Inhibition by Commercial Aminoglycosides of Human Connexin Hemichannels Expressed in Bacteria

 and
and
Abstract
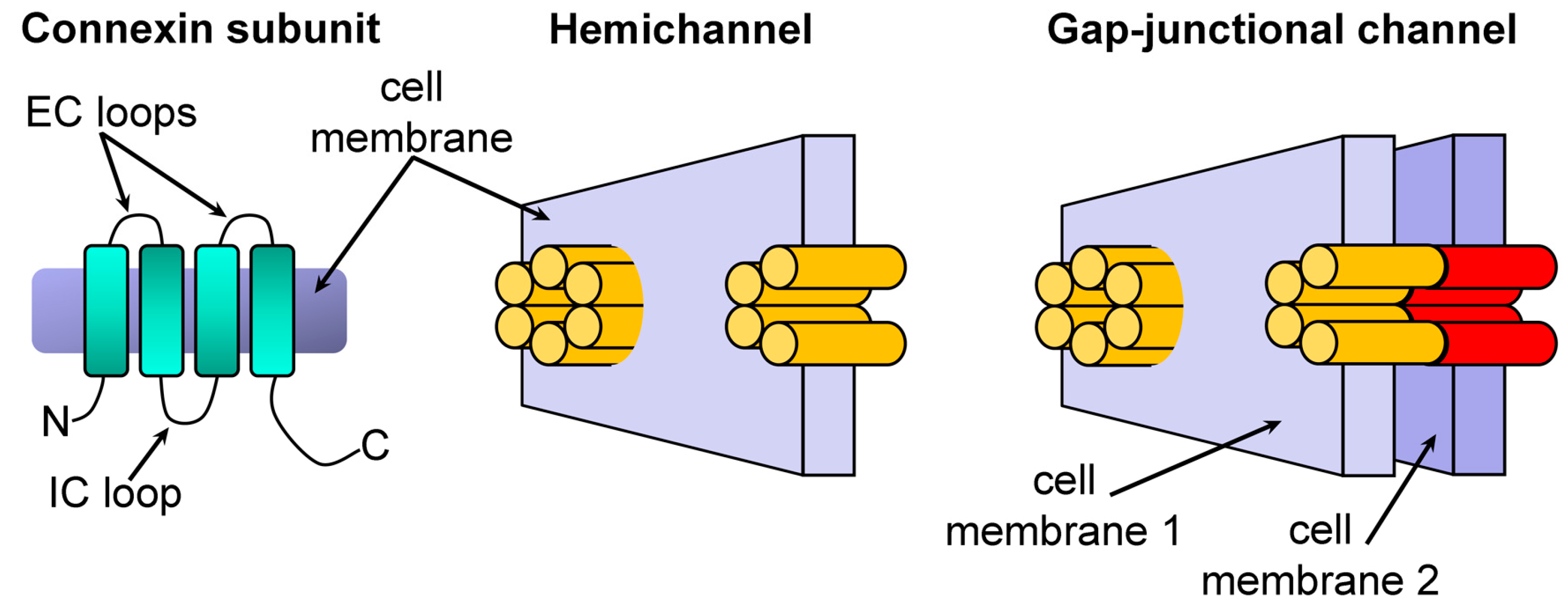
:1. Introduction
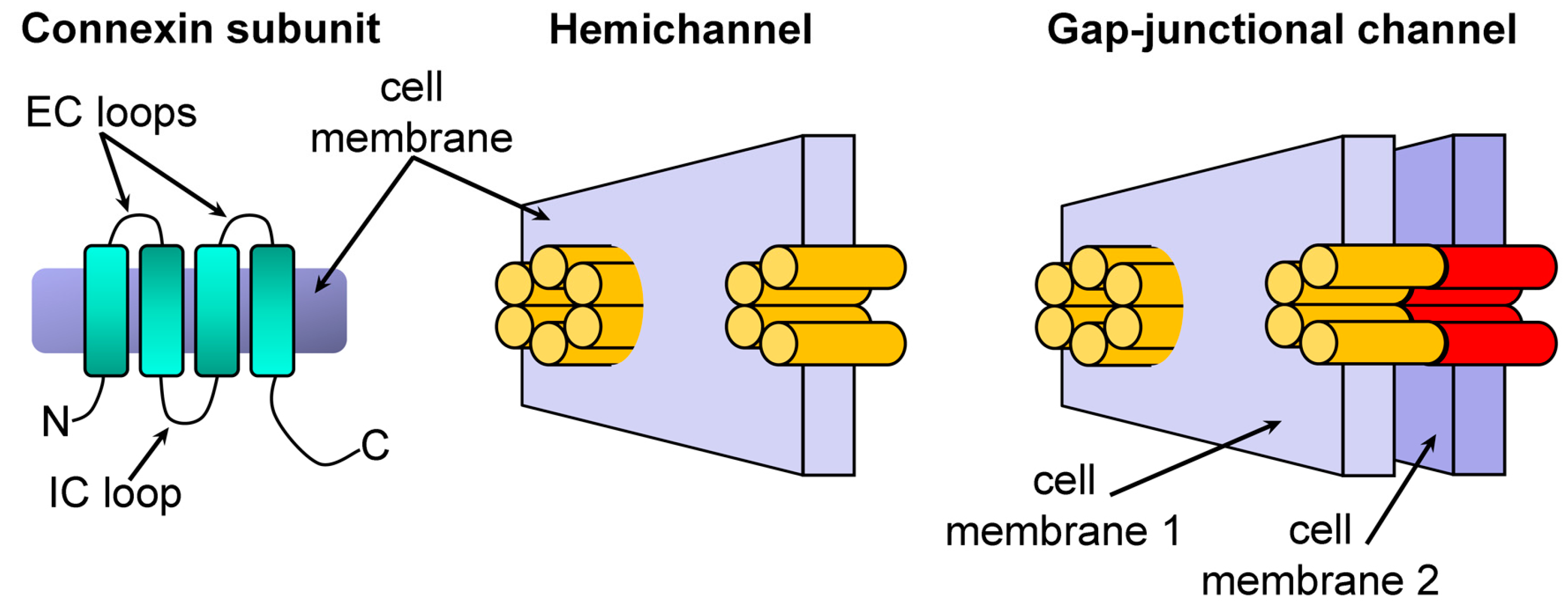
2. Results and Discussion
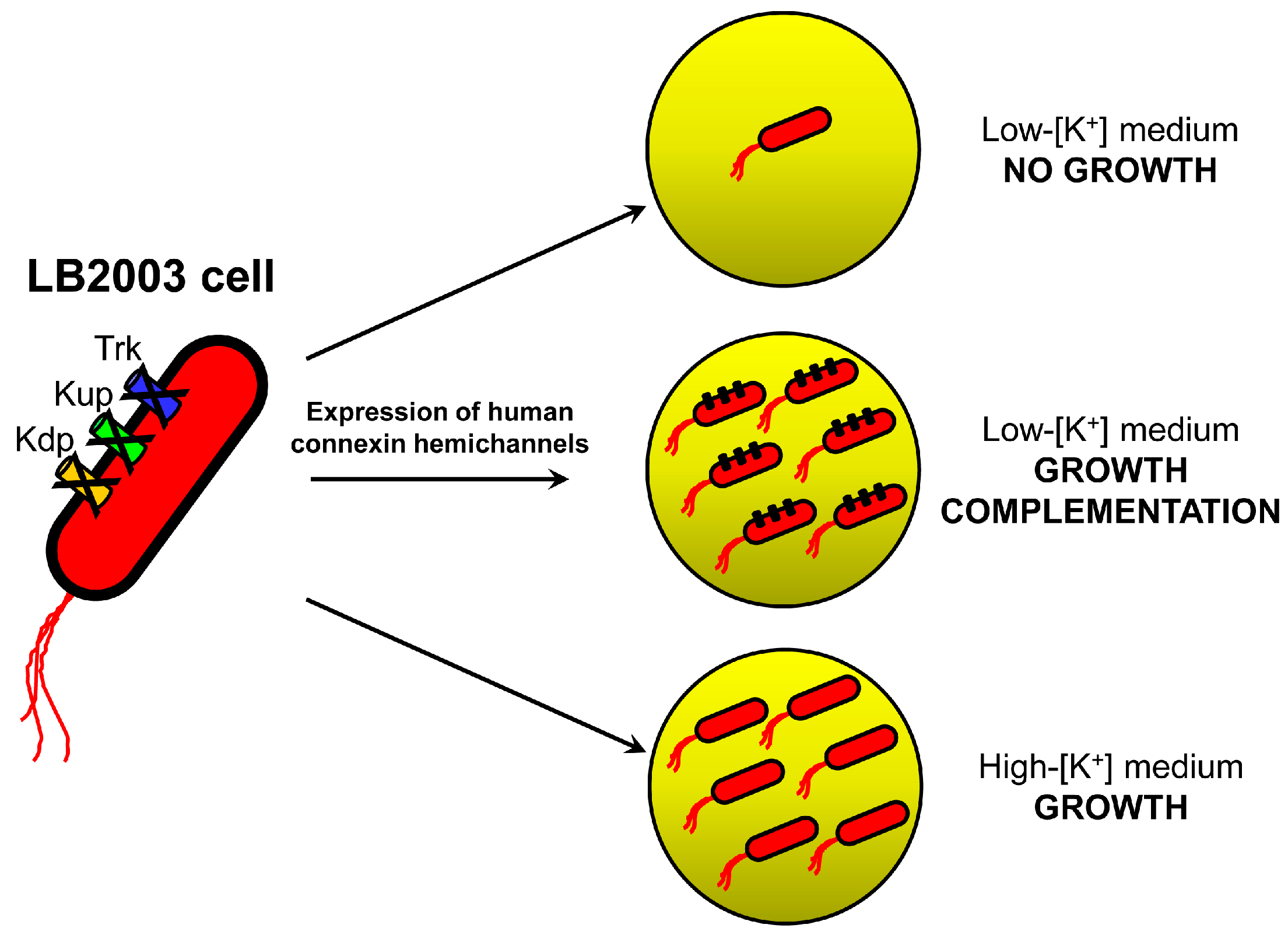
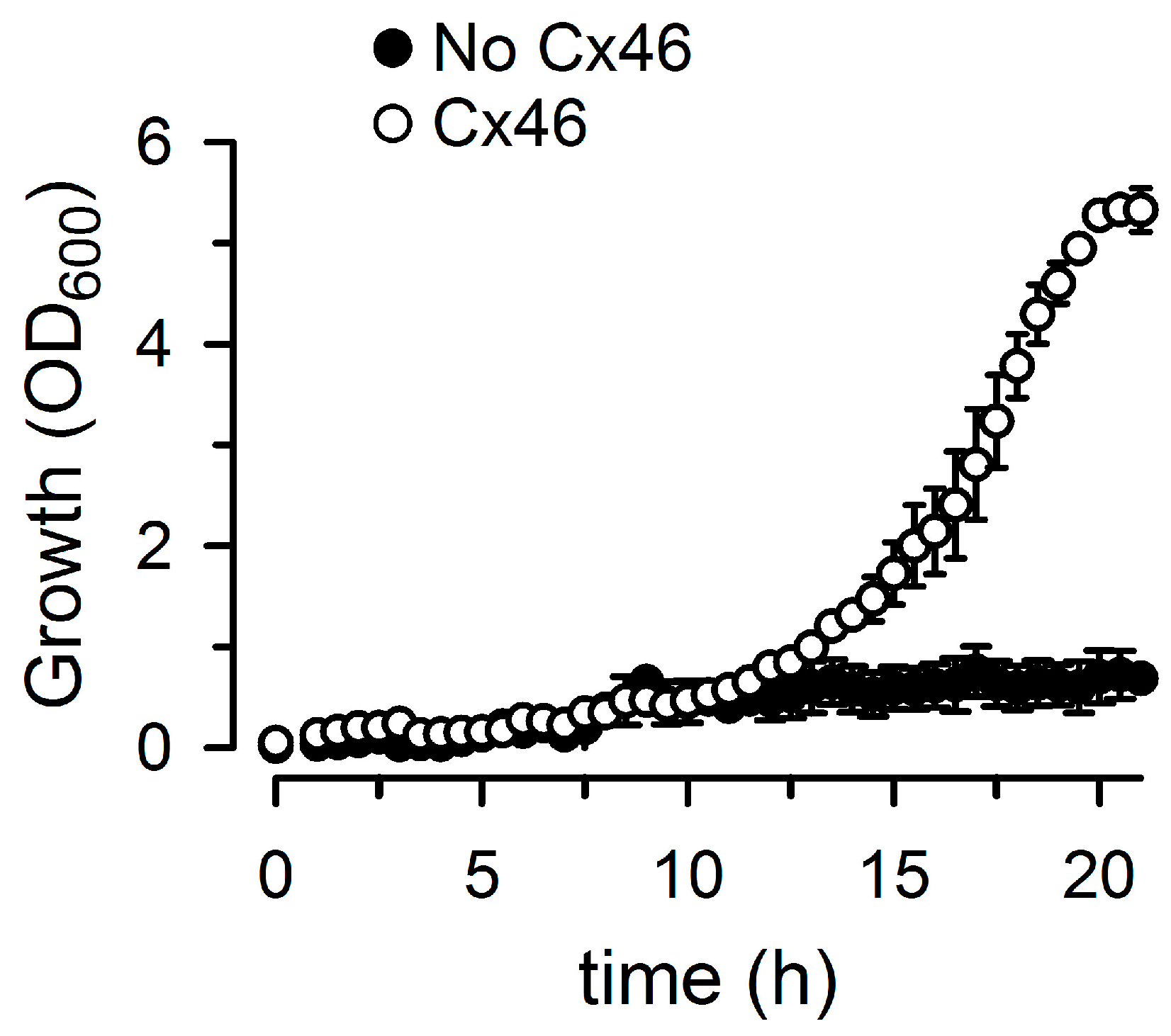
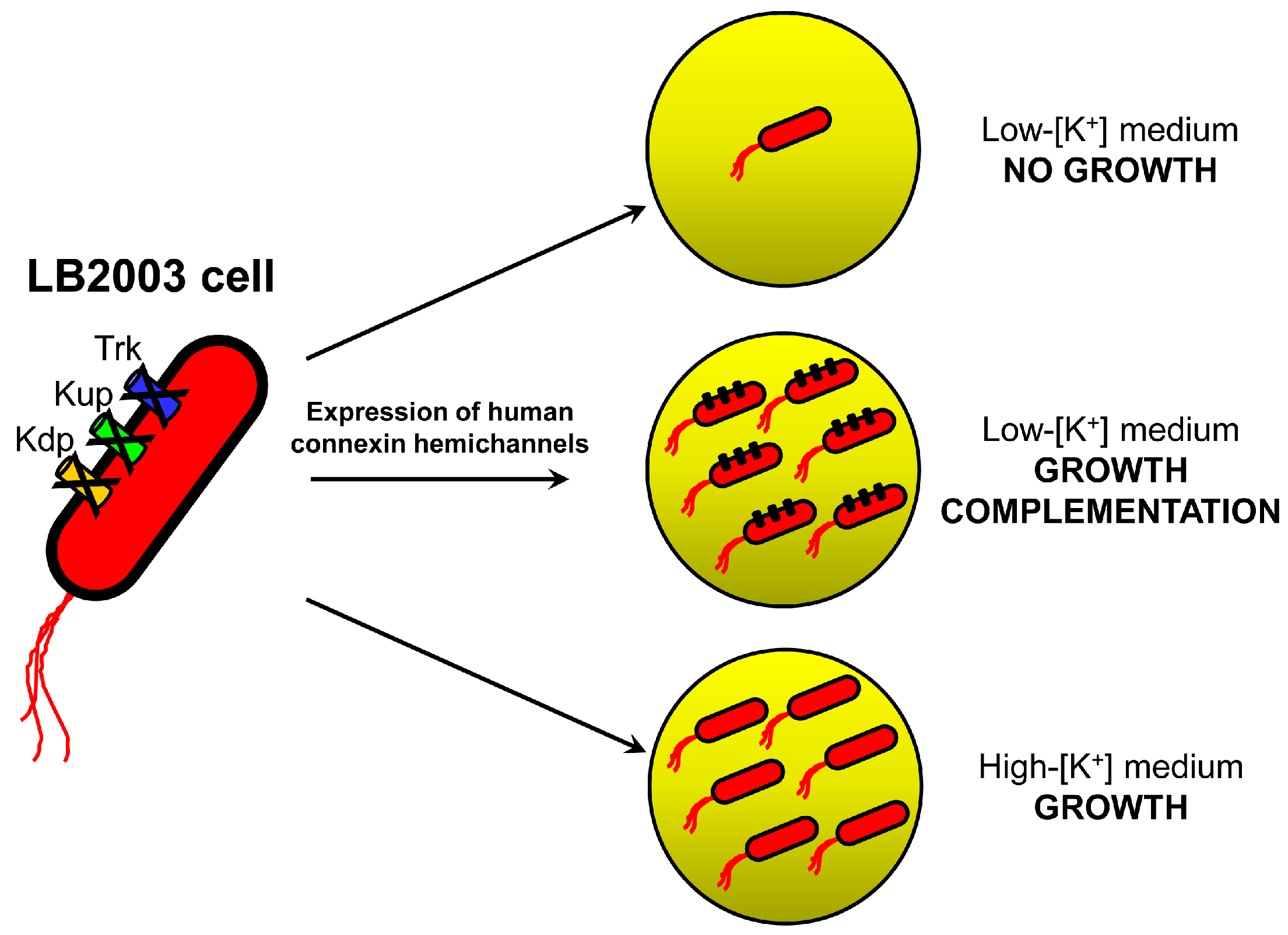
2.1. Growth Complementation by Cx26, Cx43 and Cx46
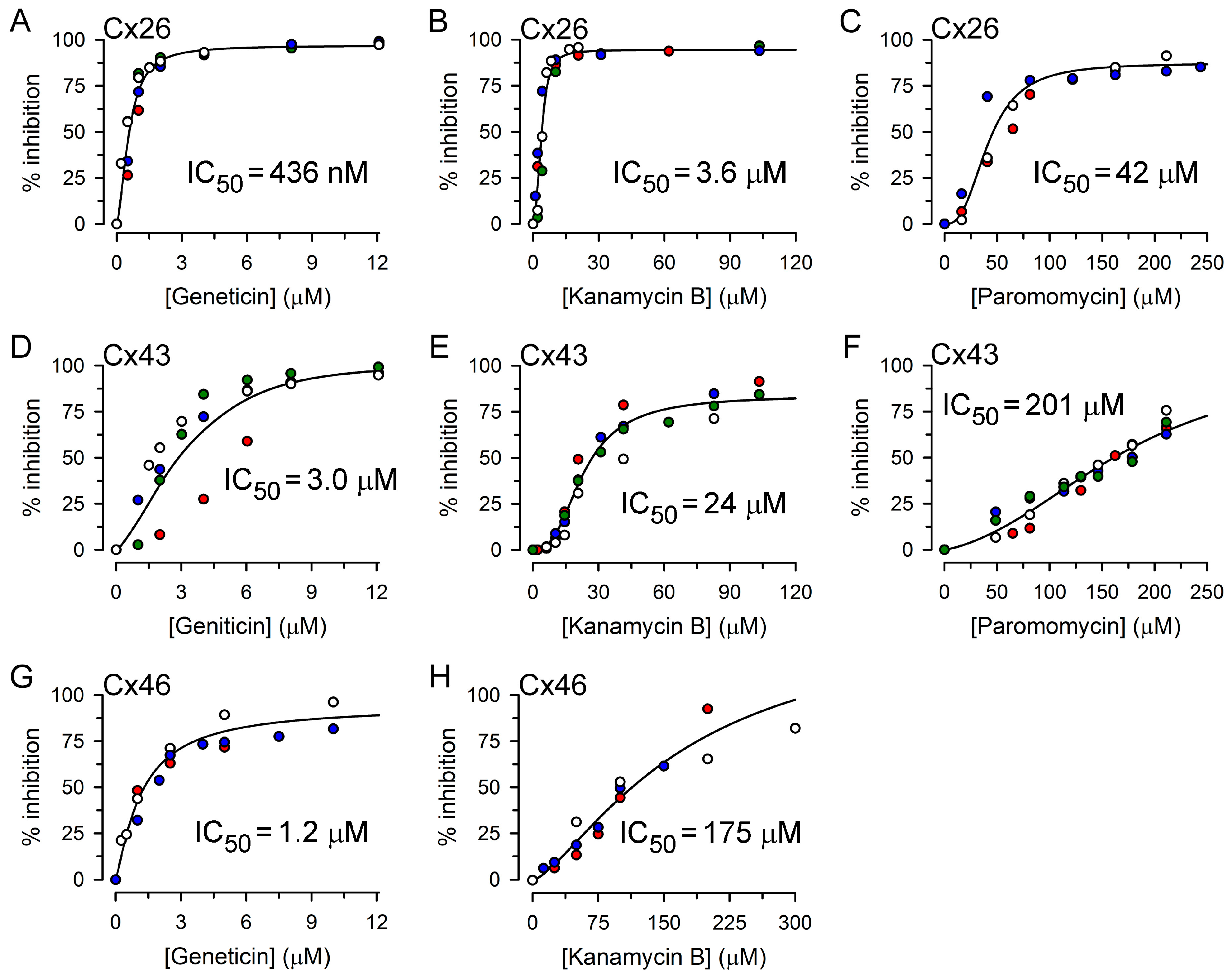
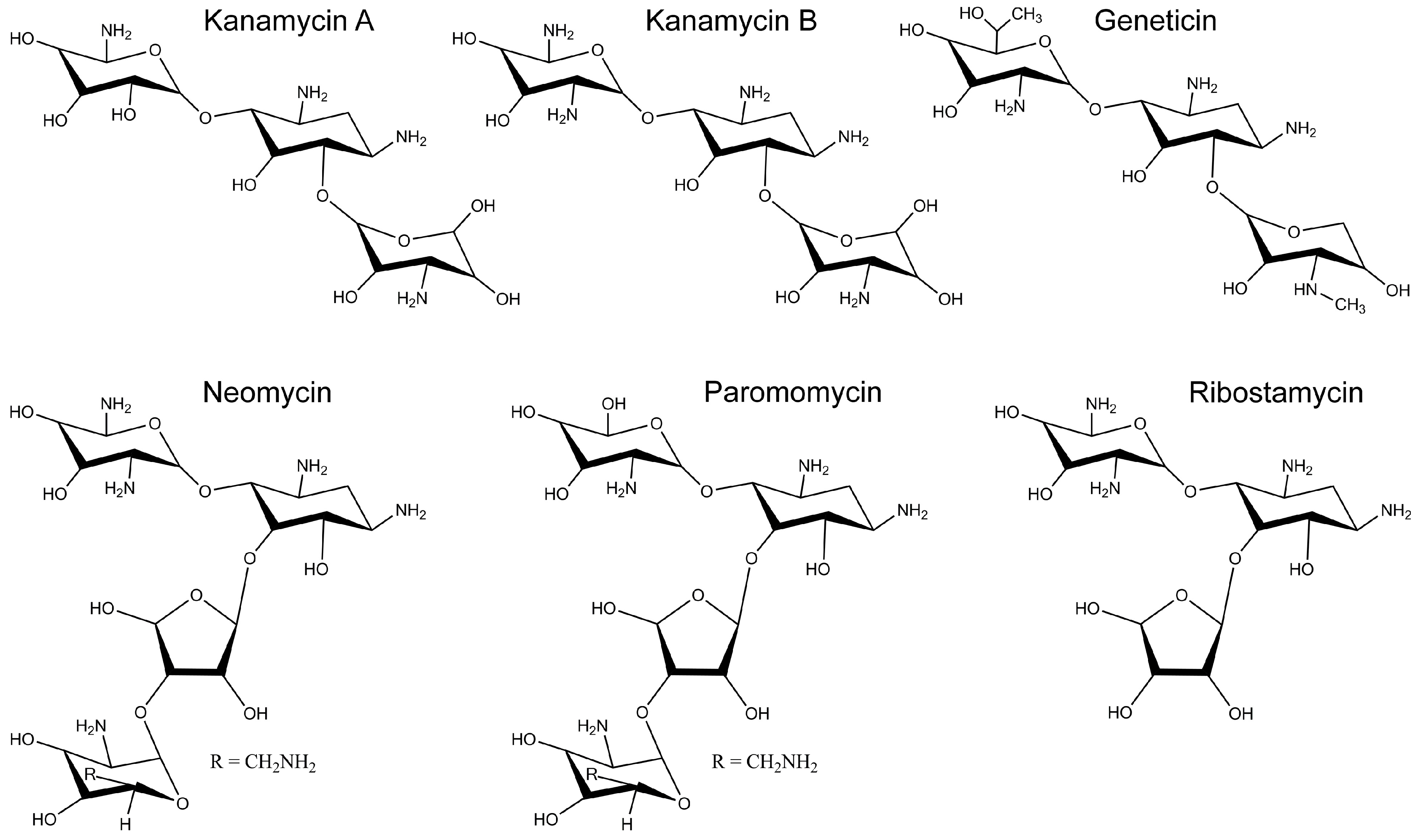
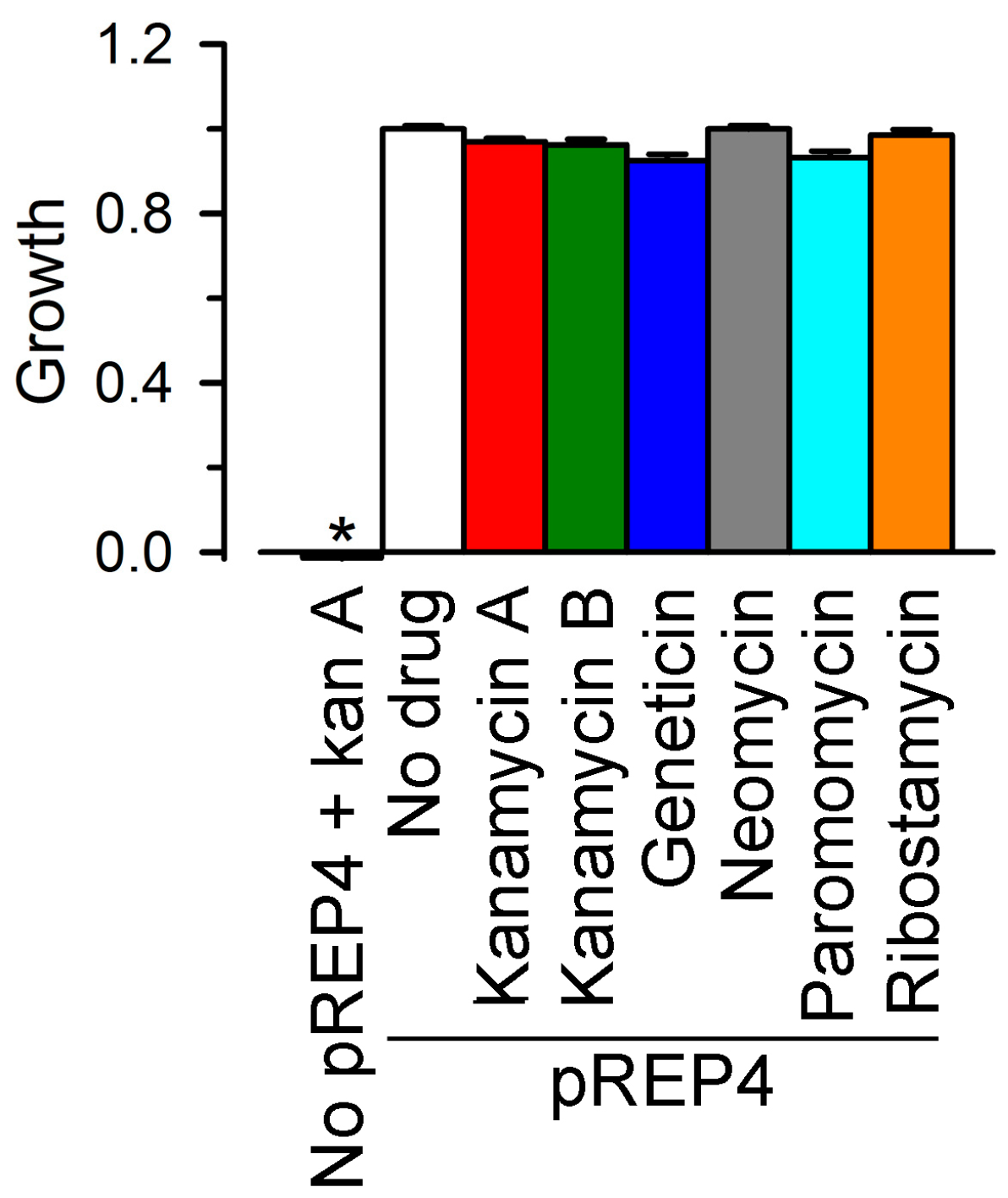
2.2. Inhibition of Cx26-, Cx43- and Cx46-Dependent Growth Complementation by AGs
3. Materials and Methods
3.1. Molecular Biology and LB2003 Cells
3.2. Growth Complementation Assay
3.3. Statistics
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abascal, F.; Zardoya, R. Evolutionary analyses of gap junction protein families. Biochim. Biophys. Acta 2013, 1828, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Nygaard Axelsen, L.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.E.; Klein, A.M.; Ghanem, A.; Geelen, T.; Coolen, B.F.; Breitbach, M.; Zimmermann, K.; Nicolay, K.; Fleischmann, B.K.; Roell, W.; et al. Embryonic