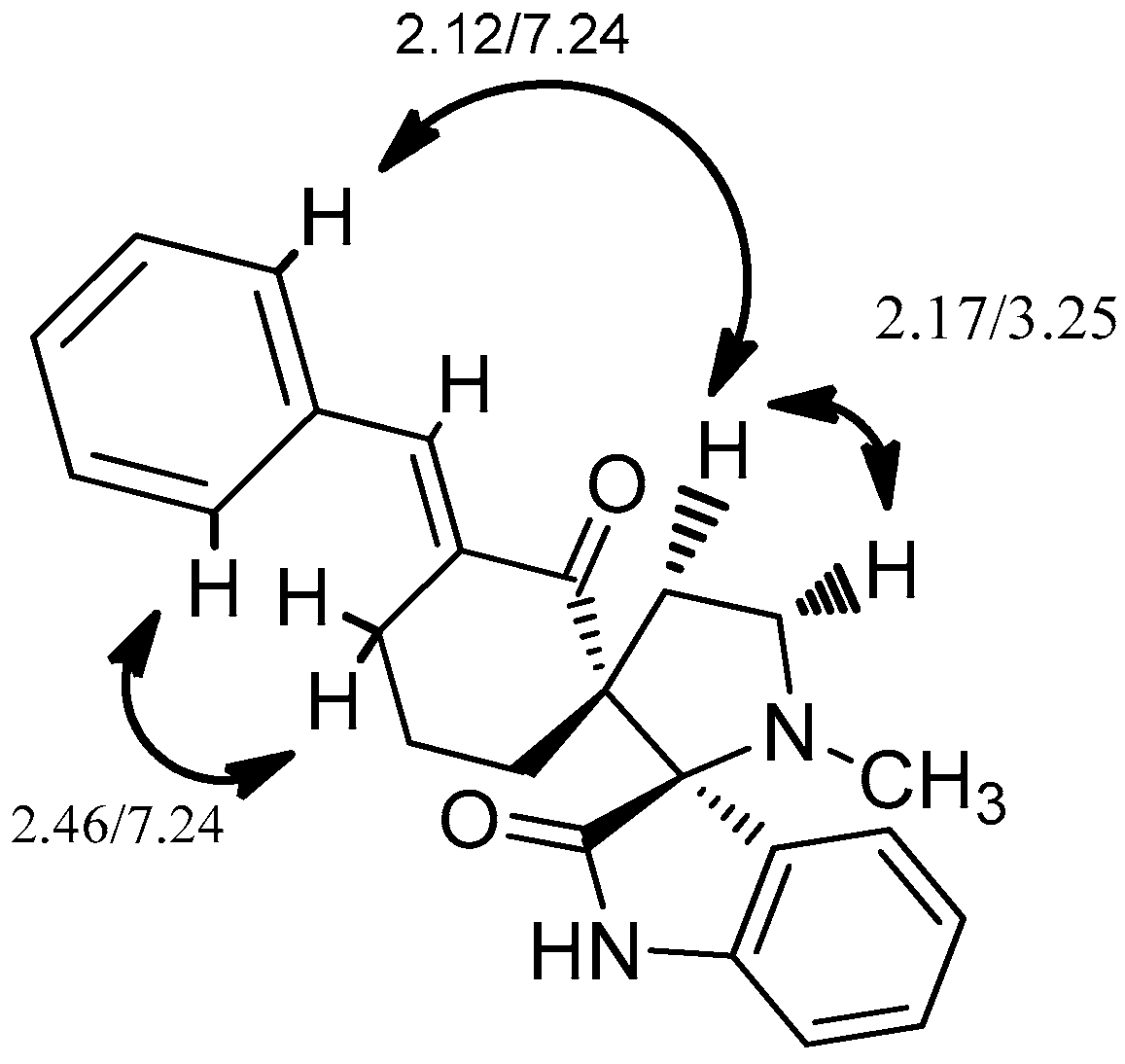
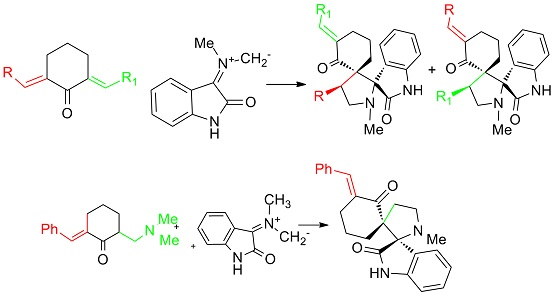
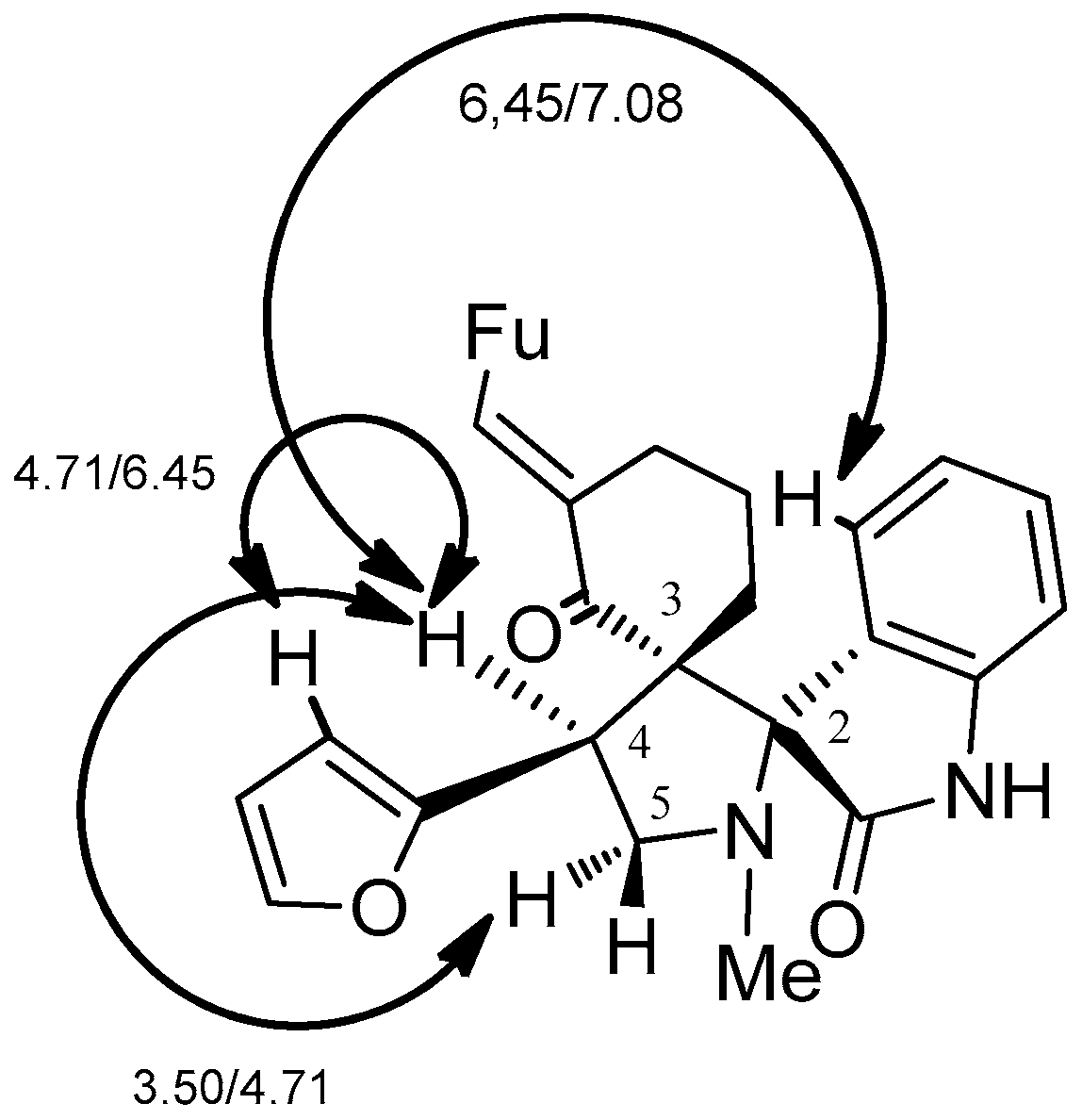
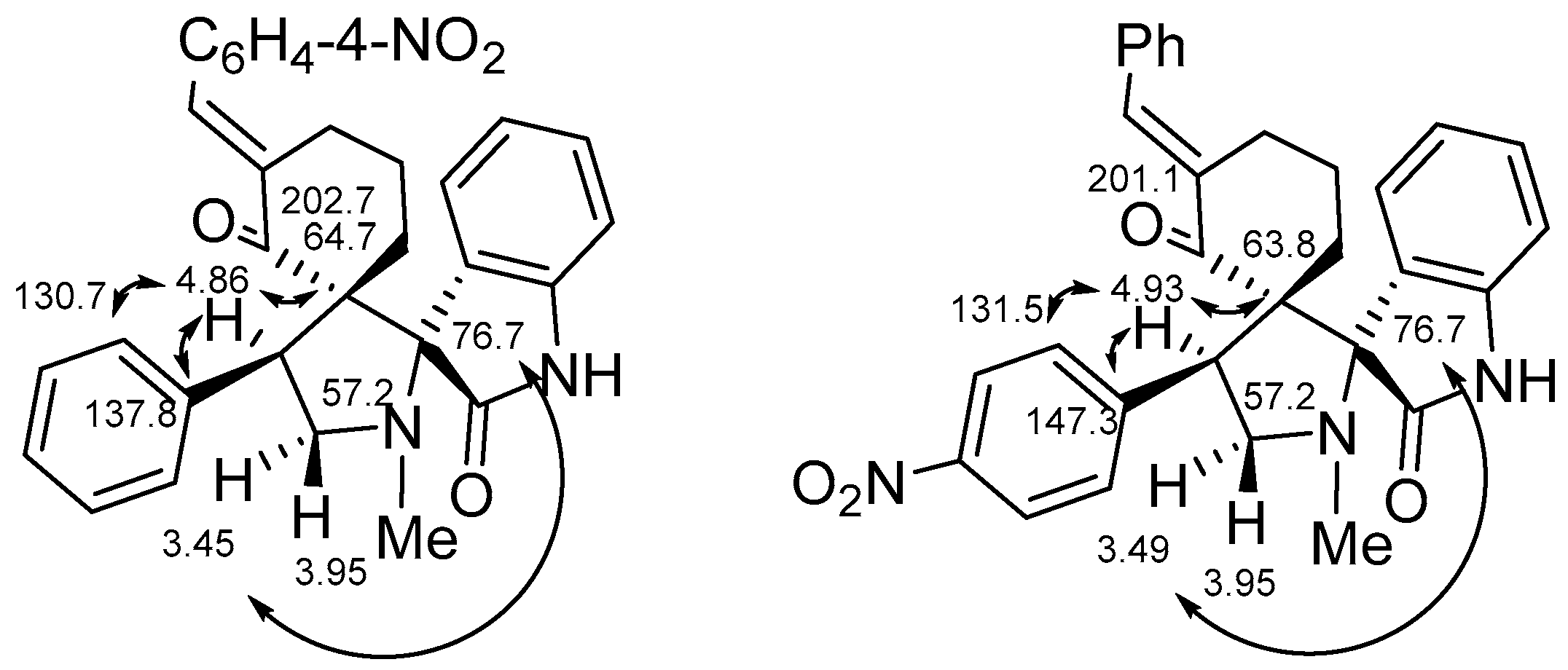
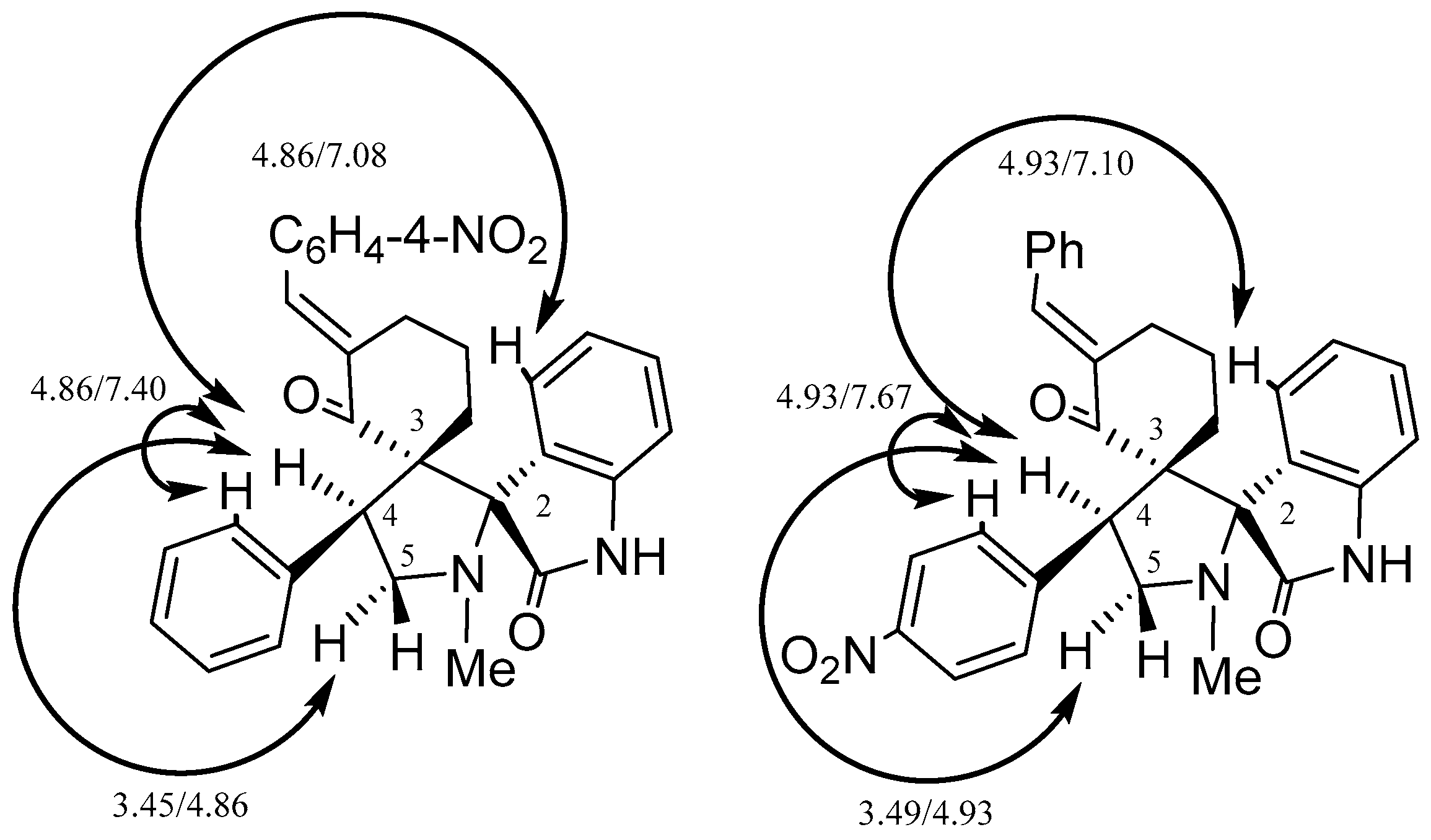
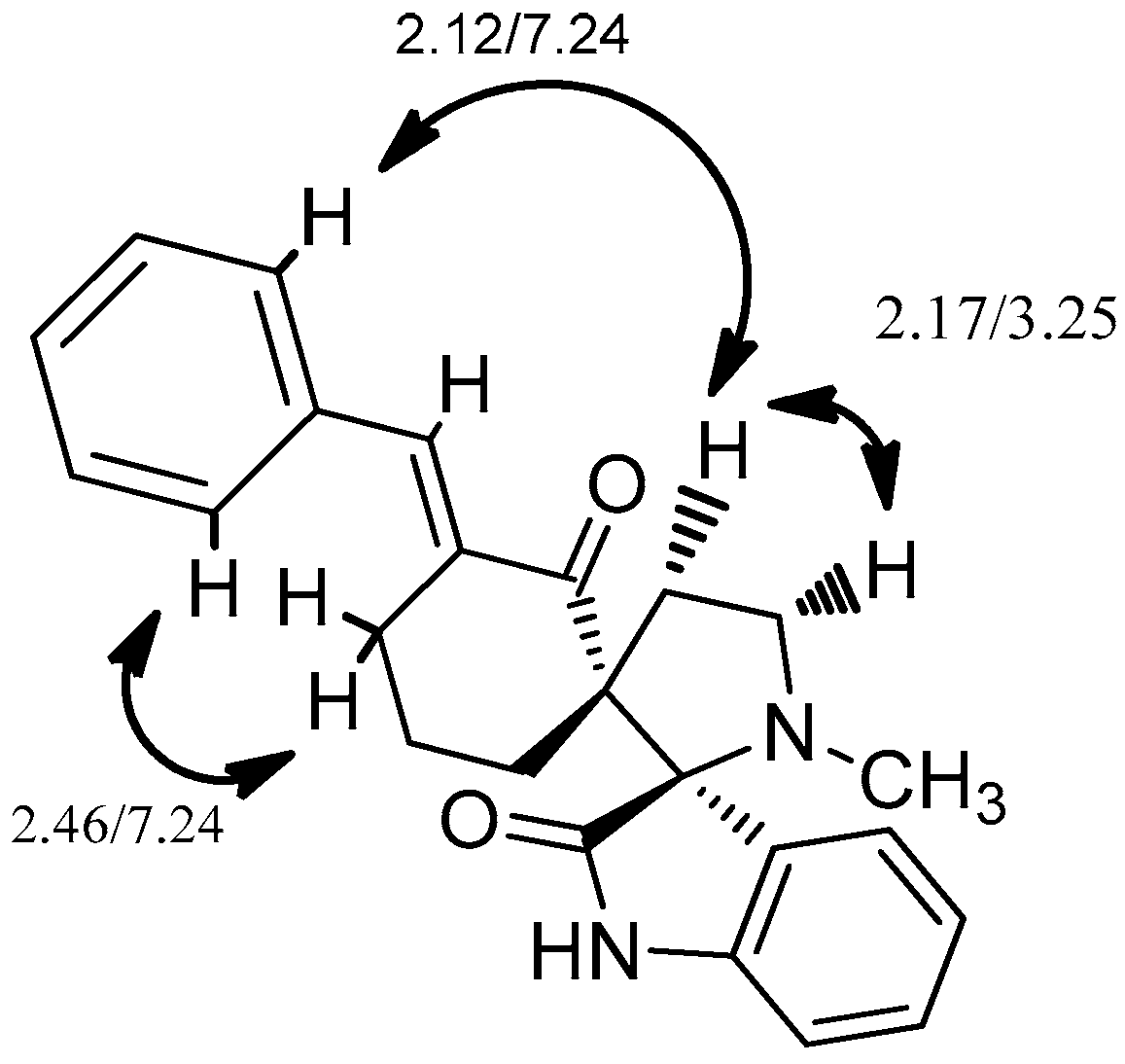
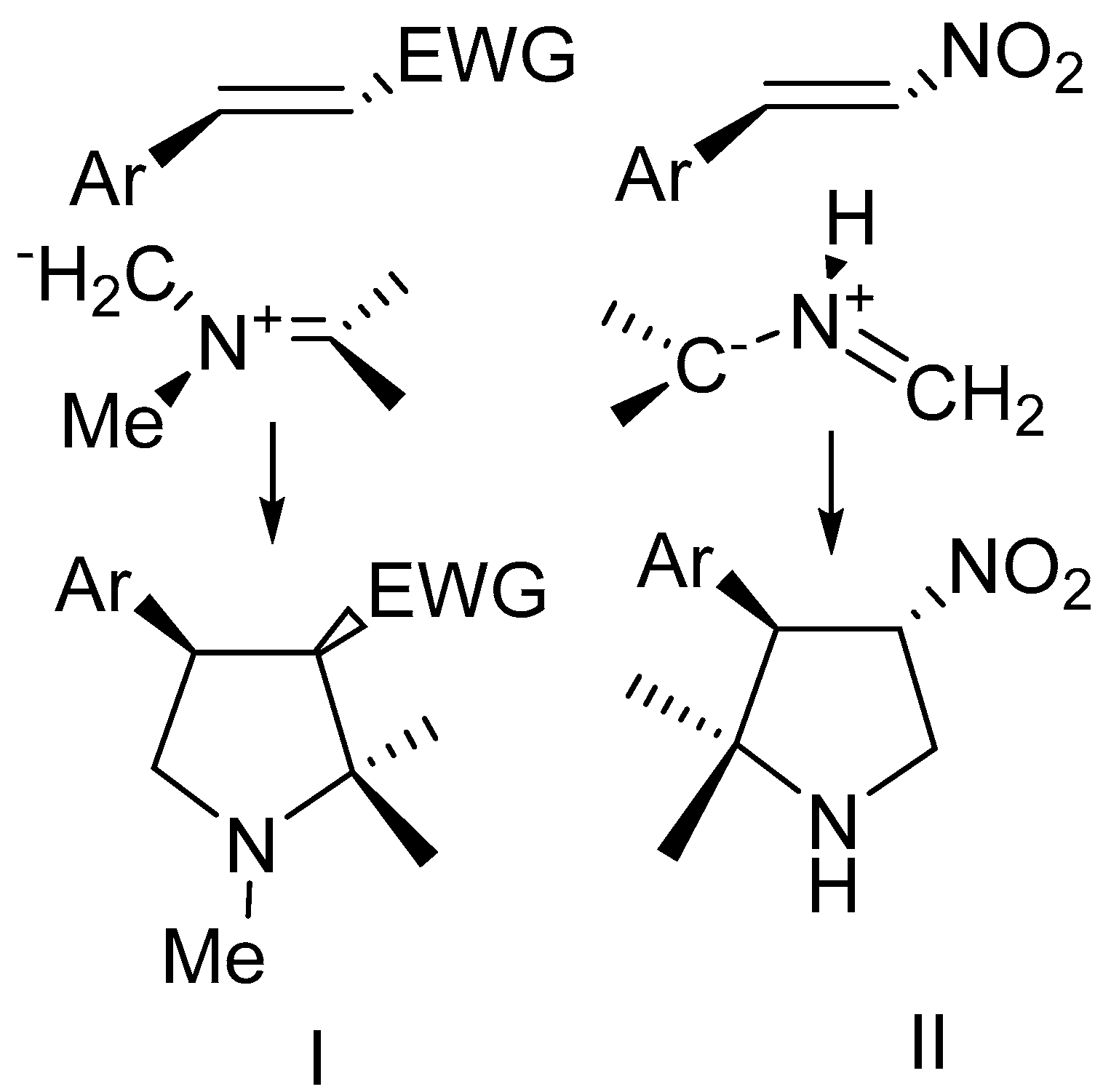
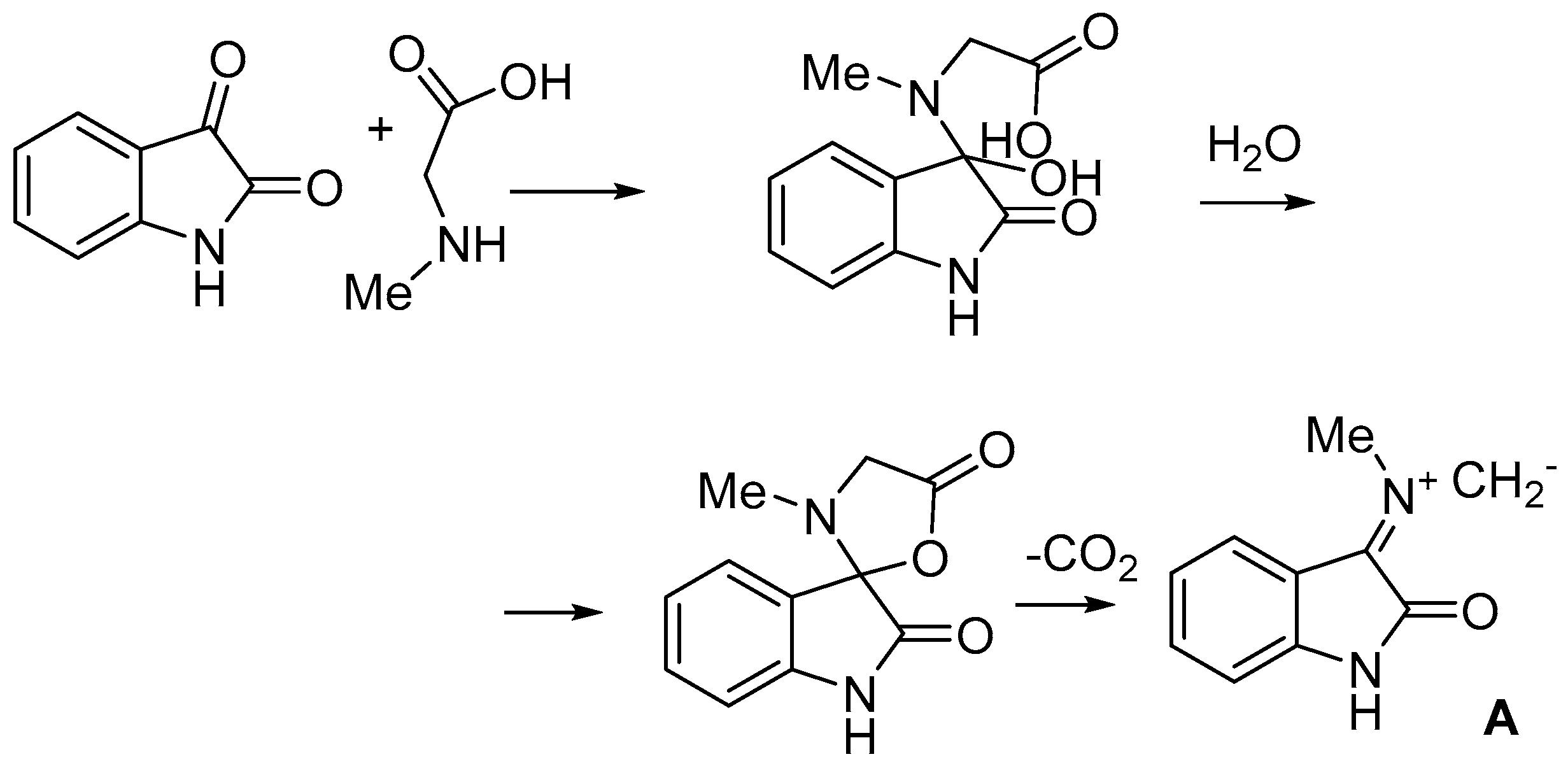
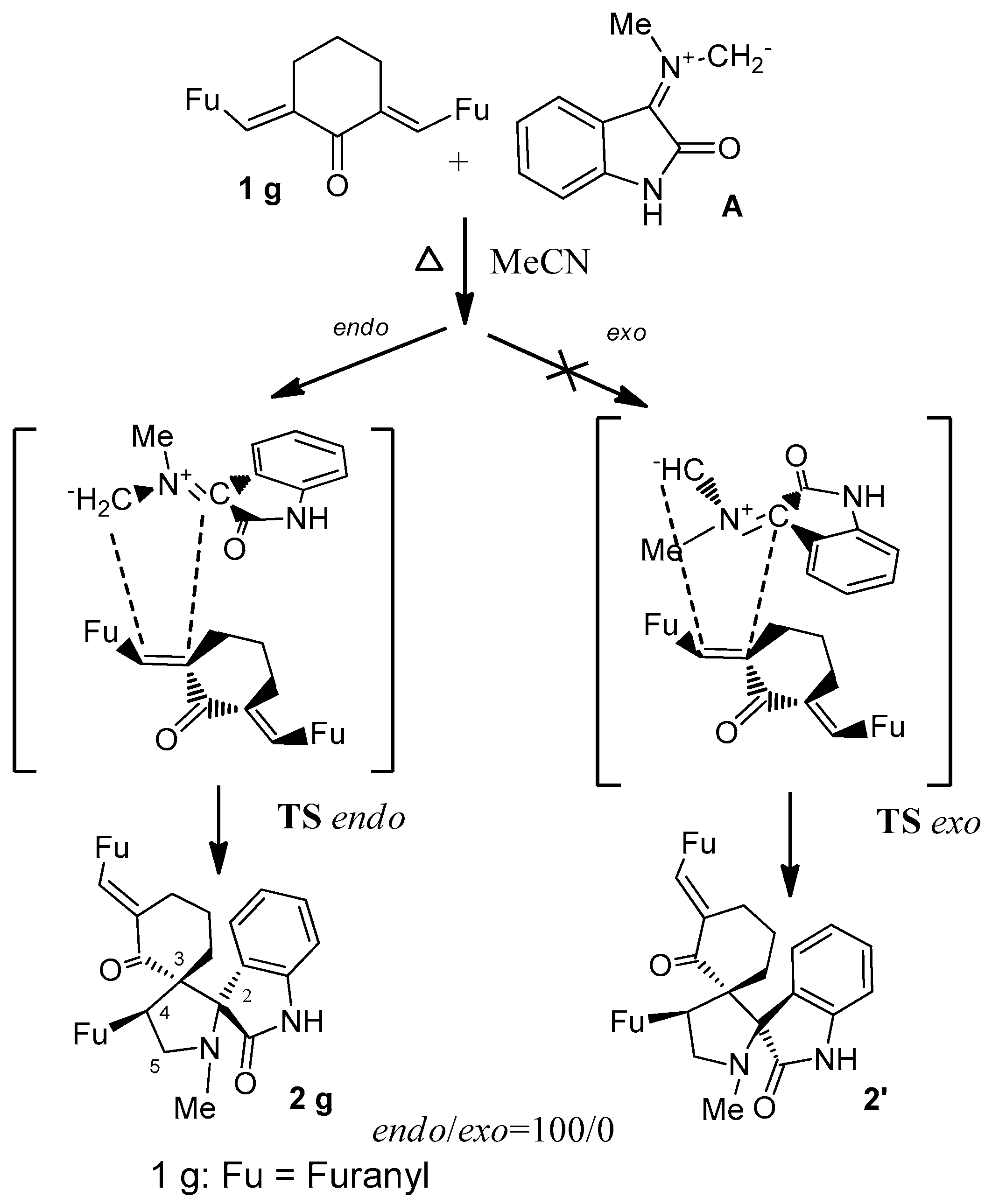
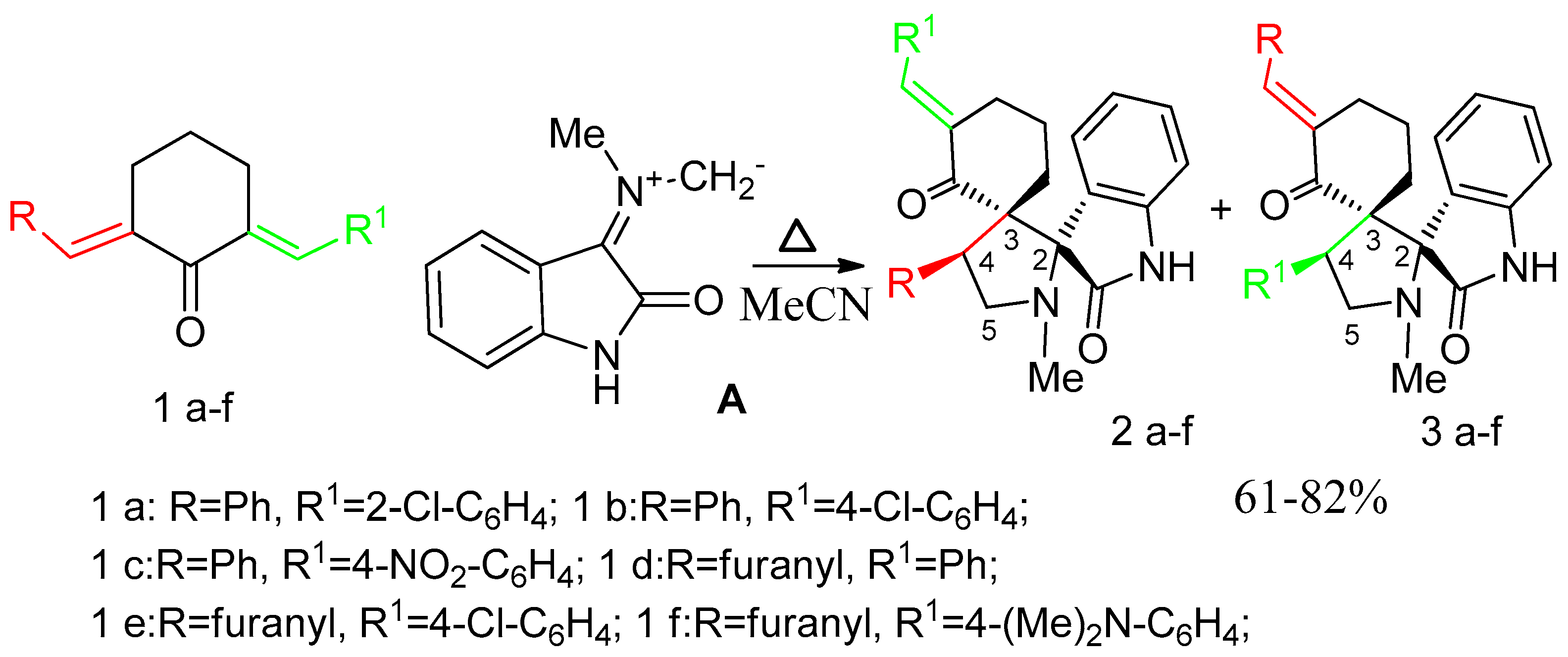
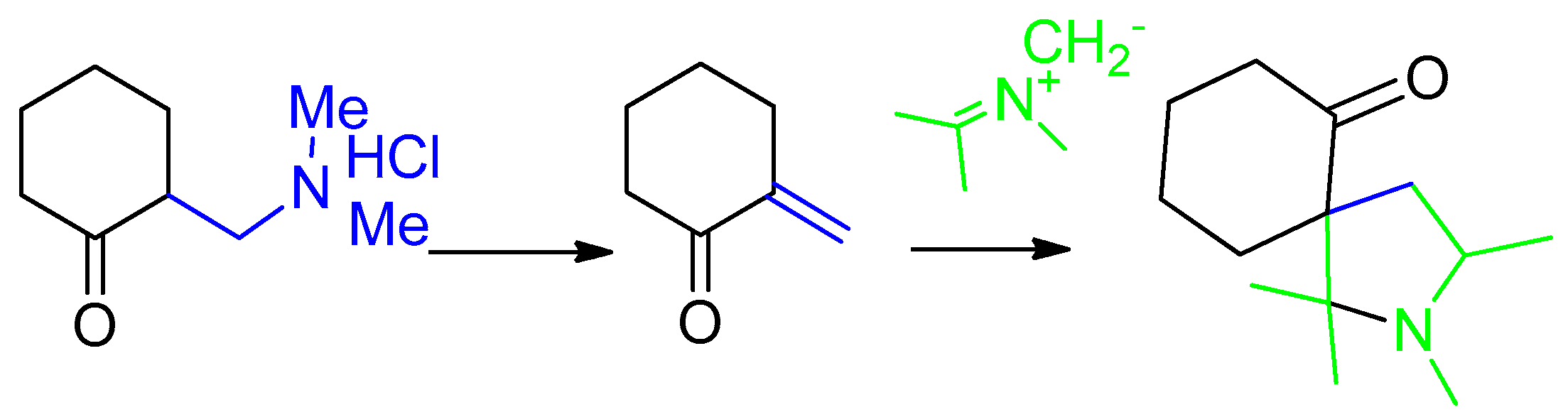
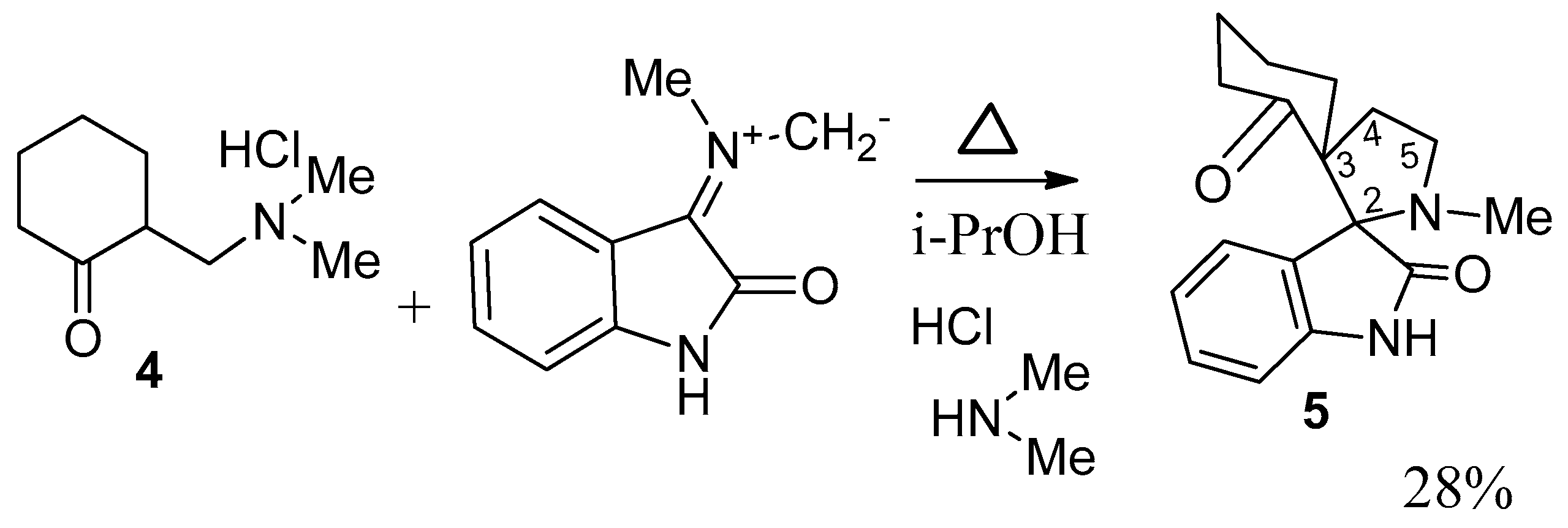
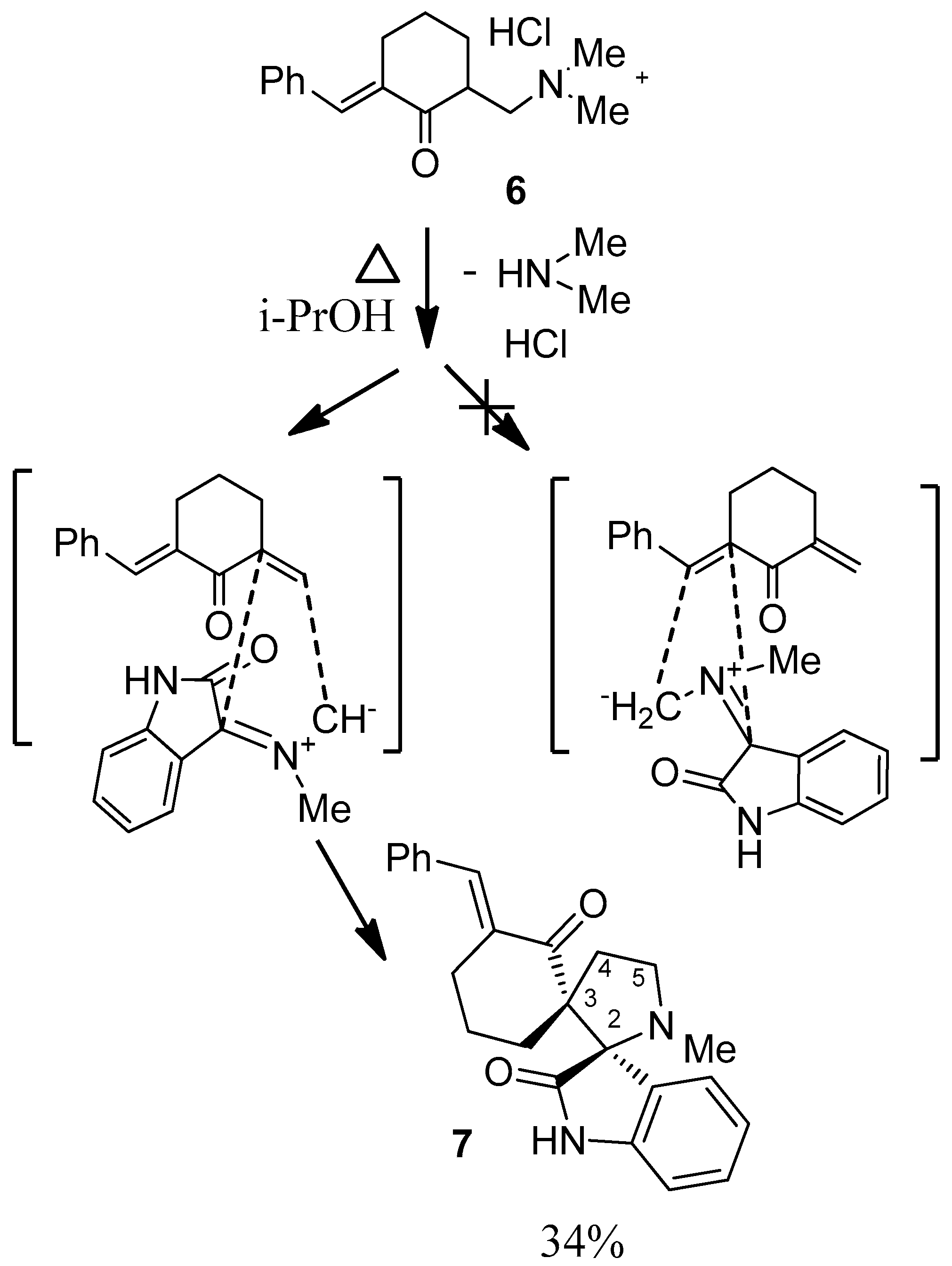
2. Results and Discussion
3. Materials and Methods
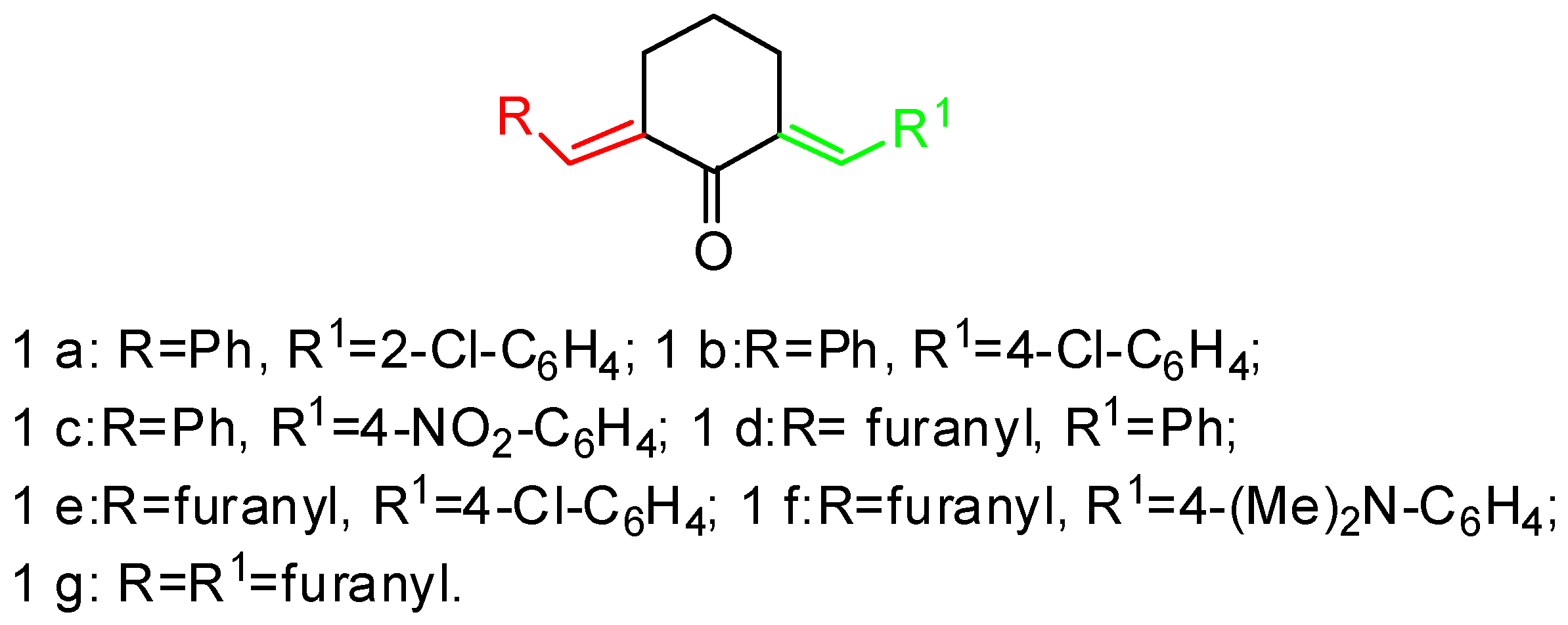
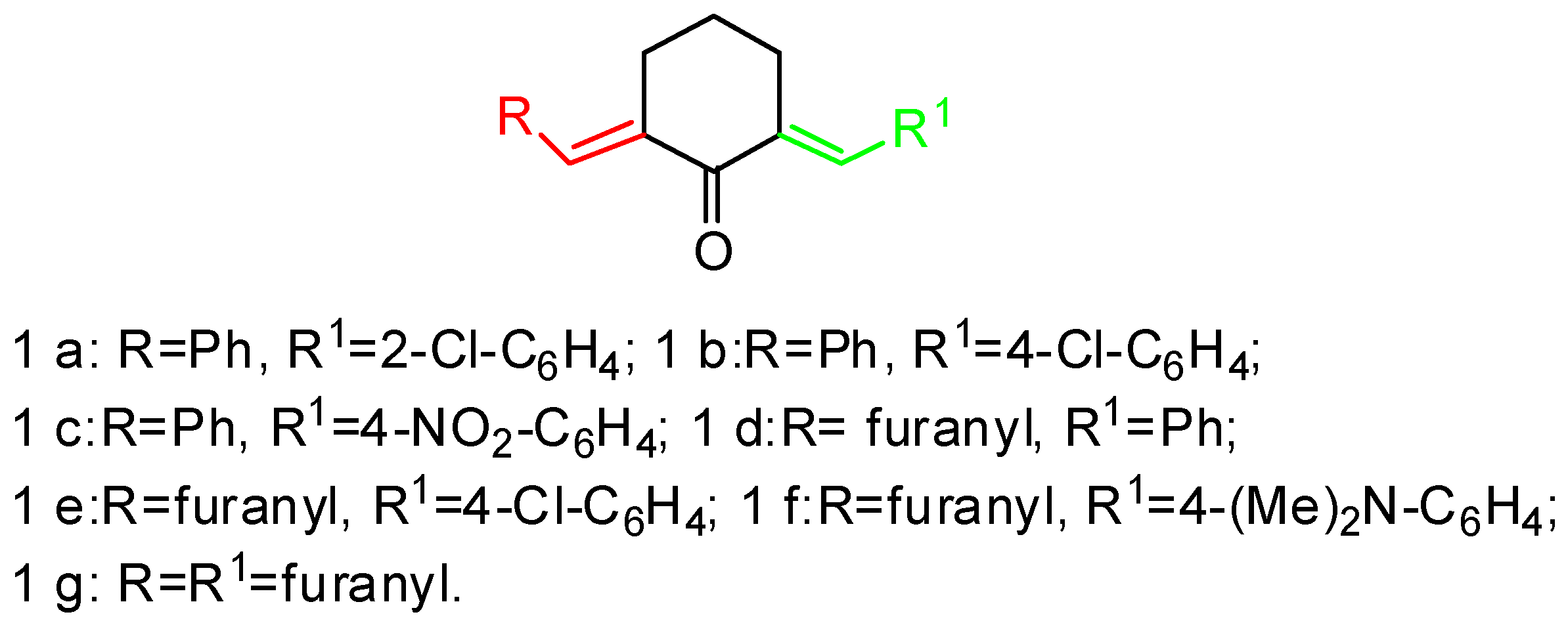
3.1. Synthesis of 1-N-Methyl-spiro[2.31]oxindole-spiro[3.211]611-arylmethylidenecyclohexanone-4-aryl-pyrrolidines (General Method). 2, 3a–g
3.2. Synthesis of 8-R-14-N-Methyl-2,14-diazaspiro[4.0.5.3]tetradecan-3,4-benzo-1,7-dione (General Method)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shankar, M.; Chowdhury, S.; Koley, S. Progress