Synthesis and In Vitro Antimycobacterial Activity of Novel N-Arylpiperazines Containing an Ethane-1,2-diyl Connecting Chain

 ,
, 
Abstract
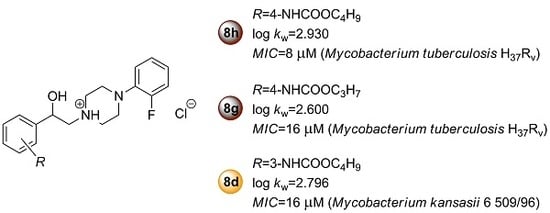
:
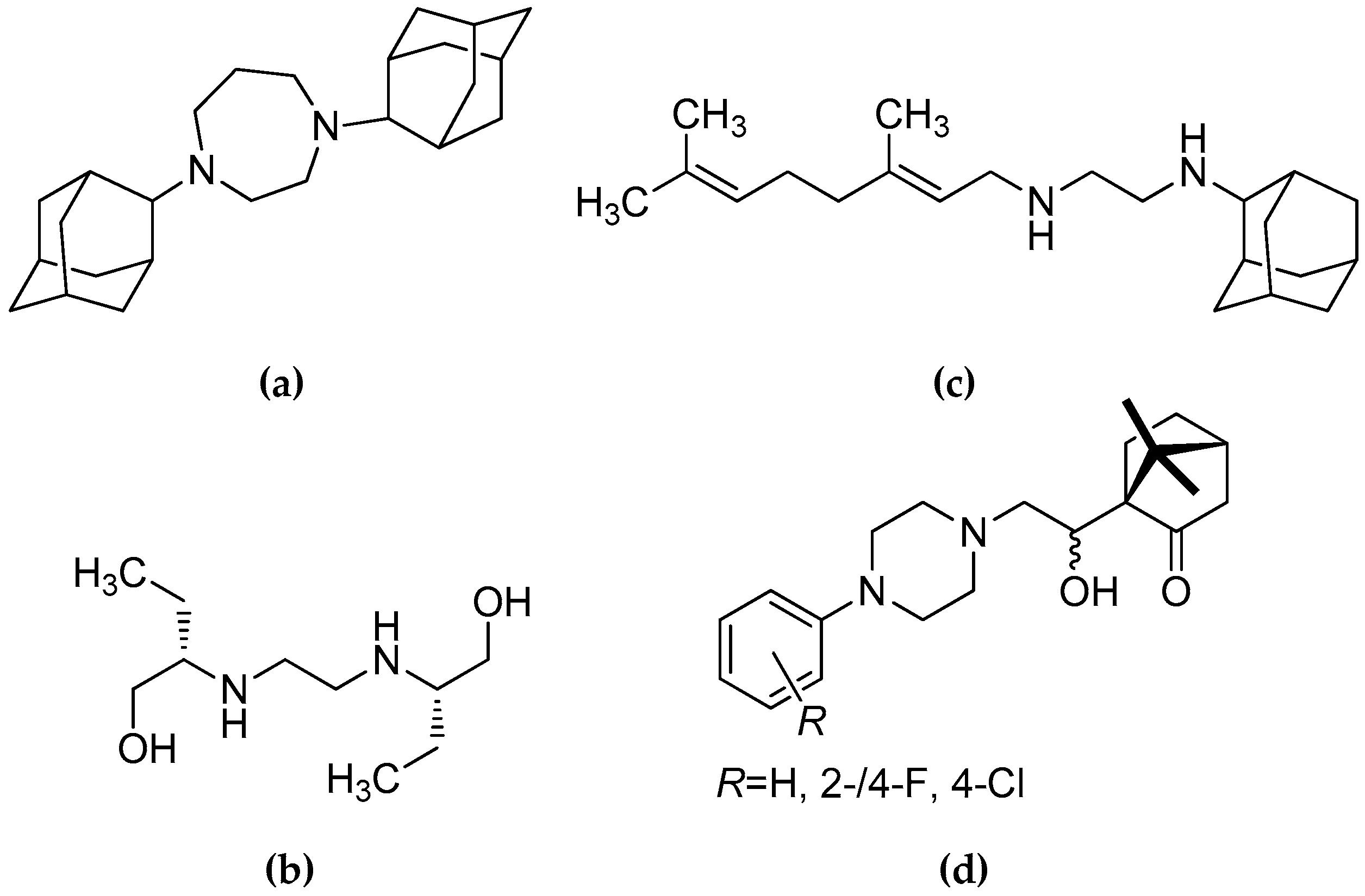
1. Introduction
2. Results and Discussion
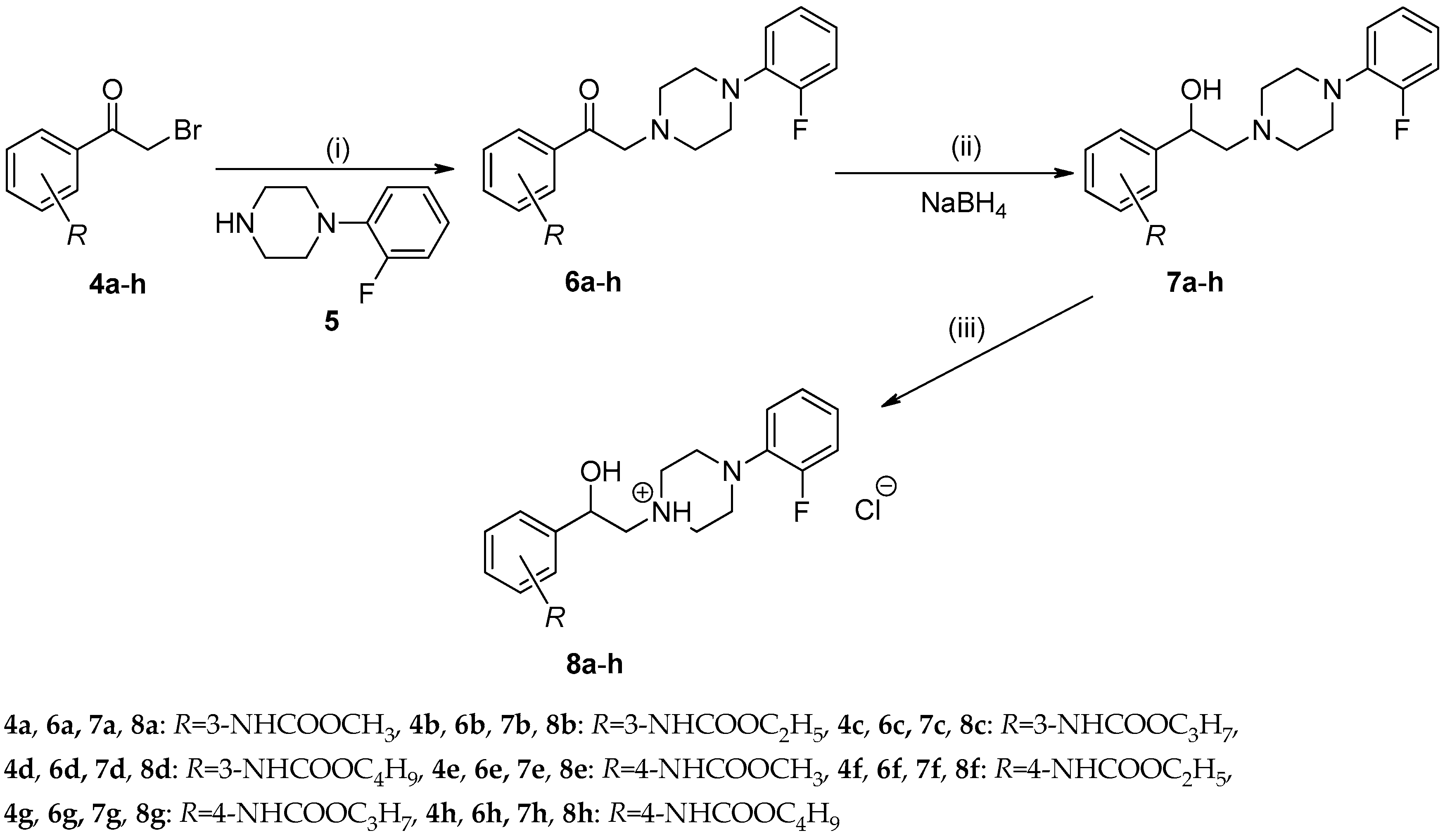
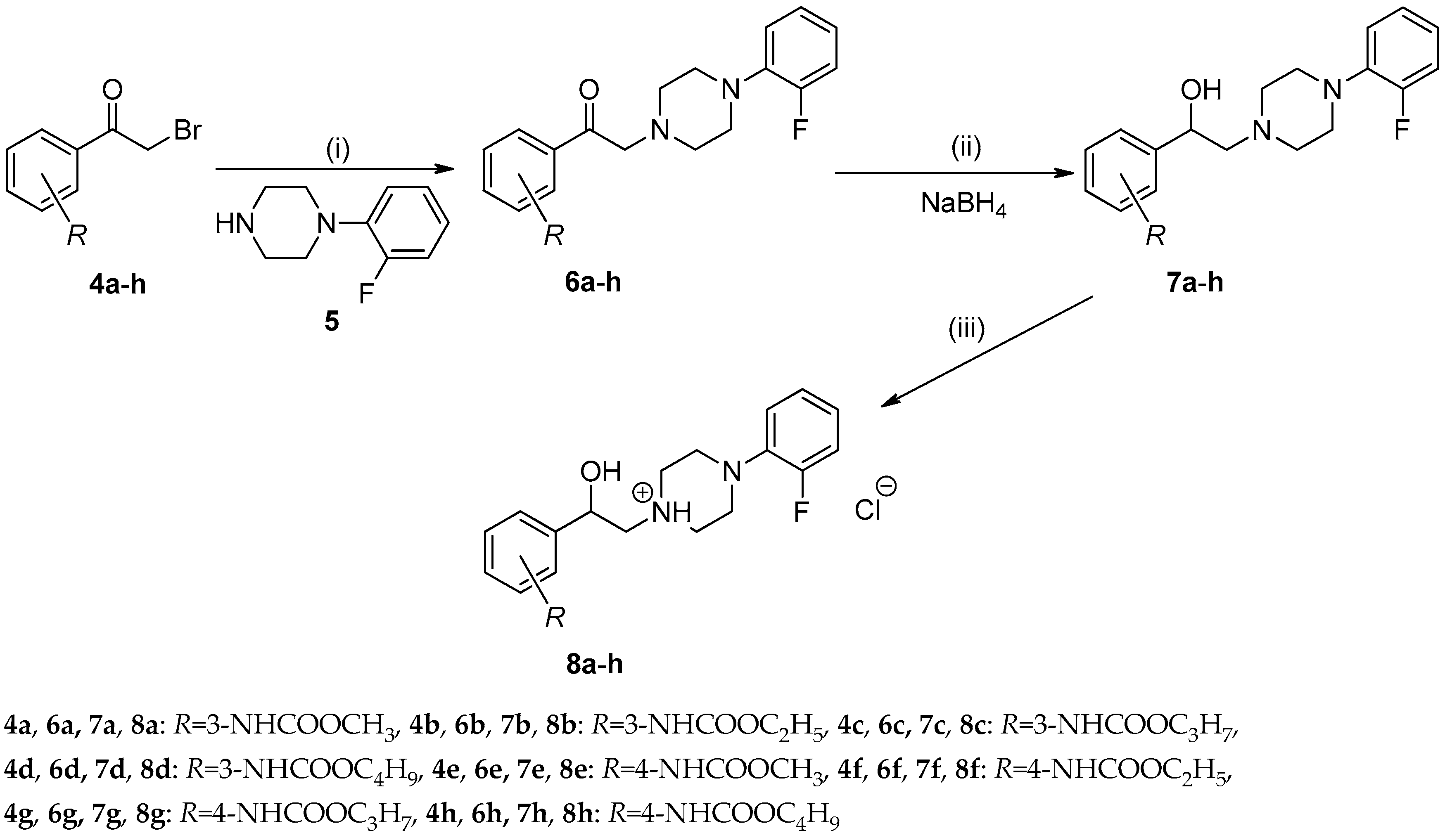
2.1. Chemistry
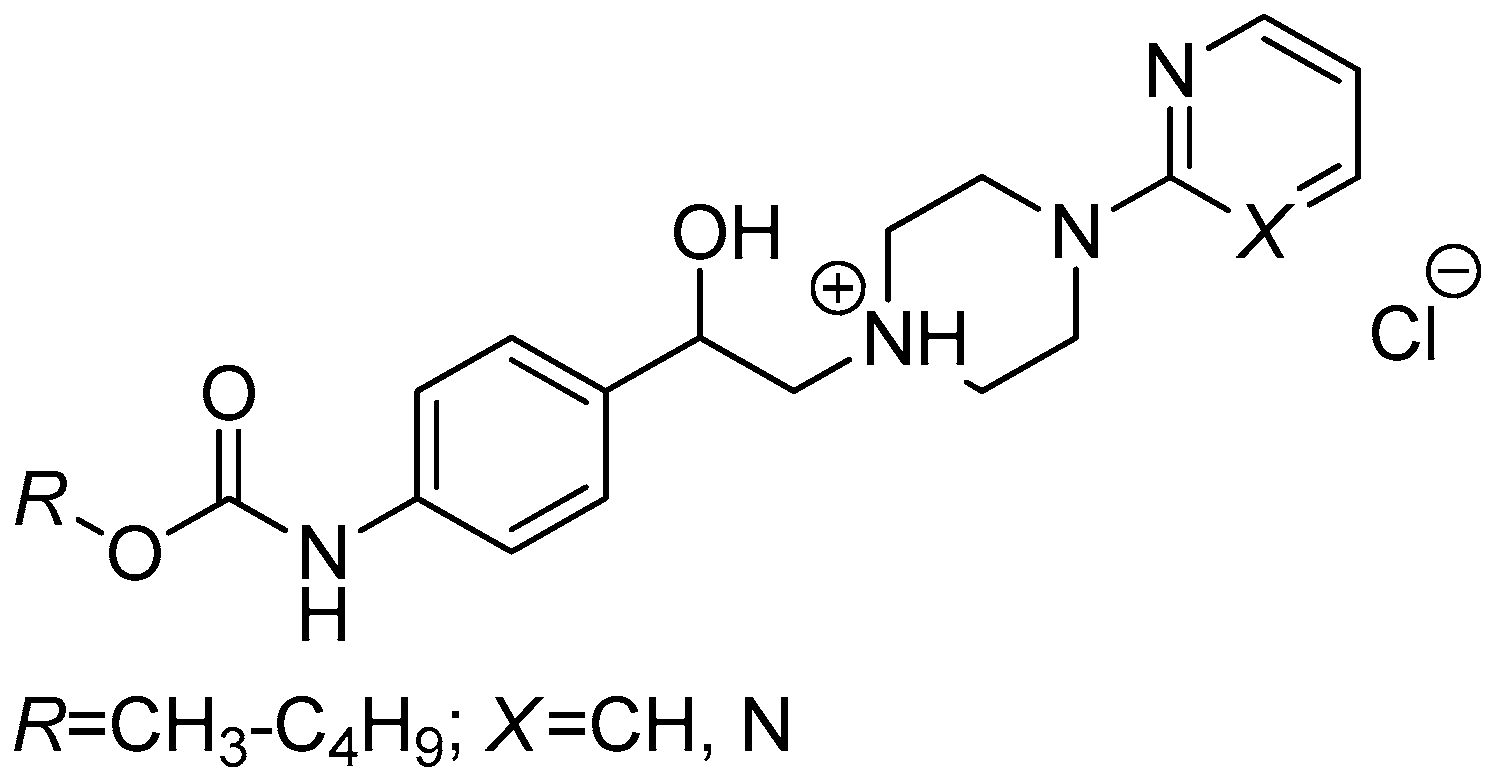
2.1.1. Synthesis and Spectral Characteristics
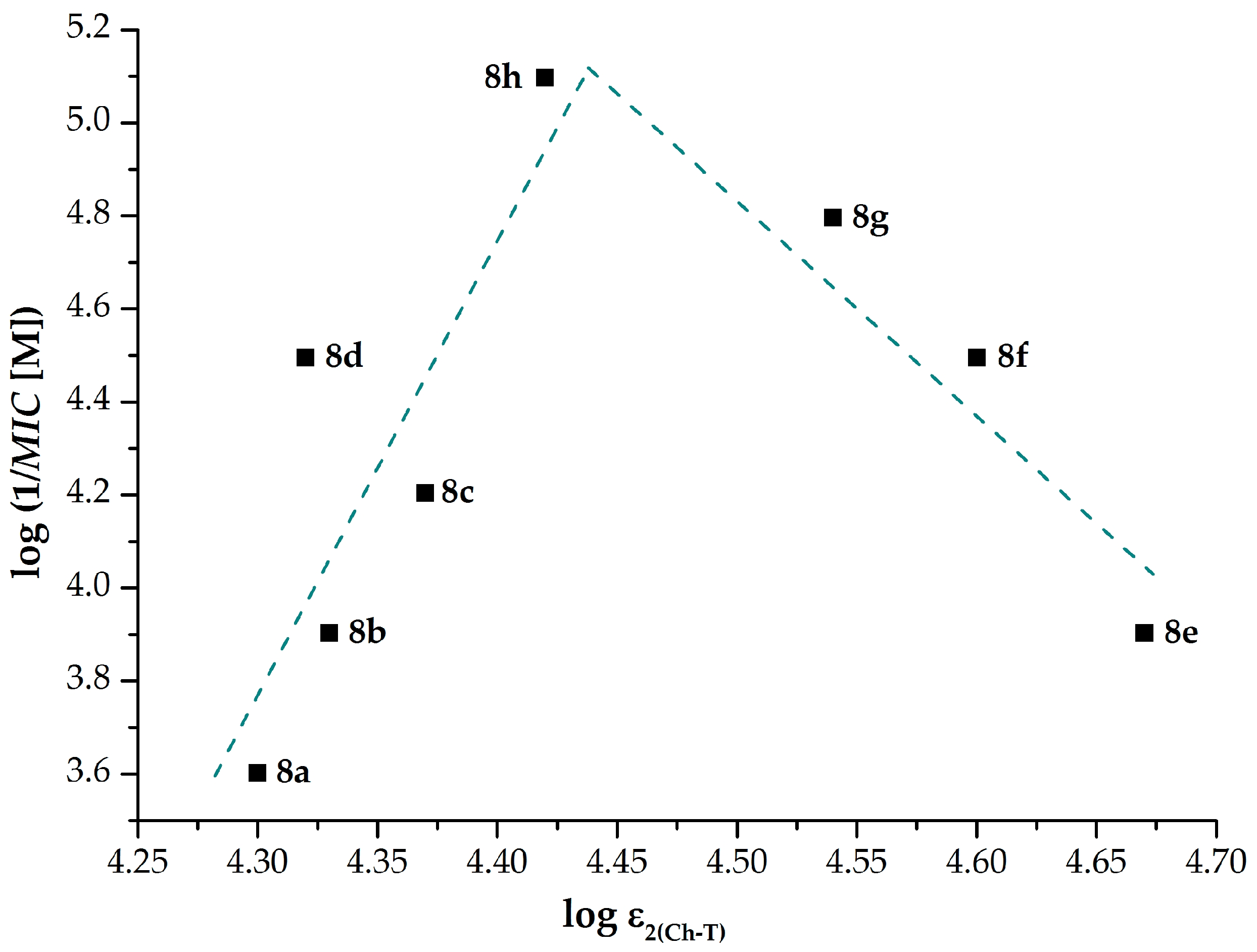
2.1.2. Lipohydrophilic Properties
NoR = 0.1409, F = 53.61, Prob > F = 0.0003, n = 8
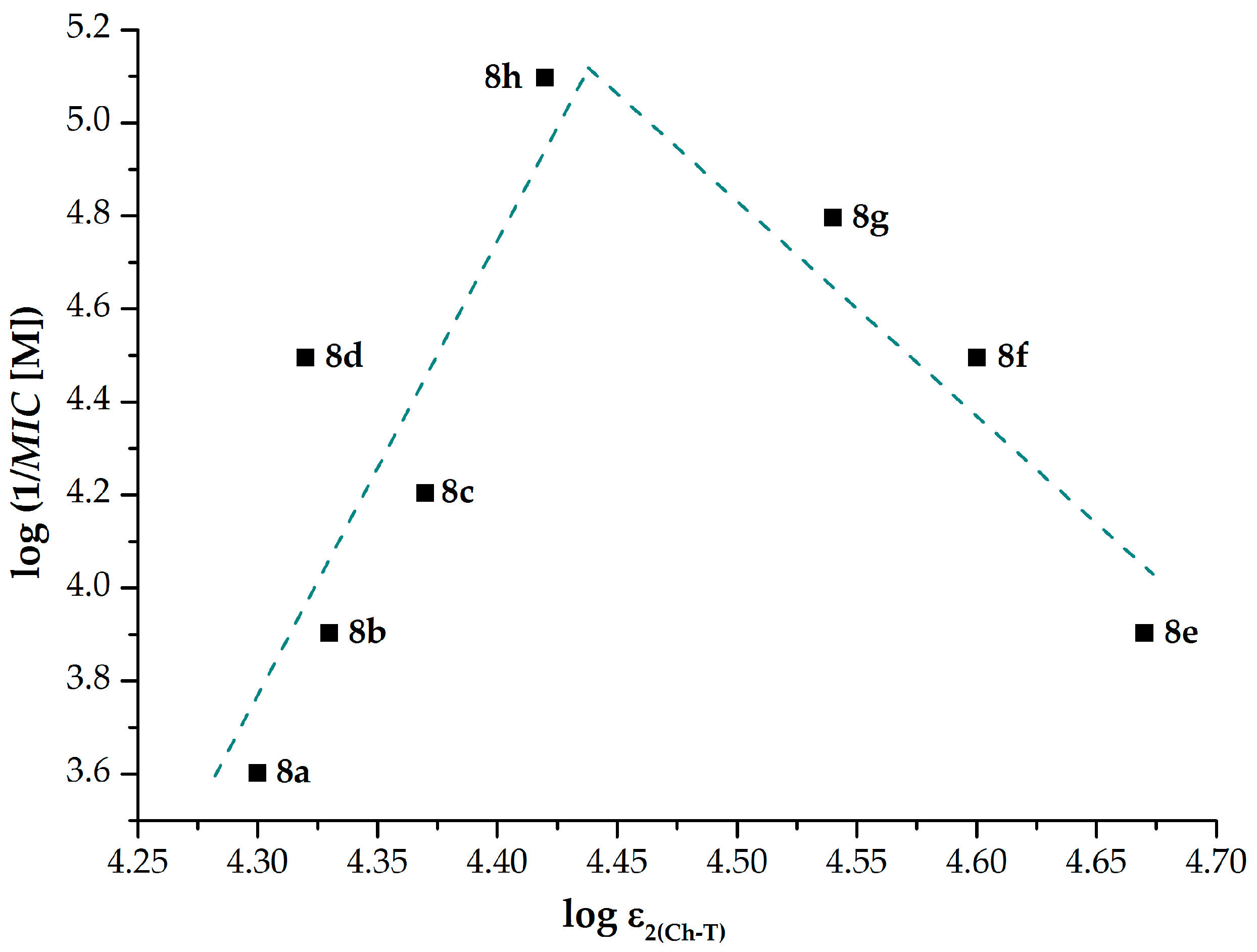
2.1.3. Electronic Properties
2.2. Biological Assays
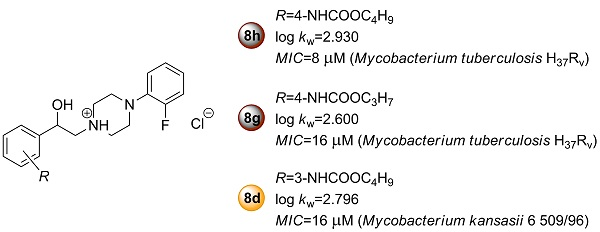
2.2.1. In Vitro Antimycobacterial Activity and Structure–Activity Relationships
2.2.2. In Vitro Cytotoxicity Screening
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Compounds
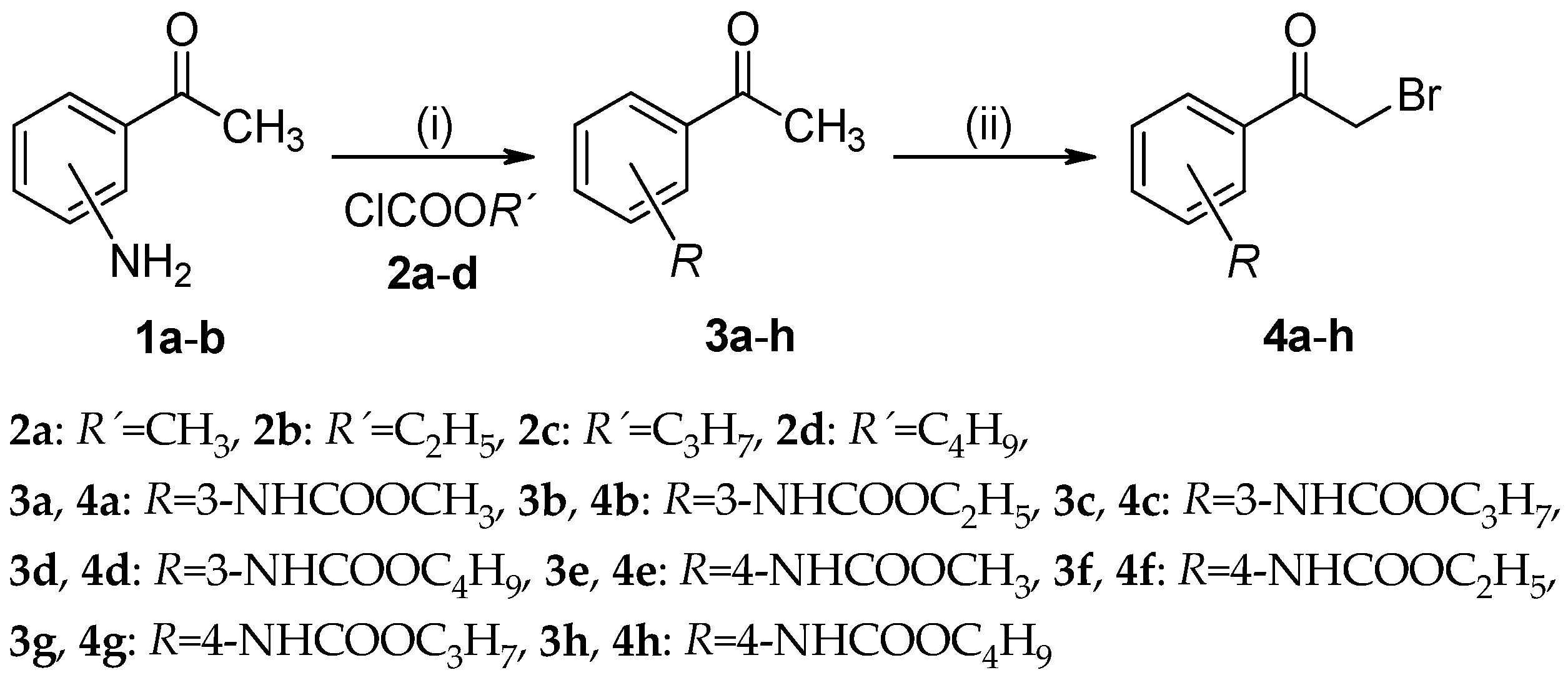
3.2.1. General Procedure For the Preparation of Alkyl (3-/4-Acetylphenyl)carbamates (3a–h)
3.2.2. General Procedure For the Preparation of Alkyl [3-/4-(Bromoacetyl)phenyl]carbamates (4a–h)
3.2.3. General Procedure For the Preparation of Alkyl {3-/4-[(4-(2-Fluorophenyl)piperazin-1-yl)-acetyl]phenyl}carbamates (6a–h)
3.2.4. General Procedure For the Preparation of Alkyl {3-/4-[2-(4-(2-Fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamates (7a–h)
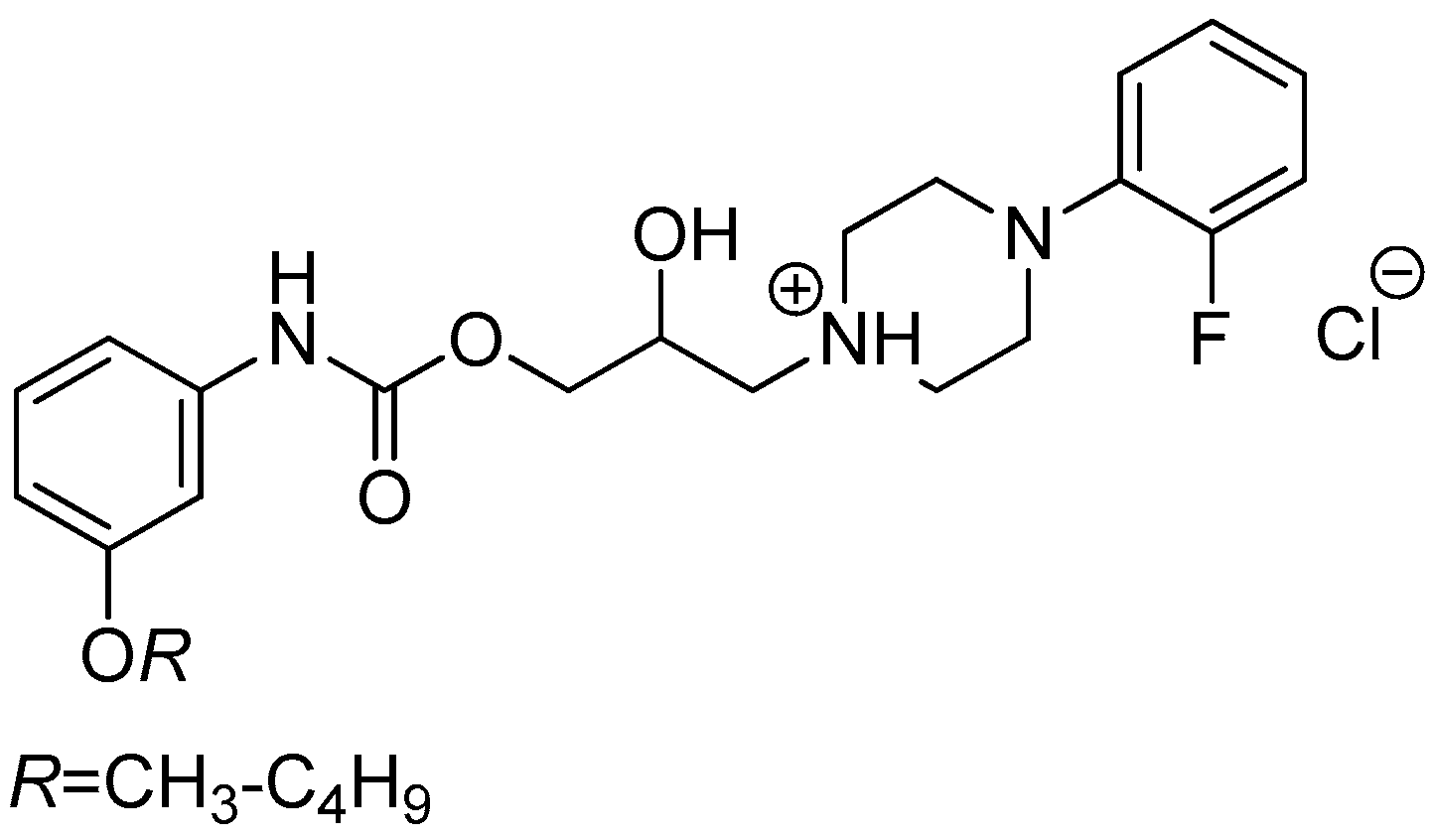
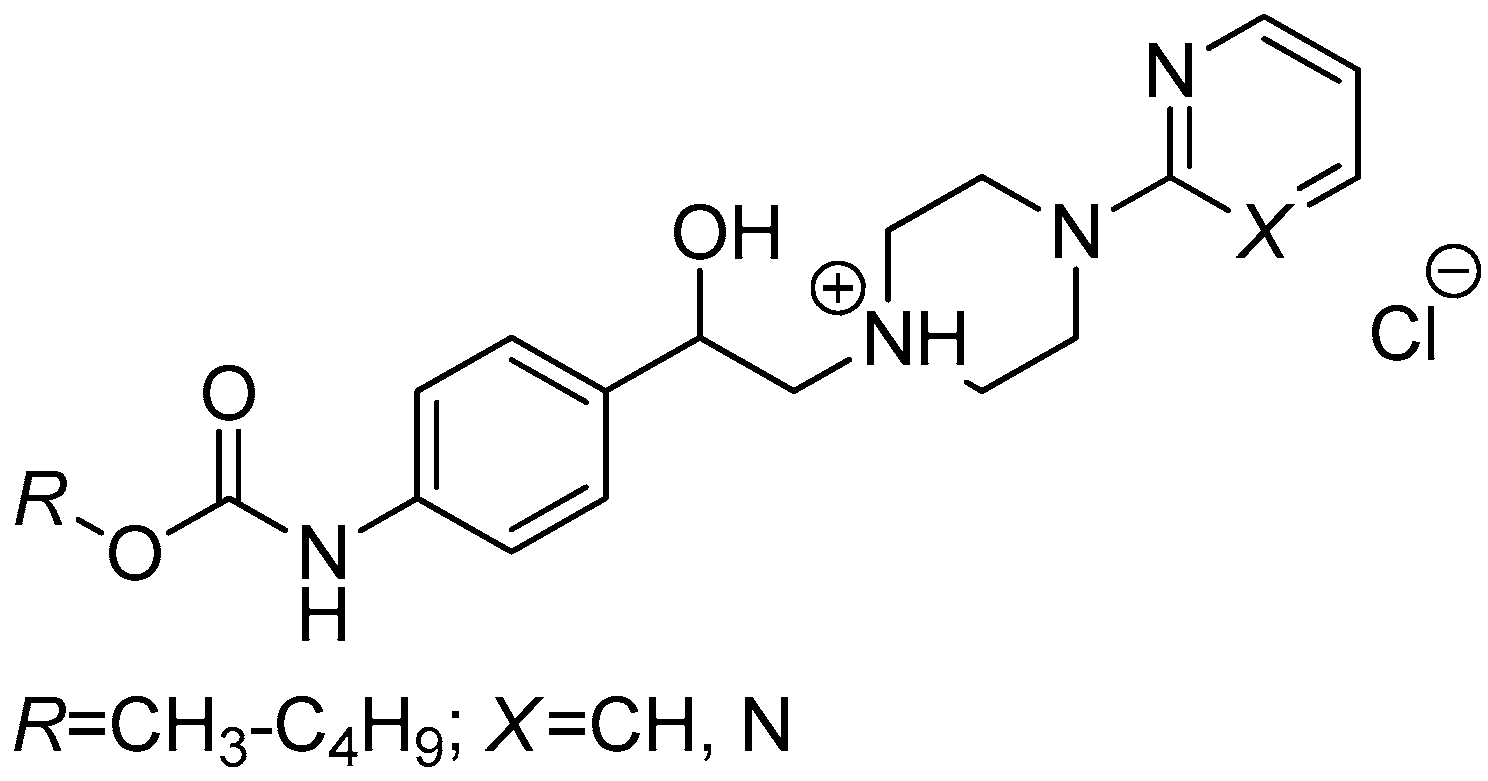
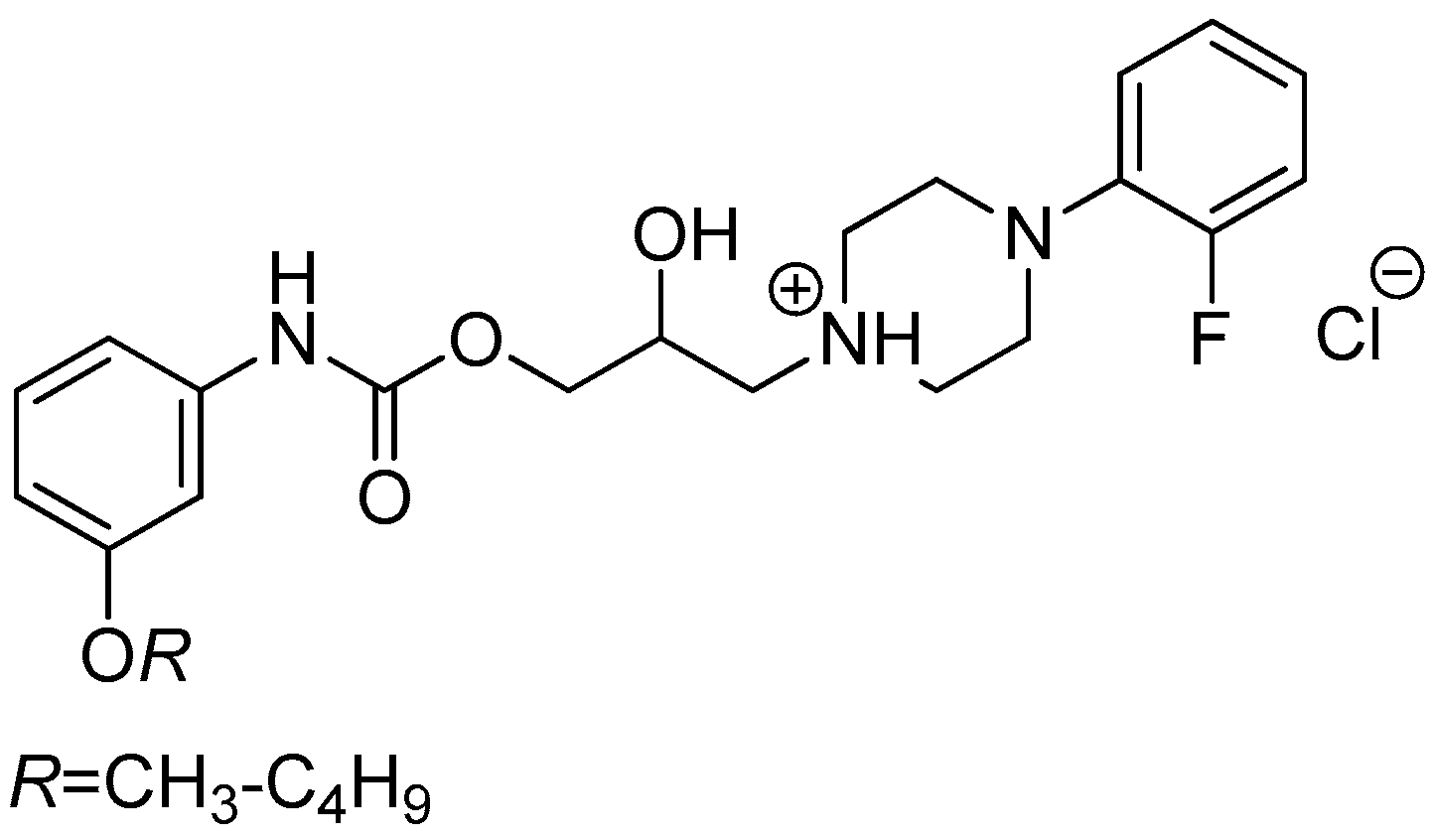
3.2.5. General Procedure For the Preparation of 1-(2-{3-/4-[(Alkoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium Chlorides (8a–h)
3.3. Lipophilicity Parameter Determination
3.3.1. Reversed-Phase Thin-Layer Chromatography (RP-TLC)
3.3.2. Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC)
3.4. Electronic Properties Determination
3.5. Biological Assays
3.5.1. In Vitro Antimycobaterial Evaluation
3.5.2. In Vitro Antiproliferative (Cytotoxicity) Screening
3.6. Calculations and Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 7-d | 7-Day incubation |
| 14-d/21-d | 14-/21-Day incubation |
| ϕM | Volume fraction of a mobile phase modifier (RP-HPLC) |
| CPX | Ciprofloxacin |
| Adj. R2 | Adjusted coefficient of determination (statistical analysis) |
| DMSO | Dimethyl sulfoxide |
| EMB | Ethambutol |
| F | Fisher´s F-test (Fisher´s significance ratio; statistical analysis) |
| INH | Isoniazid |
| k | Capacity (retention) factor (RP-HPLC) |
| log kw | Lipophilicity index; values extrapolated from intercepts of a linear relationship between the logarithm of retention factor k (log k) and volume fraction of a mobile phase modifier (ϕM; RP-HPLC) |
