An Optimized and Sensitive Pharmacokinetic Quantitative Method of Investigating Gastrodin, Parishin, and Parishin B, C and E in Beagle Dog Plasma using LC-MS/MS after Intragastric Administration of Tall Gastrodia Capsules


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Sample Preparation
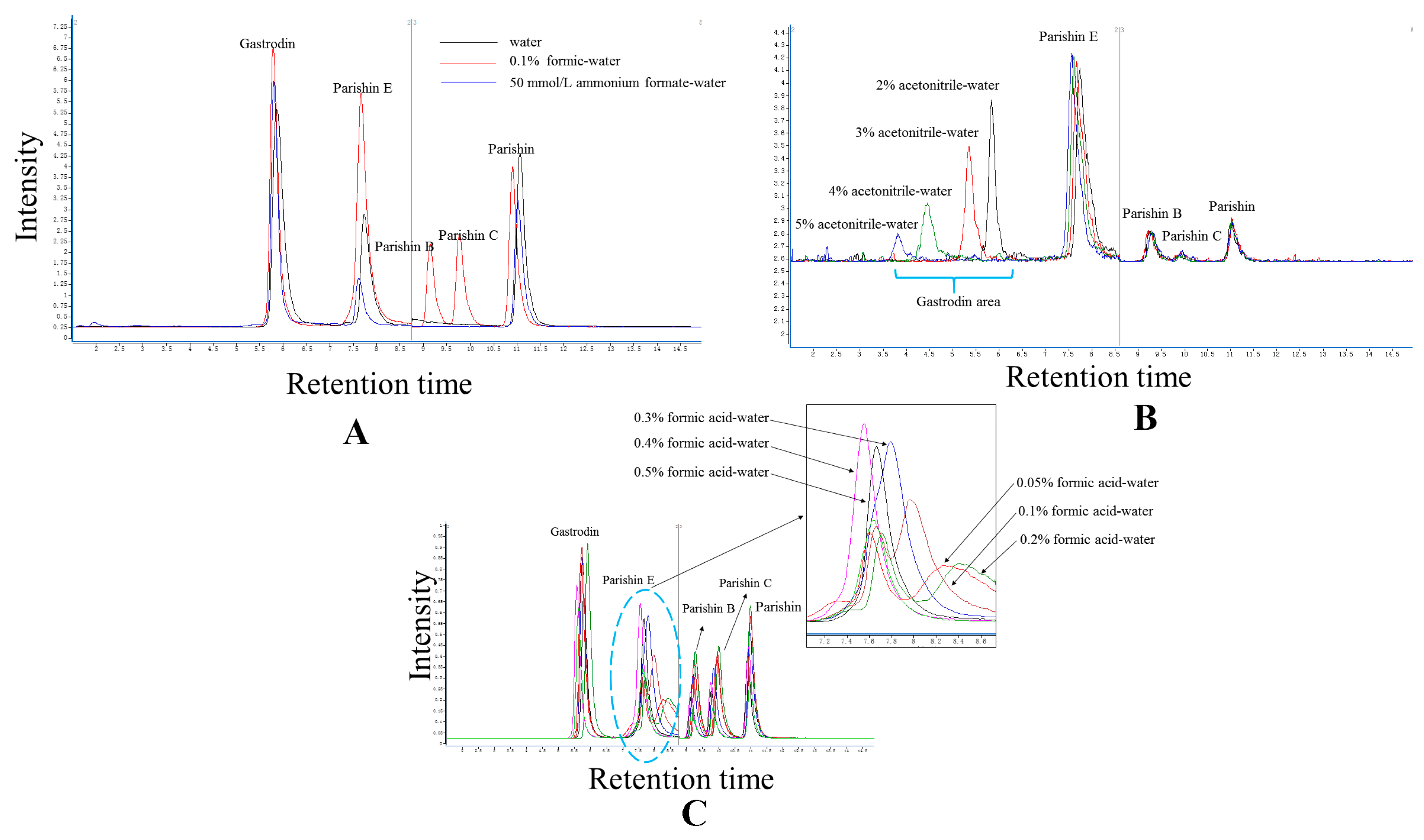
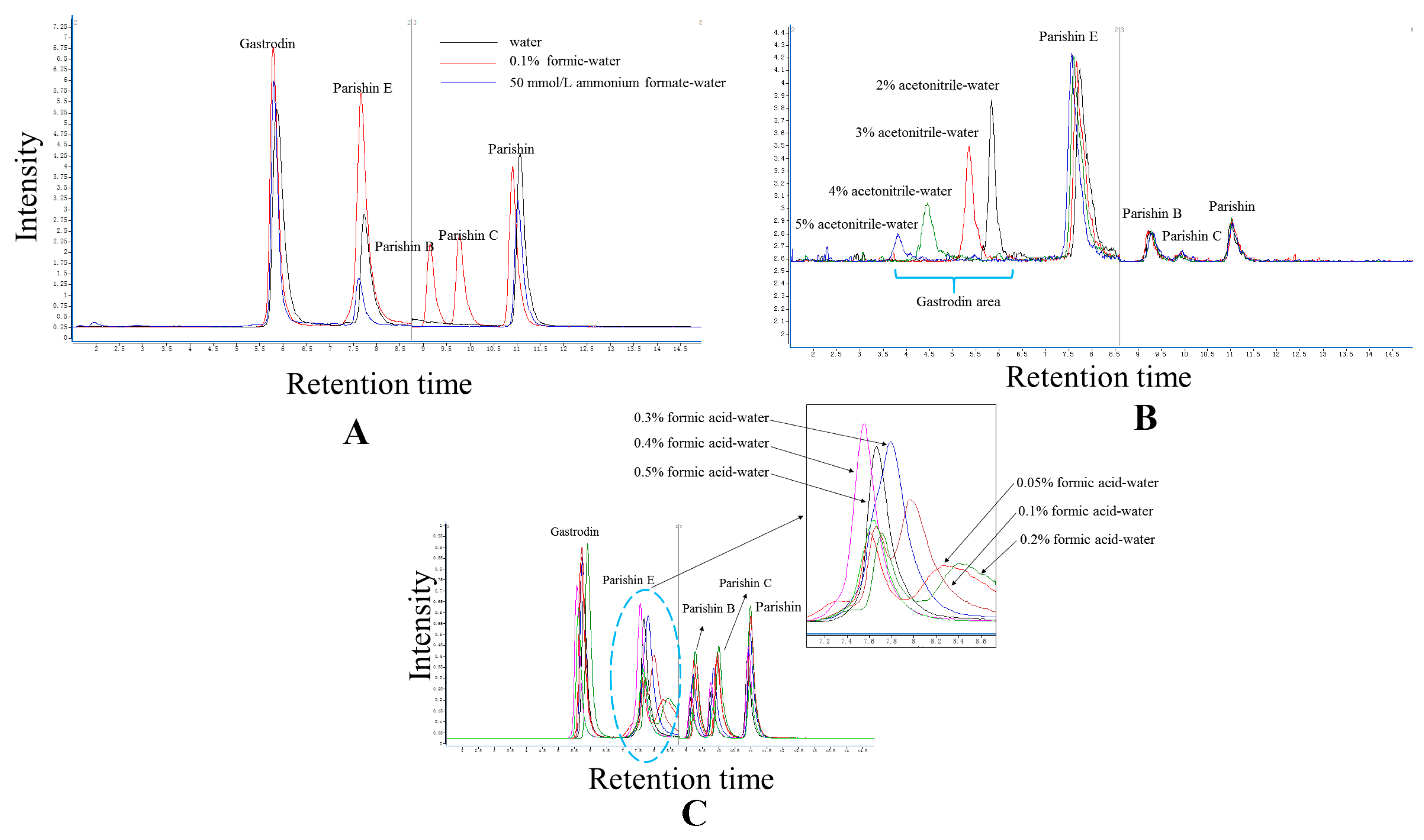
2.2. Elution System
2.3. Obtaining Compounds in Plasma Using LC-MS/MS
2.4. Method Validation
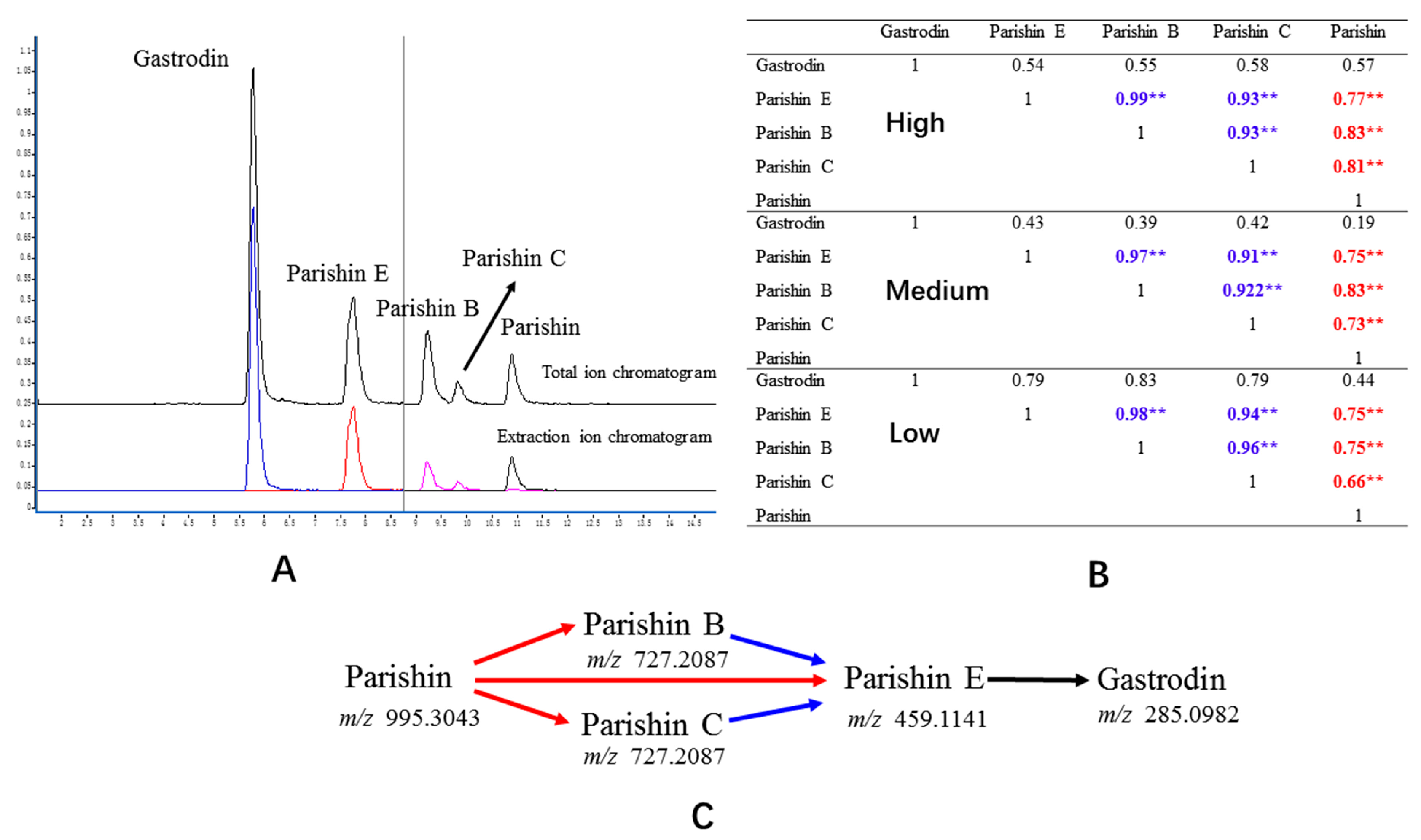
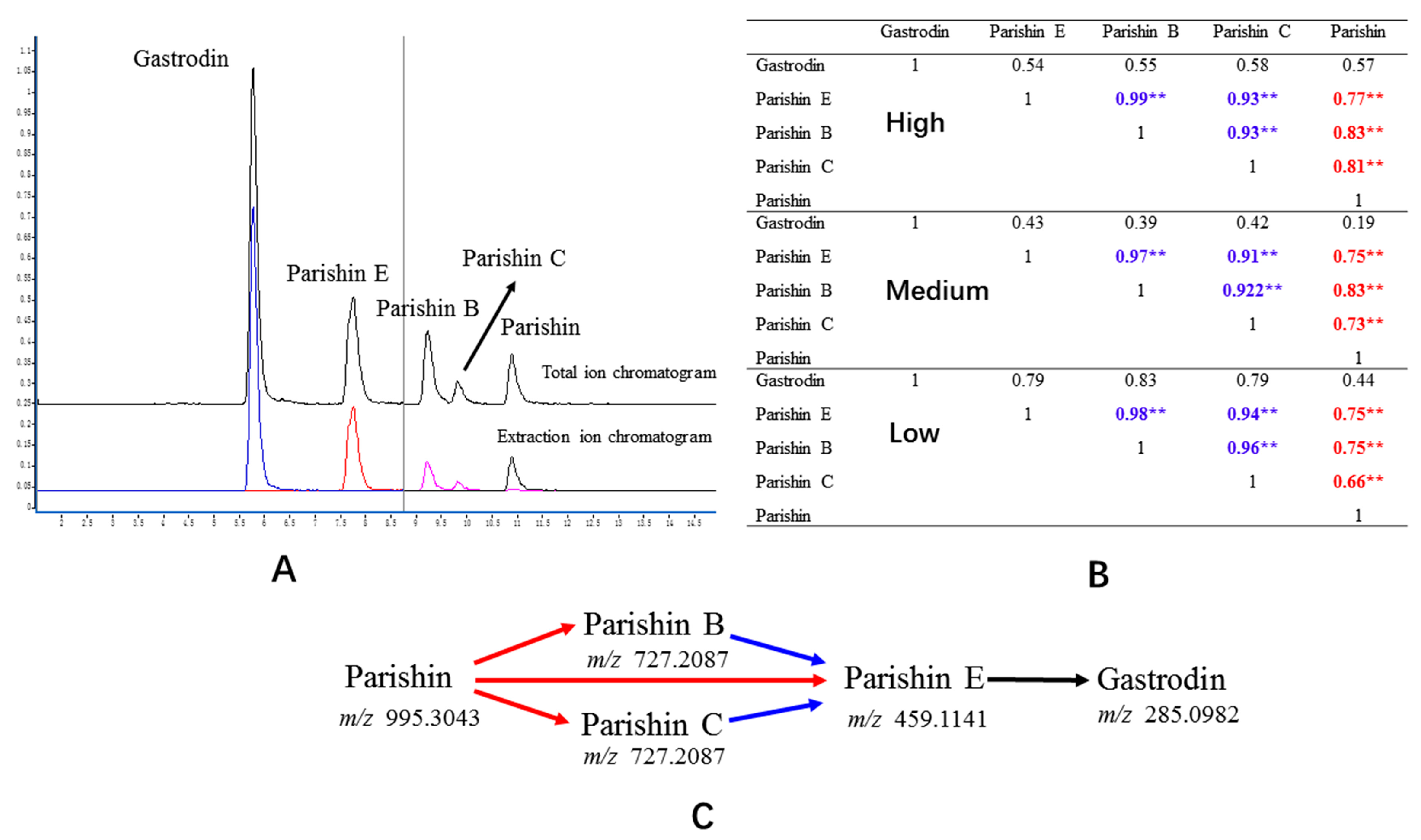
2.4.1. Separation, Selectivity, and Sensitivity
2.4.2. Recovery and Matrix Effect
2.4.3. Precision and Accuracy
2.4.4. Stability
2.5. Linearity of Calibration Curves and LLOQs
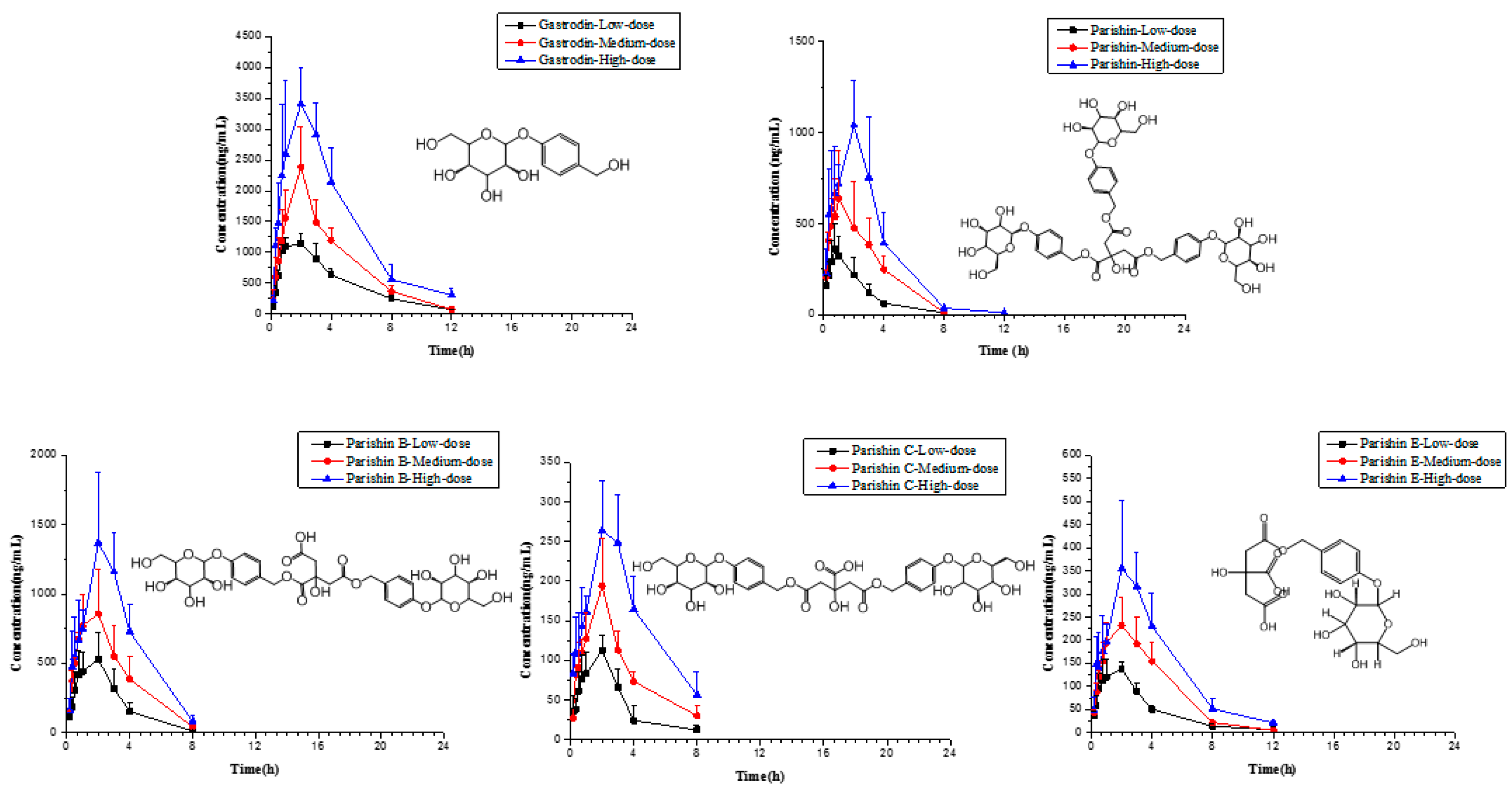
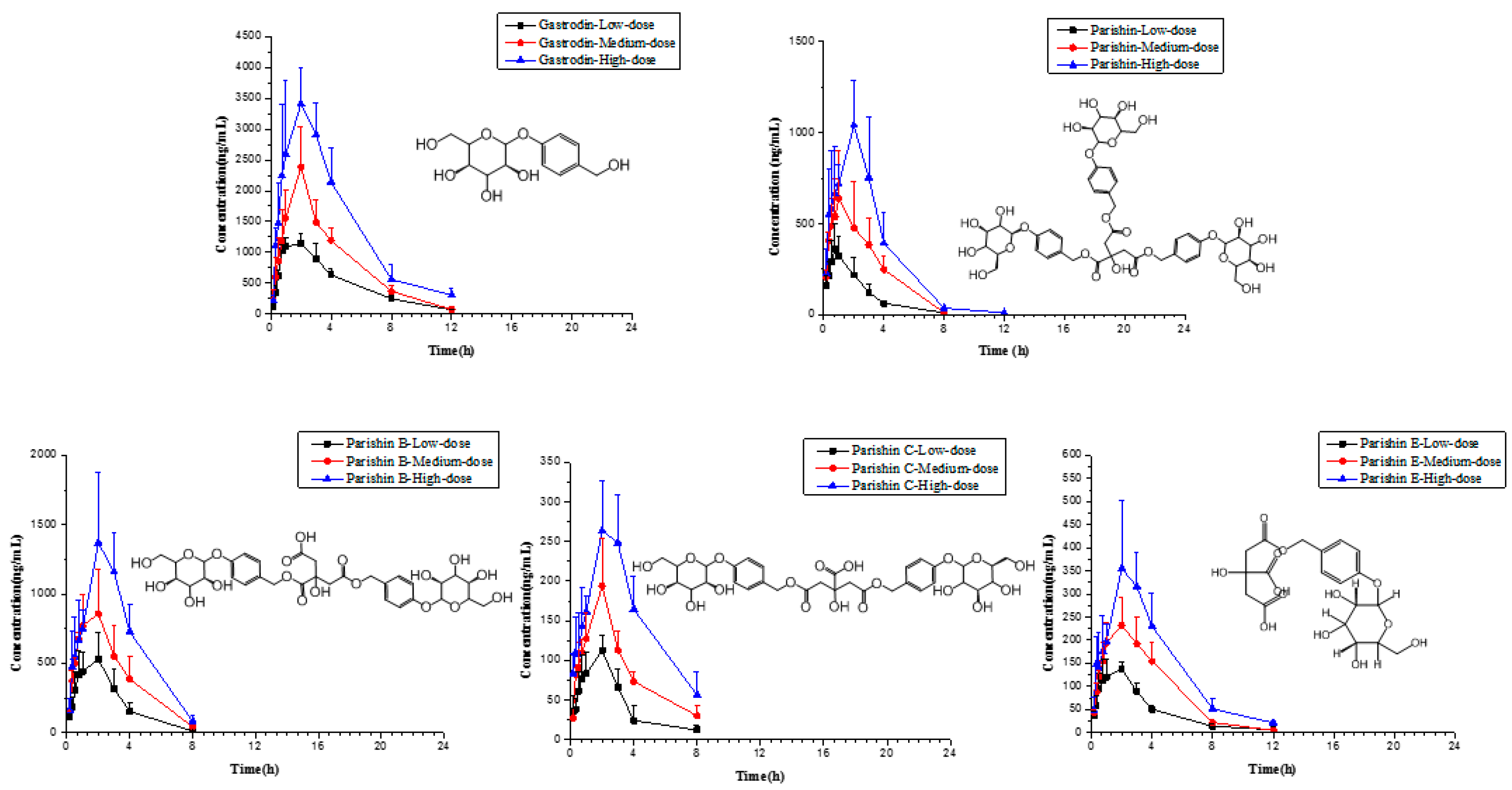
2.6. Application to Pharmacokinetic Studies
2.7. Correlation Analysis
3. Experimental
3.1. Materials and Reagents
3.2. Instruments and Analytical Conditions
3.3. Animal Experiments
3.4. Drug Administration and Blood Sampling
3.5. Experimental Condition Optimization
3.6. Plasma Sample Preparation
3.7. Validation of the Method