in 1,3-dipolar cycloadditions in the recent decade: An update to strategic development towards the arsenal of organic synthesis. Tetrahedron 2016, 72, 1603–1644. [Google Scholar]
- Rouatbi, F.; Askri, M.; Nana, F.; Kirsch, G.; Sriram, D.; Yogeeswari, P. Synthesis of new spirooxindole derivatives through 1,3-dipolar cycloaddition of azomethine ylides and their antitubercular activity. Tetrahedron Lett. 2016, 57, 163–167. [Google Scholar] [CrossRef]
- Narayanarao, M.; Koodlur, L.; Revanasiddappa, V.G.; Gopal, S.; Kamila, S. Multicomponent synthesis of spiropyrrolidine analogues derived from vinylindole/indazole by a 1,3-dipolar cycloaddition reaction. Beilstein J. Org. Chem. 2016, 12, 2893–2897. [Google Scholar] [CrossRef] [PubMed]
- Barkov, A.; Zimnitskiy, N.; Korotaev, V.; Kutyashev, I.; Moshkin, V.; Sosnovskikh, V. Highly regio- and stereoselective 1,3-dipolar cycloaddition of stabilised azomethine ylides to 3,3,3-trihalogeno-1-nitropropenes: Synthesis of trihalomethylated spiro[indoline-3,2′-pyrrolidin]-2-ones and spiro[indoline-3,3′-pyrrolizin]-2-ones. Tetrahedron 2016, 72, 6825–6836. [Google Scholar] [CrossRef]
- Mancebo-Aracil, J.; Najera, C.; Castello, L.M.; Cossíoc, P.F.; Larranagac, O.; Cozarcd, A. Regio and diastereoselective multicomponent 1,3-dipolar cycloadditions between prolinate hydrochlorides, aldehydes and dipolarophiles for the direct synthesis of pyrrolizidines. Tetrahedron 2015, 71, 9645–9661. [Google Scholar] [CrossRef]
- Almansour, N.A.A.I.; Kumar, R.S.; Menéndez, J.C.; Sultan, M.A.; Karama, U.; Ghabbour, H.A.; Fun, H. An Expedient Regio- and Diastereoselective Synthesis of Hybrid Frameworks with Embedded Spiro[9,10]dihydroanthracene [9,3′]-pyrrolidine and Spiro[oxindole-3,2′-pyrrolidine] Motifs via an Ionic Liquid-Mediated Multicomponent Reaction. Molecules 2015, 20, 16142–16153. [Google Scholar]