cardiomyocyte, but not autologous stem cell transplantation, restricts infarct expansion, enhances ventricular function, and improves long-term survival. PLoS ONE 2013, 8, e61510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, M.C.; Reuss, L.; Cuello, L.G.; Altenberg, G.A. Functional analysis and regulation of purified connexin hemichannels. Front. Physiol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Locke, D. Permeability of connexin channels. In Connexins: A Guide; Harris, A.L., Locke, D., Eds.; Humana Press: New York, NY, USA, 2009; pp. 165–206. [Google Scholar]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 2013, 1828, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; White, T.W. Connexin-26 mutations in deafness and skin disease. Expert Rev. Mol. Med. 2009, 11, e35. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Connexin hemichannels in the lens. Front. Physiol. 2014, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Gorge, P.M.; Gorbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Avendano, B.C.; Montero, T.D. Role of connexins and pannexins in ischemic stroke. Curr. Med. Chem. 2014, 21, 2165–2182. [Google Scholar] [CrossRef] [PubMed]
- Vergara, L.; Bao, X.; Bello-Reuss, E.; Reuss, L. Do connexin 43 gap-junctional hemichannels activate and cause cell damage during ATP depletion of renal-tubule cells? Acta Physiol. Scand. 2003, 179, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Stong, B.C.; Chang, Q.; Ahmad, S.; Lin, X. A novel mechanism for connexin 26 mutation linked deafness: Cell death caused by leaky gap junction hemichannels. Laryngoscope 2006, 116, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Chen, Y.; Lee, S.H.; Lee, S.C.; Reuss, L.; Altenberg, G.A. Membrane transport proteins with complete replacement of transmembrane helices with polyalanine sequences remain functional. J. Biol. Chem. 2005, 280, 8647–8650. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Zampighi, G.A.; Leung, D.W.; Wright, E.M.; Loo, D.D. Inhibition of gap junction hemichannels by chloride channel blockers. J. Membr. Biol. 2002, 185, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M. Pharmacology of connexin channels. In Connexins: A Guide; Harris, A.L., Locke, D., Eds.; Humana Press: New York, NY, USA, 2009; pp. 207–223. [Google Scholar]