3.8. Pharmacokinetic Study and Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jia, Y.; Li, X.; Xie, H.; Shen, J.; Luo, J.; Wang, J.; Wang, K.D.; Liu, Q.; Kong, L. Analysis and pharmacokinetics studies of gastrodin and p-hydroxybenzyl alcohol in dogs using ultra fast liquid chromatography-tandem mass spectrometry method. J. Pharm. Biomed. Anal. 2014, 99, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.L.; Wang, L.; Liu, X.X.; Cheng, M.; Qu, Y.; Xiao, H.B. Comparative pharmacokinetics of gastrodin in rats after intragastric administration of free gastrodin, parishin and Gastrodia elata extract. J. Ethnopharmacol. 2015, 176, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, X.J.; Zhou, X.; Kang, Z.J. Relative bioavailability of gastrodin and parishin from extract and powder of Gastrodiae Rhizoma in rat. J. Pharm. Biomed. Anal. 2014, 100, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.L.; Wang, L.; Li, J.J.; Liu, X.X.; Cheng, M.C.; Xiao, H.B. Analysis of the metabolic profile of parishin by ultra-performance liquid chromatography/quadrupole-time of flight mass spectrometry. Biomed. Chromatogr. 2015, 29, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.K.; Chern, Y.; Fang, J.M.; Lin, C.I.; Chen, W.P.; Lin, Y.L. Neuroprotective principles from Gastrodia elata. J. Nat. Prod. 2007, 70, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Chiang, S.Y.; Cheng, K.S.; Lin, Y.H.; Tang, N.Y.; Lee, C.J.; Pon, C.Z.; Hsieh, C.T. Anticonvulsive and free radical scavenging activities of Gastrodia elata Bl. in kainic acid-treated rats. Am. J. Chin. Med. 2001, 29, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.W.; Lee, J.Y.; Kim, C.J. Anti-asthmatic activity of phenolic compounds from the roots of Gastrodia elata Bl. Int. Immunopharmacol. 2010, 10, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wang, T.; Duan, J.A.; Zhou, W.; Hua, Y.Q.; Tang, Y.P.; Yu, L.; Qian, D.W. Anti-inflammatory and analgesic activity of different extracts of Commiphora myrrha. J. Ethnopharmacol. 2011, 134, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.M.; Lee, Y.J.; Kang, D.G.; Lee, H.S. Anti-inflammatory effect of Gastrodia elata rhizome in human umbilical vein endothelial cells. Am. J. Chin. Med. 2009, 37, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.K.; Jeon, H.J.; Lim, E.J.; Jung, H.J.; Park, E.H. Anti-inflammatory and anti-angiogenic activities of Gastrodia elata Blume. J. Ethnopharmacol. 