- Mathusalini, S.; Arasakumar, T.; Lakshmi, K.; Lin, C.; Subramaniam, M.P.; Gogul Ramnathc, M.; Thirugnanasampandan, R. Synthesis and biological evaluation of new spirooxindoles with embedded pharmacophores. New J. Chem. 2016, 40, 5164–5169. [Google Scholar] [CrossRef]
- Onishi, T.; Sebahar, P.R.; Williams, R.M. Concise, Asymmetric Total Synthesis of Spirotryprostatin A. Org. Lett. 2003, 5, 3135–3137. [Google Scholar] [CrossRef] [PubMed]
- Anis’kov, A.A.; Kamneva, Y.I.; Zheleznova, A.M.; Yegorova, A.Y. Reaction of arylmethylidene derivatives of 3H-furan-2-ones with azomethine ylide. Chem. Heterocycl. Compd. 2015, 51, 709–712. [Google Scholar] [CrossRef]
- Klochkova, I.N.; Shchekina, M.P.; Anis’kov, A.A. Synthesis of Spiropyrrolidines and Spiropyrrolizidines from Azomethine Ylides. Chem. Heterocycl. Compd. 2014, 50, 479–488. [Google Scholar] [CrossRef]
- Izmest’ev, N.A.; Gazieva, G.A.; Sigay, N.V.; Serkov, S.A.; Karnoukhova, V.A.; Kachala, V.V.; Shashkov, A.S.; Zanin, I.E.; Kravchenko, A.N.; Makhova, N.N. An effective one-pot access to polynuclear dispiroheterocyclic