- Verselis, V.K.; Srinivas, M. Connexin channel modulators and their mechanisms of action. Neuropharmacology 2013, 75, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Maes, M.; Crespo Yanguas, S.; Vinken, M. Inhibitors of connexin and pannexin channels as potential therapeutics. Pharmacol. Ther. 2017, 180, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Dhein, S. Peptides targeting gap junctional structures. Curr. Pharm. Des. 2010, 16, 3056–3070. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, E.; Boengler, K.; Antoons, G.; Sipido, K.R.; Schulz, R.; Leybaert, L. Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br. J. Pharmacol. 2011, 163, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.C.; Krishnan, S.; Cortes, D.M.; Retamal, M.A.; Reuss, L.; Altenberg, G.A.; Cuello, L.G. Functional hemichannels formed by human connexin 26 expressed in bacteria. Biosci. Rep. 2015, 35, e00177. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Fiori, M.C.; Whisenant, T.E.; Cortes, D.M.; Altenberg, G.A.; Cuello, L.G. An Escherichia coli-based assay to assess the function of recombinant human hemichannels. SLAS Discov. 2017, 22, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, S.; Bakker, E.P. Requirement of a large K+-uptake capacity and of extracytoplasmic protease activity for protamine resistance of Escherichia coli. Arch. Microbiol. 1997, 167, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Buurman, E.T.; McLaggan, D.; Naprstek, J.; Epstein, W. Multiple paths for nonphysiological transport of K+ Escherichia coli. J. Bacteriol. 2004, 186, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Ptak, C.P.; Cuello, L.G.; Perozo, E. Electrostatic interaction of a K+ channel RCK domain with charged membrane surfaces. Biochemistry 2005, 44, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Hellmer, J.; Zeilinger, C. MjK1, a K+ channel from M. jannaschii, mediates K+ uptake and K+ sensitivity in E. coli. FEBS Lett. 2003, 547, 165–169. [Google Scholar] [CrossRef]
- Dalamon, V.; Fiori, M.C.; Figueroa, V.A.; Oliva, C.A.; Del Rio, R.; Gonzalez, W.; Canan, J.; Elgoyhen, A.B.; Altenberg, G.A.; Retamal, M.A. Gap-junctional channel and hemichannel activity of two recently identified connexin 26 mutants associated with deafness. Pflugers Arch.-Eur. J. Physiol. 