2007, 110, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jang, Y.W.; Kang, H.S.; Moon, H.; Sim, S.S.; Kim, C.J. Anti-inflammatory action of phenolic compounds from Gastrodia elata root. Arch. Pharm. Res. 2006, 29, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Ouyang, H.; Lu, Y.; Qiu, Y.; Feng, Y.; Jiang, H.; Zhou, X.; Yang, S. A novel dereplication strategy for the identification of two new trace compounds in the extract of Gastrodia elata using UHPLC/Q-TOF-MS/MS. J. Chromatogr. B 2015, 988, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, J.Q.; Xiao, H.; Zhang, J.; Liu, A. Simultaneous Qualitative assessment and quantitative analysis of metabolites (phenolics, nucleosides and amino acids) from the roots of fresh Gastrodia elata using UPLC-ESI-Triple quadrupole ion MS and ESI-linear ion trap high-resolution MS. PLoS ONE 2016, 11, e0150647. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Zhao, J.; Zheng, W.P.; Zhao, Y. Neuroprotective effect of 4-hydroxybenzyl alcohol against transient focal cerebral ischemia via anti-apoptosis in rats. Brain Res. 2010, 1308, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.N.; Zong, Y.; Zhong, L.M.; Li, Y.M.; Zhang, W.; Bian, L.G.; Ai, Q.L.; Liu, Y.D.; Sun, J.; Lu, D. Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PLoS ONE 2011, 6, e21891. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Guan, H.; Cui, C.; Tian, S.; Yang, D.; Wang, X.; Zhang, S.; Wang, L.; Jiang, H. Gastrodin inhibits cell proliferation in vascular smooth muscle cells and attenuates neointima formation in vivo. Int. J. Mol. Med. 2012, 30, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Chen, C.; Zhang, D.P.; Guo, H.; Zhou, H.; Zong, J.; Bian, Z.; Dong, X.; Dai, J.; Zhang, Y.; et al. Gastrodin protects against cardiac hypertrophy and fibrosis. Mol. Cell. Biochem. 2012, 359, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, R.; Qiao, Y.; Xue, F.; Nie, H.; Zhang, Z.; Wang, Y.; Peng, Z.; Tan, Q. Gastrodin ameliorates depression-like behaviors and up-regulates proliferation of hippocampal-derived neural stem cells in rats: Involvement of its anti-inflammatory action. Behav. Brain