| MDR | Multi-drug resistant |
| MeOH | Methanol |
| MA | Mycobacterium avium |
| MIC | Minimum inhibitory concentration (in the μΜ units) |
| MK | Mycobacterium kansasii |
| Mp/mp | Melting point |
| MT | Mycobacterium tuberculosis |
| NoR | Norm of residuals (statistical analysis) |
| OFLX | Ofloxacin |
| Prob > F | Probability of obtaining the F Ratio (statistical analysis) |
| PZA | Pyrazinamide |
| RM | Lipophilicity index (RP-TLC) |
| RMSE | Root mean squared error (statistical analysis) |
| RSS | Residual sum of squares (statistical analysis) |
| S | Slope (RP-HPLC) |
| SAR | Structure–activity relationship(s) |
| tr | Retention time of a compound (RP-HPLC) |
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Bobesh, K.A.; Renuka, J.; Srilakshmi, R.R.; Yellanki, S.; Kulkarni, P.; Yogeeswari, P.; Sriram, D. Replacement of cardiotoxic aminopiperidine linker with piperazine moiety reduces cardiotoxicity? Mycobacterium tuberculosis novel bacterial topoisomerase inhibitors. Bioorg. Med. Chem. 2016, 24, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, S.; Feng, L.-S.; Li, X.-N.; Huang, G.-Ch.; Chai, Y.; Lv, Z.-S.; Guo, H.-Y.; Liu, M.-L. Synthesis and in vitro antimycobacterial and antibacterial activity of 8-OMe ciprofloxacin-hydrozone/azole hybrids. Molecules 2017, 22, 1171. [Google Scholar] [CrossRef] [PubMed]
- Kayukova, L.A.; Orazbaeva, M.A.; Bismilda, V.L.; Chingisova, L.T. Synthesis and antituberculosis activity of O-aroyl-β-(4-phenylpiperazin-1-yl)propioamidooximes. Pharm. Chem. J. 2010, 44, 17–20. [Google Scholar] [CrossRef]
- Keng Yoon, Y.; Ashraf Ali, M.; Choon, T.S.; Ismail, R.; Chee Wei, A.; Suresh Kumar, R.; Osman, H.; Beevi, F. Antituberculosis: Synthesis and antimycobacterial activity of novel benzimidazole derivatives. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Senthilkumar, P.; Sangaraju, D.; Nelli, R.; Banerjee, D.; Bhat, P.; Manjashetty, T.H. Synthesis and antimycobacterial evaluation of novel phthalazin-4-ylacetamides against log- and starved phase cultures. Chem. Biol. Drug. Des. 2010, 75, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Malinka, W.; Świątek, P.; Śliwińska, M.; Szponar, B.; Gamian, A.; Karczmarzyk, Z.; Fruziński, A. Synthesis of novel isothiazolopyridines and their in vitro evaluation against Mycobacterium and Propionibacterium acnes. Bioorg. Med. Chem. 2013, 21, 5282–5291. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, E.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Barbosa, F.; Einck, L.; Nacy, C.A.; Protopopova, M. Identification of new diamine scaffolds with activity against Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 3045–3048. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.G.; Baughn, C.; Cantrall, M.L.; Goodstein, B.; Thomas, J.P.; Wilkinson, R.G. Structure–activity studies leading to ethambutol, a new type of antituberculous compound. Ann. N. Y. Acad. Sci. 1966, 135, 686–710. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Protopopova, M.; Crooks, E.; Slayden, R.A.; Terrot, M.; Barry, C.E., III. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003, 5, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Stavrakov, G.; Valcheva, V.; Philipova, I.; Doytchinova, I. Novel camphane-based anti-tuberculosis agents with nanomolar activity. Eur. J. Med. Chem. 2013, 70, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Petkova, Z.; Valcheva, V.; Momekov, G.; Petrov, P.; Dimitrov, V.; Doytchinova, I.; Stavrakov, G.; Stoyanova, M. Antimycobacterial activity of chiral aminoalcohols with camphane scaffold. Eur. J. Med. Chem. 2014, 81, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed]
- Moraczewski, A.L.; Banaszynski, L.A.; From, A.M.; White, C.E.; Smith, B.D. Using hydrogen bonding to control carbamate C−N rotamer equilibria. J. Org. Chem. 1998, 63, 7258–7262. [Google Scholar] [CrossRef] [PubMed]
- Kečkéšová, S.; Sedlárová, E.; Čižmárik, J.; Garaj, V.; Csöllei, J.; Mokrý, P.; Andriamainty, F.; Malík, I.; Kaustová, J. Antimycobacterial activity of novel derivatives of arylcarbonyloxyaminopropanols. Čes. Slov. Farm. 2009, 58, 203–207. [Google Scholar]
- Tengler, J.; Kapustíková, I.; Peško, M.; Govender, R.; Keltošová, S.; Mokrý, P.; Kollár, P.; O’Mahony, J.; Coffey, A.; Kráľová, K.; et al. Synthesis and biological evaluation of 2-hydroxy-3-[(2-aryloxyethyl)amino]propyl-4-[(alkoxycarbonyl)amino]benzoates. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Maruniak, M.; Sedlárová, E.; Csöllei, J.; Kapustíková, I.; Mokrý, P.; Malík, I.; Havranová Sichrovská, Ľ.; Stanzel, L. Study of physicochemical properties and antimycobacterial activity of phenylcarbamic acid derivatives. In Advances in Pharmaceutical Chemistry, 1st ed.; Sedlárová, E., Malík, I., Garaj, V., Maruniak, M., Eds.; KO and KA Company: Bratislava, Slovak Republic, 2016; pp. 68–76. [Google Scholar]
- Waisser, K.; Dražková, K.; Čižmárik, J.; Kaustová, J. Antimycobacterial activity of basic ethylesters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2003, 48, 45–50. [Google Scholar] [CrossRef]
- Waisser, K.; Dražková, K.; Čižmárik, J.; Kaustová, J. Antimycobacterial activity of piperidinylpropyl esters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2003, 48, 585–587. [Google Scholar] [CrossRef]