structures comprising pyrrolidinyloxindole and imidazothiazolotriazine moieties via a 1,3-dipolar cycloaddition strategy. Beilstein J. Org. Chem. 2016, 12, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Sobhi, C.; Khorief Nacereddine, A.; Djerourou, A.; Ríos-Gutiérrez, M. A DFT study of the mechanism and selectivities of the [3 + 2] cycloaddition reaction between 3-(benzylideneamino)oxindole and trans-β-nitrostyrene. J. Phys. Org. Chem. 2017, 30, 3637–3646. [Google Scholar] [CrossRef]
- Jayashankaran, J.; Manian, R.D.R.S.; Venkatesan, R.; Raghunathan, R. A regioselective synthesis of dispiro[oxindole-cyclohexanone]pyrrolidines and dispiro[oxindole-hexahydroindazole]pyrrolidines by sequential 1,3-dipolar cycloaddition and annulation through a microwave induced solvent-free approach. Tetrahedron 2005, 61, 5595–5598. [Google Scholar] [CrossRef]
- Sumana, K.; Thennarasu, S. Acetic acid promoted tandem cyclization of in situ generated 1,3-dipoles: Stereoselective synthesis of dispiroimidazolidinyl and dispiropyrrolidinyl oxindoles with multiple chiral stereocenters. RSC Adv. 2015, 5, 79413–79422. [Google Scholar] [CrossRef]
- Peng, C.; Ren, J.; Xiao, J.; Zhang, H.; Yang, H.; Luo, Y. Additive-assisted regioselective 1,3-dipolar cycloaddition of azomethine ylides with benzylideneacetone. Beilstein J. Org. Chem. 2014, 10, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Bugaev, A.A.; Golikov, A.G.; Kriven’ko, A.P. Synthesis of substituted hexahydroindazoles. Chem. Heterocycl. Compd. 2005, 41, 831–834. [Google Scholar] [CrossRef]
- Amal Raj, A.; Raghunathan, R.; SrideviKumarib, M.R.; Raman, N. Synthesis, Antimicrobial and antifungal activity of a new class of spiro pyrrolidines. Bioorg. Med. Chem. 2003, 11, 407–419. [Google Scholar]
- Shchekina, M.P.; Tumskii, R.S.; Klochkova, I.N.; Anis’kov, A.A. Synthesis of pyrimidinethiones and spiropyrans proceeding from Mannich ketones. Russ. J. Org. Chem. 2017, 53, 263–269. [Google Scholar] [CrossRef]