2016, 468, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, V.A.; Retamal, M.A.; Cea, L.A.; Salas, J.D.; Vargas, A.A.; Verdugo, C.A.; Jara, O.; Martinez, A.D.; Saez, J.C. Extracellular gentamicin reduces the activity of connexin hemichannels and interferes with purinergic Ca2+ signaling in HeLa cells. Front. Cell. Neurosci. 2014, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, S.; Tsuchiya, T. Total synthesis and chemical modification of the aminoglycoside antibiotics. In Aminoglycoside Antibiotics; Umezawa, S., Hooper, I.R., Eds.; Springer: New York, NY, USA, 1982; pp. 37–110. [Google Scholar]
- Wang, J.; Chang, C.-W.T. Design, chemical synthesis, and antibacterial activity of kanamycin and neomycin class aminoglycoside antibiotics. In Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery; Arya, D.P., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 141–180. [Google Scholar]
- Mingeot-Leclercq, M.P.; Tulkens, P.M. Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 1999, 43, 1003–1012. [Google Scholar] [PubMed]
- Nickel, R.; Forge, A. Gap junctions and connexins in the inner ear: Their roles in homeostasis and deafness. Curr. Opin. Otolaryngol. Head Neck Surg. 2008, 16, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; von Bernhardi, R.; Giaume, C.; Saez, J.C. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev. Neurosci. 2012, 23, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Basilio, D.; Saez, J.C.; Orellana, J.A.; Raine, C.S.; Bukauskas, F.; Bennett, M.V.; Berman, J.W. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J. Neuroimmune Pharmacol. 2012, 7, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Fiori, M.C.; Cuello, L.G.; Altenberg, G.A. A cell-based assay to assess hemichannel function. Yale J. Biol. Med. 2017, 90, 87–95. [Google Scholar] [PubMed]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hernandez, J.M.; de Miguel, M.; Larrosa, B.; Gonzalez, D.; Barrio, L.C. Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 16030–16035. [Google Scholar] [CrossRef] [PubMed]
- Lopez, W.; Ramachandran, J.; Alsamarah, A.; Luo, Y.; Harris, A.L.; Contreras, J.E. Mechanism of gating by calcium in connexin hemichannels. Proc. Natl. Acad. Sci. USA 2016, 113, E7986–E7995. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aminoglycoside | Growth Complementation Dependent on | ||
|---|---|---|---|