- Waisser, K.; Dražková, K.; Čižmárik, J.; Kaustová, J. A new group of potential antituberculotics: Hydrochlorides of piperidinylalkyl esters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2004, 49, 265–268. [Google Scholar] [CrossRef]
- Hansch, C.; Clayton, J.M. Lipophilic character and biological activity of drugs II. The parabolic case. J. Pharm. Sci. 1973, 62, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Balgavý, P.; Devínsky, F. Cut-off effects in biological activities of surfactants. Adv. Colloid Interface Sci. 1996, 12, 23–63. [Google Scholar] [CrossRef]
- Waisser, K.; Dražková, K.; Čižmárik, J.; Kaustová, J. Influence of lipophilicity on the antimycobacterial activity of the hydrochlorides of piperidinylethyl esters of ortho-substituted phenylcarbamic acids. Sci. Pharm. 2004, 72, 43–49. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 13, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Vandavasi, J.K.; Kardile, R.A.; Lahore, S.V.; Dixit, S.S.; Deokar, H.S.; Shinde, P.D.; Sarmah, M.P.; Chattopadhyaya, J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- Parai, M.K.; Panda, G.; Chaturvedi, V.; Manju, Y.K.; Sinha, S. Thiophene containing triarylmethanes as antitubercular agents. Bioorg. Med. Chem. Lett. 2008, 18, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Kettmann, V.; Csöllei, J.; Račanská, E.; Švec, P. Synthesis and structure–activity relationships of new β-adrenoreceptor antagonists. Evidence for the electrostatic requirements for β-adrenoreceptor antagonists. Eur. J. Med. Chem. 1991, 26, 843–851. [Google Scholar] [CrossRef]
- Kiss, A.; Potor, A.; Hell, Z. Heterogeneous catalytic solvent-free synthesis of quinoline derivatives via the Friedländer Reaction. Catal. Lett. 2008, 125, 250–253. [Google Scholar] [CrossRef]
- Broutin, P.-E.; Hilty, P.; Thomas, A.W. An efficient synthesis of ortho-N-Boc-arylmethyl ketone derivatives. Tetrahedron Lett. 2003, 44, 6429–6432. [Google Scholar] [CrossRef]
- Kolosov, M.A.; Orlov, V.D. 5-Thiazolyl derivatives of 4-aryl-3,4-dihydropyrimidin-2(1H)-ones. Chem. Heterocycl. Compd. 2008, 44, 1418–1420. [Google Scholar] [CrossRef]
- Hu, B.; Ellingboe, J.; Han, S.; Largis, E.; Lim, K.; Malamas, M.; Mulvey, R.; Niu, C.; Oliphant, A.; Pelletier, J.; et al. Novel (4-piperidin-1-yl)-phenyl sulfonamides as potent and selective human β3 agonists. Bioorg. Med. Chem. 2001, 9, 2045–2059. [Google Scholar] [CrossRef]
- Pan, Y.; Li, P.; Xie, S.; Tao, Y.; Chen, D.; Dai, M.; Hao, H.; Huang, L.; Wang, Y.; Wang, L.; et al. Synthesis, 3D-QSAR analysis and biological evaluation of quinoxaline 1,4-di-N-oxide derivatives as antituberculosis agents. Bioorg. Med. Chem. Lett. 2016, 26, 4146–4153. [Google Scholar] [CrossRef] [PubMed]