- Kriven’ko, A.P.; Bugaev, A.A.; Golikov, A.G. Synthesis and configuration of 6-arylidene-2-furfurylidene-cyclohexanones. Chem. Heterocycl. Compd. 2005, 41, 163–167. [Google Scholar] [CrossRef]
- Mannich, C.; Braun, R. Über die Synthese von β-Ketobasen aus Aminsalzen, Formaldehyd und cyclo-Hexanon. Eur. J. Inorg. Chem. 1920, 53, 1874–1880. [Google Scholar] [CrossRef]
- Lorand, T.; Kocsis, B.; Sohar, P.; Nagy, G.; Kispal, G.; Krane, H.-G.; Schmitt, H.; Weckert, E. Synthesis and antibacterial study of unsaturated Mannich ketones. Eur. J. Med. Chem. 2001, 36, 705–717. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2, 3c,e,f, 5 and 7 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ratio 2/3 | Yields, % | Ratio Endo/Exo |
|---|---|---|---|
| a | 64/36 | 78 | 100/0 |
| b | 50/50 | 72 | 100/0 |
| c | 55/45 | 82 | 100/0 |
| d | 80/20 | 65 | 100/0 |
| e | 80/20 | 68 | 100/0 |
| f | 70/30 | 64 | 100/0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anis’kov, A.; Klochkova, I.; Tumskiy, R.; Yegorova, A. A Diastereoselective Synthesis of Dispiro[oxindole-cyclohexanone]pyrrolidines by 1,3-Dipolar Cycloaddition. Molecules 2017, 22, 2134. https://doi.org/10.3390/molecules22122134
Anis’kov A, Klochkova I, Tumskiy R, Yegorova A. A Diastereoselective Synthesis of Dispiro[oxindole-cyclohexanone]pyrrolidines by 1,3-Dipolar Cycloaddition. Molecules. 2017; 22(12):2134. https://doi.org/10.3390/molecules22122134
Chicago/Turabian StyleAnis’kov, Alexander, Irina Klochkova, Roman Tumskiy, and Alevtina Yegorova. 2017. "A Diastereoselective Synthesis of Dispiro[oxindole-cyclohexanone]pyrrolidines by 1,3-Dipolar Cycloaddition" Molecules 22, no. 12: 2134. https://doi.org/10.3390/molecules22122134
APA StyleAnis’kov, A., Klochkova, I., Tumskiy, R., & Yegorova, A. (2017). A Diastereoselective Synthesis of Dispiro[oxindole-cyclohexanone]pyrrolidines by 1,3-Dipolar Cycloaddition. Molecules, 22(12), 2134. https://doi.org/10.3390/molecules22122134