| Cx26 | Cx43 | Cx46 | |
| Kanamycin A | 11.5 ± 1.8 μM (3) | 48 ± 2 μM (4) | 112 ± 5 μM (3) |
| Kanamycin B | 3.6 ± 0.9 μM (4) | 24 ± 2 μM (4) | 175 ± 9 μM (3) |
| Geneticin | 0.44 ± 0.06 μM (5) | 3.0 ± 0.8 μM (4) | 1.2 ± 0.2 μM (3) |
| Neomycin | 7.4 ± 1.3 μM (5) | 45 ± 9 μM (4) | 16 ± 2 μM (3) |
| Paromomycin | 42 ± 9 μM (3) | 201 ± 17 μM (4) | >200 μM (3) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiori, M.C.; Krishnan, S.; Kjellgren, A.; Cuello, L.G.; Altenberg, G.A. Inhibition by Commercial Aminoglycosides of Human Connexin Hemichannels Expressed in Bacteria. Molecules 2017, 22, 2063. https://doi.org/10.3390/molecules22122063
Fiori MC, Krishnan S, Kjellgren A, Cuello LG, Altenberg GA. Inhibition by Commercial Aminoglycosides of Human Connexin Hemichannels Expressed in Bacteria. Molecules. 2017; 22(12):2063. https://doi.org/10.3390/molecules22122063
Chicago/Turabian StyleFiori, Mariana C., Srinivasan Krishnan, Abbey Kjellgren, Luis G. Cuello, and Guillermo A. Altenberg. 2017. "Inhibition by Commercial Aminoglycosides of Human Connexin Hemichannels Expressed in Bacteria" Molecules 22, no. 12: 2063. https://doi.org/10.3390/molecules22122063