- Pancholia, S.; Dhameliya, T.M.; Shah, P.; Jadhavar, P.S.; Sridevi, J.P.; Yogeshwari, P.; Sriram, D.; Chakraborti, A.K. Benzo[d]thiazol-2-yl(piperazin-1-yl)methanones as new anti-mycobacterial chemotypes: Design, synthesis, biological evaluation and 3D-QSAR studies. Eur. J. Med. Chem. 2016, 116, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Rajkhowa, S.; Deka, R.C. DFT Based QSAR/QSPR models in the development of novel anti-tuberculosis drugs targeting Mycobacterium tuberculosis. Curr. Pharm. Des. 2014, 20, 4455–4473. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.D.; More, U.A.; Aminabhavi, T.M.; Badiger, A.M. Two- and three-dimensional QSAR studies on a set of antimycobacterial pyrroles: CoMFA, topomer CoMFA, and HQSAR. Med. Chem. Res. 2014, 23, 107–126. [Google Scholar] [CrossRef]
- Pliška, V.; Testa, B.; van de Waterbeemd, H. Lipophilicity in drug action and toxicology. In Methods and Principles of Medicinal Chemistry; Mannhold, R., Kubinyi, H., Timmerman, H., Eds.; Wiley-VCh Publishers: Weinheim, Germany, 1996; Volume 4, pp. 1–6. [Google Scholar]
- Ottaviani, M.F.; Leonardis, I.; Cappiello, A.; Cangiotti, M.; Mazzeo, R.; Trufelli, H.; Palma, P. Structural modifications and adsorption capability of C18-silica/binary solvent interphases studied by EPR and RP-HPLC. J. Colloid Interface Sci. 2010, 352, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Dolan, J.W. Initial experiments in high-performance liquid chromatographic method development I. Use of a starting gradient run. J. Chromatogr. A. 1996, 721, 3–14. [Google Scholar] [CrossRef]
- Du, Ch.M.; Valko, K.; Bevan, Ch.; Reynolds, D.; Abraham, M.H. Rapid method for estimating octanol–water partition coefficient (log Poct) from isocratic RP-HPLC and a hydrogen bond acidity term (A). J. Liqud Chromatogr. Relat. Technol. 2001, 24, 635–649. [Google Scholar] [CrossRef]
- Terada, H. Determination of log Poct by high-performance liquid chromatography, and its application in the study of Quantitative Structure–Activity Relationships. Quant. Struct. Act. Relat. 1986, 5, 81–88. [Google Scholar] [CrossRef]
- Snyder, L.R.; Dolan, J.W.; Grant, J.R. Gradient elution in high-performance liquid chromatography: I. Theoretical basis for reversed-phase systems. J. Chromatogr. A 1979, 165, 3–30. [Google Scholar] [CrossRef]
- Valkó, K.; Snyder, L.R.; Glajch, J.L. Retention in reversed-phase liquid chromatography as a function of mobile-phase composition. J. Chromatogr. A 1993, 656, 501–520. [Google Scholar] [CrossRef]
- Soczewiński, E. Mechanistic molecular model of liquid–solid chromatography: Retention–eluent composition relationships. J. Chromatogr. A 2002, 965, 109–116. [Google Scholar] [CrossRef]
- Vrakas, D.; Panderi, I.; Hadjipavlou-Litina, D.; Tsantili-Kakoulidou, A. Investigation of the relationships between log P and various chromatographic indices for a series of substituted coumarins. Evaluation of their similarity/dissimilarity using multivariate statistics. QSAR Comb. Sci. 2005, 24, 254–260. [Google Scholar] [CrossRef]
- Sztanke, K.; Markowski, W.; Świeboda, R.; Polak, B. Lipophilicity of novel antitumour and analgesic active 8-aryl-2,6,7,8-tetrahydroimidazo[2,1-c][1,2,4]triazine-3,4-dione derivatives determined by reversed-phase HPLC and computational methods. Eur. J. Med. Chem. 2010, 45, 2644–2649. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.D.S. Ultraviolet and visible spectroscopy. In Organic Spectroscopy; Yadav, L.D.S., Ed.; Springer: Amsterdam, The Netherlands, 2005; pp. 7–51. [Google Scholar]
- Férriz, J.M.; Vávrová, K.; Kunc, F.; Imramovský, A.; Stolaříková, J.; Vavříková, E.; Vinšová, J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010, 18, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard, 8th ed.; CLSI Document M11-A8; CLSI: Wayne, NJ, USA, 2012; pp. 10–56. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing, 24th ed.; Informational Supplement M100-S24; CLSI: Wayne, NJ, USA, 2014; pp. 106–211. [Google Scholar]
- Waisser, K.; Doležal, R.; Čižmárik, J.; Malík, I.; Kaustová, J. The potential antituberculotics of the series of 2-hydroxy-3-(4-phenylpiperazin-1-yl)-propylphenylcarbamates. Folia Pharm. Univ. Carol. 2007, 35–36, 45–48. [Google Scholar]
- Doležal, M.; Zitko, J.; Kešetovičová, D.; Kuneš, J.; Svobodová, M. Substituted N-phenylpyrazine-2-carboxamides: Synthesis and antimycobacterial evaluation. Molecules 2009, 14, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Čižmárik, J.; Waisser, K.; Doležal, R. QSAR Study of antimicrobial activity of esters of substituted phenylcarbamic acid. Acta Fac. Pharm. Univ. Comen. 2008, 55, 90–95. [Google Scholar]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Forbes, M.; Kuck, N.A.; Peets, E.A. Mode of action of ethambutol. J. Bacteriol. 1962, 84, 1099–1103. [Google Scholar] [PubMed]
- Jena, L.; Waghmare, P.; Kashikar, S.; Kumar, S.; Harinath, B.C. Computational approach to understanding the mechanism of action of isoniazid, an anti-TB drug. Int. J. Mycobacteriol. 2014, 3, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Kuck, N.A.; Peets, E.A.; Forbes, M. Mode of action of ethambutol on Mycobacterium tuberculosis, strain H37Rv. Am. Rev. Respir. Dis. 1963, 87, 905–906. [Google Scholar] [PubMed]
- Mikusová, K.; Slayden, R.A.; Besra, G.S.; Brennan, P.J. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 1995, 39, 2484–2489. [Google Scholar] [CrossRef] [PubMed]
- Lata, M.; Sharma, D.; Kumar, B.; Deo, N.; Tiwari, P.K.; Bisht, D.; Venkatesan, K. Proteome analysis of ofloxacin and moxifloxacin induced Mycobacterium tuberculosis isolates by proteomic approach. Protein Pept. Lett. 2015, 22, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.; Pan, X.S.; Fisher, L.M.; Jarlier, V.; Cambau, E. Mycobacterium tuberculosis DNA gyrase: Interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 2004, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- De Wijs, H.; Jollès, P. Cell walls of three strains of mycobacteria (Mycobacterium phlei, Mycobacterium fortuitum and Mycobacterium kansasii): Preparation, analysis and digestion by lysozymes of different origins. Biochim. Biophys. Acta 1964, 83, 326–332. [Google Scholar] [CrossRef]
- Suffness, M.; Douros, J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Witek, S.; Bielawski, J.; Bielawska, A. Synthesis of N-(formylphenyl)- and N-(acetophenyl) derivatives of urea and carbamic acid. J. Prakt. Chem. 1979, 321, 804–812. [Google Scholar] [CrossRef]
- Takeuchi, H.; Mastubara, E. Electrophilic aromatic N-substitution by ethoxycarbonylnitrenium ion generated from ethyl azidoformate in the presence of trifluoroacetic acid. J. Chem. Soc. Perkin Trans. 1984, 1, 981–985. [Google Scholar] [CrossRef]
- Park, Ch.-H.; Givens, R.S. New photoactivated protecting groups. 6. p-Hydroxyphenacyl: A phototrigger for chemical and biochemical probes. J. Am. Chem. Soc. 1997, 119, 2453–2463. [Google Scholar] [CrossRef]
- Basterfield, S.; Woods, E.L.; Wright, H.N. Studies in urethans. III. The preparation of various substituted urethans. J. Am. Chem. Soc. 1926, 48, 2371–2375. [Google Scholar] [CrossRef]
- Smith Broadbent, H.; Chu, C.-Y. The carbethoxylation products of p-aminoacetophenone and p-dimethylaminoacetophenone. J. Am. Chem. Soc. 1953, 75, 226–227. [Google Scholar] [CrossRef]
- Sigman, E.M.; Autrey, T.; Schuster, G.B. Aroylnitrenes with singlet ground states: Photochemistry of acetyl-substituted aroyl and aryloxycarbonyl azides. J. Am. Chem. Soc. 1988, 110, 4297–4305. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Angelina, E.; Lima, S.; Gonec, T.; Otevrel, J.; Marvanova, P.; Padrtova, T.; Mokry, P.; Bobal, P.; Acosta, L.M.; et al. An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Bietti, G.; Cereda, E.; Donetti, A.; del Soldato, P.; Giachetti, A.; Micheletti, R. Guanidino-heterocyclyl-phenyl-amidines and Salts Thereof. U.S. Patent No. US4548944 A. Available online: https://encrypted.google.com/patents/US4548944?cl=un (accessed on 12 November 2017).
- Rather, J.B.; Reid, E.E. The identification of acids. IV. Phenacyl esters. J. Am. Chem. Soc. 1919, 41, 75–83. [Google Scholar] [CrossRef]
- Dross, K.; Rekker, R.F.; de Vries, G.; Mannhold, R. The lipophilic behaviour of organic compounds: 3. The search for interconnections between reversed-phase chromatographic data and log Pfoct values. Quant. Struct. Act. Relat. 1999, 18, 549–557. [Google Scholar] [CrossRef]
- Kulig, K.; Malawska, B. Estimation of the lipophilicity of antiarrhythmic and antihypertensive active 1-substituted pyrrolidin-2-one and pyrrolidine derivatives. Biomed. Chromatogr. 2003, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Özden, S.; Atabey, D.; Yıldız, S.; Göker, H. Synthesis, potent anti-staphylococcal activity and QSARs of some novel 2-anilinobenzazoles. Eur. J. Med. Chem. 2008, 43, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]benz-amides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; O’Mahony, J.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.A.; Tatar, J.F. Calculation of the residual sum of squares for all possible regressions. Technometrics 1972, 14, 317–325. [Google Scholar] [CrossRef]
- Cheng, B.; Tong, H. On residual sums of squares in non-parametric autoregression. Stoch. Process. Their Appl. 1983, 48, 157–174. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches. In Methods and Principles in Medicinal Chemistry; Mannhold, R., Krogsgaard-Larsen, P., Timmerman, H., Eds.; Wiley-VCh Verlag: Weinheim, Germany, 1993; Volume 1, pp. 22–56. [Google Scholar]
- Weisberg, S. Multiple Regression. In Applied Linear Regression, 3rd ed.; Weisberg, S., Ed.; Wiley-Interscience (John Wiley and Sons): Hoboken, NJ, USA, 2005; pp. 47–68. [Google Scholar] [CrossRef]
- Nakagawa, S.; Schielzeth, H. General and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 2013, 4, 133–142. [Google Scholar] [CrossRef]
- Mevik, B.H.; Cederkvist, H.R. Mean squared error of prediction (MSEP) estimates for principal component regression (PCR) and partial least squares regression (PLSR). J. Chemom. 2004, 18, 422–429. [Google Scholar] [CrossRef]
- Ying, X.; Yang, L.; Zha, H. A fast algorithm for multidimensional ellipsoid-specific fitting by minimizing a new defined vector norm of residuals using semidefinite programming. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 34, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 8a–h are available from the authors Tomáš Goněc and Ivan Malík. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. | 1 RM | Mobile phase MeOH/water (v/v) | |||||||
| 60:40 | 70:30 | 80:20 | 85:15 | ||||||
| tr (min) | log k | tr (min) | log k | tr (min) | log k | tr (min) | log k | ||
| 8a | −0.55 | 6.593 | 0.612 | 3.587 | 0.248 | 2.493 | −0.034 | 2.200 | −0.156 |
| 8b | −0.35 | 7.707 | 0.695 | 4.893 | 0.444 | 2.907 | 0.095 | 2.433 | −0.056 |
| 8c | −0.16 | 8.933 | 0.771 | 5.907 | 0.552 | 3.193 | 0.166 | 2.620 | 0.010 |
| 8d | 0.01 | 10.021 | 0.829 | 7.820 | 0.702 | 3.586 | 0.245 | 2.830 | 0.074 |
| 8e | −0.02 | 5.307 | 0.491 | 3.180 | 0.163 | 2.360 | −0.085 | 2.120 | −0.196 |
| 8f | 0.19 | 7.153 | 0.656 | 3.953 | 0.312 | 2.620 | 0.010 | 2.275 | −0.121 |
| 8g | 0.39 | 8.280 | 0.732 | 5.320 | 0.492 | 3.033 | 0.128 | 2.533 | −0.020 |
| 8h | 0.63 | 11.035 | 0.881 | 7.287 | 0.665 | 3.543 | 0.240 | 2.814 | 0.069 |
| Comp. | log kw | 1 S | 2 RSS | 3 R | 4 Adj. R2 | 5 RMSE | 6 NoR | 7 F | 8 Prob > F |
|---|---|---|---|---|---|---|---|---|---|
| 8a | 2.430 | 3.0678 | 0.0023 | 0.9967 | 0.9902 | 0.0337 | 0.0477 | 305.70 | 0.0033 ** |
| 8b | 2.546 | 3.0529 | 0.0017 | 0.9975 | 0.9925 | 0.0294 | 0.0415 | 398.96 | 0.0025 ** |
| 8c | 2.679 | 3.1244 | 0.0051 | 0.9930 | 0.9791 | 0.0504 | 0.0713 | 141.72 | 0.0070 ** |
| 8d | 2.796 | 3.1641 | 0.0208 | 0.9730 | 0.9201 | 0.1019 | 0.1441 | 35.58 | 0.0270 * |
| 8e | 2.113 | 2.7386 | 0.0019 | 0.9965 | 0.9896 | 0.0311 | 0.0440 | 285.13 | 0.0035 ** |
| 8f | 2.512 | 3.1156 | 0.0009 | 0.9988 | 0.9964 | 0.0208 | 0.0294 | 826.63 | 0.0012 ** |
| 8g | 2.600 | 3.0739 | 0.0027 | 0.9962 | 0.9885 | 0.0367 | 0.0519 | 259.09 | 0.0038 ** |
| 8h | 2.930 | 3.3441 | 0.0081 | 0.9903 | 0.9710 | 0.0637 | 0.0901 | 101.50 | 0.0097 ** |
| Comp. | λ1 (nm) | log ε1 | λ2(Ch-T) (nm) | 1 log ε2(Ch-T) | λ3 (nm) | log ε3 |
|---|---|---|---|---|---|---|
| 8a | 210 | 4.30 | 238 | 4.30 | 276 | 3.45 |
| 8b | 210 | 4.31 | 238 | 4.33 | 276 | 3.40 |
| 8c | 210 | 4.30 | 238 | 4.37 | 276 | 3.42 |
| 8d | 210 | 4.31 | 238 | 4.32 | 276 | 3.49 |
| 8e | 210 | 4.61 | 240 | 4.67 | 274 | 3.67 |
| 8f | 208 | 4.59 | 240 | 4.60 | 274 | 3.60 |
| 8g | 210 | 4.47 | 240 | 4.54 | 274 | 3.52 |
| 8h | 208 | 4.34 | 240 | 4.42 | 274 | 3.42 |
| Comp. | MIC (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MT My 331/88 | MK My 235/80 | MK 6 509/96 | MA My 330/88 | |||||||
| 1 14 d | 2 21 d | 3 7 d | 14 d | 21 d | 7 d | 14 d | 21 d | 14 d | 21 d | |
| 8a | 250 | 250 | 125 | 500 | 1000 | 125 | 500 | 500 | 500 | 500 |
| 8b | 125 | 125 | 62.5 | 250 | 250 | 62.5 | 250 | 250 | 250 | 250 |
| 8c | 62.5 | 62.5 | 62.5 | 125 | 125 | 32 | 125 | 125 | 125 | 250 |
| 8d | 32 | 32 | 32 | 62.5 | 62.5 | 16 | 32 | 62.5 | 62.5 | 62.5 |
| 8e | 125 | 125 | 125 | 500 | 500 | 125 | 500 | 500 | 250 | 500 |
| 8f | 32 | 62.5 | 125 | >250 | >250 | 62.5 | >125 | >125 | >250 | >250 |
| 8g | 16 | 16 | 125 | >250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 8h | 8 | 8 | 62.5 | >125 | >125 | 125 | >250 | >250 | >250 | 250 |
| INH | 0.5 | 0.5 | >250 | >250 | >250 | 4 | 8 | 8 | >250 | >250 |
| EMB | 1 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 16 | 16 |
| OFLX | 1 | 2 | 0.5 | 1 | 1 | 0.5 | 0.5 | 1 | 32 | 62.5 |
| Comp. | MIC (μM) | |||
|---|---|---|---|---|
| MK | MAP | MAI | MAH | |
| CIT11/06 | CIT03 | ATCC 13950 | CIT10/08 | |
| 8a | >610 | >610 | >610 | >610 |
| 8b | 295 | >590 | >590 | >590 |
| 8c | >571 | >571 | >571 | >571 |
| 8d | >553 | > 53 | >553 | >553 |
| 8e | >610 | >610 | >610 | >610 |
| 8f | 295 | >590 | >590 | >590 |
| 8g | >571 | >571 | >71 | >571 |
| 8h | >553 | >553 | >553 | >553 |
| INH | >1823 | >1823 | >1823 | >1823 |
| CPX | >91 | 181 | 181 | 181 |
| PZA | >2031 | >2031 | >2031 | 487 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goněc, T.; Malík, I.; Csöllei, J.; Jampílek, J.; Stolaříková, J.; Solovič, I.; Mikuš, P.; Keltošová, S.; Kollár, P.; O’Mahony, J.; et al. Synthesis and In Vitro Antimycobacterial Activity of Novel N-Arylpiperazines Containing an Ethane-1,2-diyl Connecting Chain. Molecules 2017, 22, 2100. https://doi.org/10.3390/molecules22122100
Goněc T, Malík I, Csöllei J, Jampílek J, Stolaříková J, Solovič I, Mikuš P, Keltošová S, Kollár P, O’Mahony J, et al. Synthesis and In Vitro Antimycobacterial Activity of Novel N-Arylpiperazines Containing an Ethane-1,2-diyl Connecting Chain. Molecules. 2017; 22(12):2100. https://doi.org/10.3390/molecules22122100
Chicago/Turabian StyleGoněc, Tomáš, Ivan Malík, Jozef Csöllei, Josef Jampílek, Jiřina Stolaříková, Ivan Solovič, Peter Mikuš, Stanislava Keltošová, Peter Kollár, Jim O’Mahony, and et al. 2017. "Synthesis and In Vitro Antimycobacterial Activity of Novel N-Arylpiperazines Containing an Ethane-1,2-diyl Connecting Chain" Molecules 22, no. 12: 2100. https://doi.org/10.3390/molecules22122100