Res. 2014, 266, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, N.; Zhen, Y.; Ge, M.; Li, Y.; Yu, B.; He, H.; Shao, R.G. Protective effect of gastrodin on bile duct ligation-induced hepatic fibrosis in rats. Food Chem. Toxicol. 2015, 86, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Dai, J.; Huang, Z.; Du, Q.; Lin, J.; Wang, Y. Simultaneous determination of gastrodin and puerarin in rat plasma by HPLC and the application to their interaction on pharmacokinetics. J. Chromatogr. B 2013, 915, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.L.; Wang, L.; Liu, X.X.; Cheng, M.C.; Xiao, H.B. Pharmacokinetic study of Gastrodia elata in rats. Anal. Bioanal. Chem. 2015, 407, 8903–8910. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, S.; Zhang, L.; Zhang, K.; Zheng, X. A study of the neuroprotective effect of the phenolic glucoside gastrodin during cerebral ischemia in vivo and in vitro. Planta Med. 2006, 72, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Xing, G.H.; Hong, B.; Li, X.M.; Zou, Y.; Zhang, X.J.; Dong, M.X. Gastrodin prevents motor deficits and oxidative stress in the MPTP mouse model of Parkinson's disease: Involvement of ERK1/2-Nrf2 signaling pathway. Life Sci. 2014, 114, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Zhao, L.X.; Wan, J.Y.; Zhang, L.; Mao, X.N.; Long, F.Y.; Zhang, S.; Chen, C.; Du, J.R. Pharmacological characterization of a novel gastrodin derivative as a potential anti-migraine agent. Fitoterapia 2015, 109, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zheng, X.; Gong, X.; Chao, Z.; Xin, Z.; Yang, Z.; Yan, Y. Relative tissue distribution and excretion studies of gastrodin and parishin from powder and extract of Gastrodiae Rhizoma in rat by UHPLC-ESI-MS/MS. Biomed. Chromatogr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, A.M.; Chi, M.Y.; Wang, Y.L. Quality evaluation of tall Gastrodia capsules by simultaneous determination of the contents of multi-constituents. Chin. Pharm. J. 2012, 5, 380–383. [Google Scholar]
- Tang, C.L.; Wang, L.; Cheng, M.C.; Zhang, X.Z.; Liu, X.Y.; Xiao, H.B. Rapid and sensitive analysis of parishin and its metabolites in rat plasma using ultra high performance liquid chromatography-fluorescence detection. J. Chromatogr. B 2014, 973, 104–109. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds gastrodin, parishin, parishin B, parishin C, parishin E and bergenin are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition 1 | Condition 2 | Condition 3 | Condition 4 | Condition 5 | Condition 6 | |
|---|---|---|---|---|---|---|
| Drying gas Temperature (°C) | 300 | 300 | 300 | 300 | 300 | 300 |
| Drying gas flow rates (L/min) | 5 | 8 | 8 | 8 | 8 | 8 |
| Sheath gas temperature (°C) | 250 | 400 | 300 | 300 | 350 | 350 |
| Sheath gas flow rates (L/min) | 9 | 12 | 12 | 12 | 12 | 12 |
| Nozzle voltage (kV) | 0.5 | 1.0 | 1.0 | 0.5 | 0.5 | 1.0 |
| Spiked Conc. (ng/mL) | Recovery (%) | RSD (%) | Matrix Effect (%) | RSD (%) | |
|---|---|---|---|---|---|
| Gastrodin | 3620 | 98.9 | 0.84 | 101 | 2.89 |
| 723 | 99.5 | 0.81 | 107 | 1.90 | |
| 144 | 98.1 | 1.46 | 99.9 | 4.45 | |
| Parishin | 840 | 98.4 | 2.83 | 103 | 1.91 |
| 168 | 101 | 2.43 | 107 | 3.82 | |
| 33.6 | 97.0 | 7.65 | 102 | 8.02 | |
| Parishin B | 1040 | 95.3 | 3.08 | 103 | 2.24 |
| 208 | 95.3 | 4.81 | 96.6 | 2.00 | |
| 41.6 | 98.5 | 5.19 | 103 | 4.64 | |
| Parishin C | 200 | 98.9 | 2.09 | 101 | 4.72 |
| 40 | 96.5 | 5.55 | 100 | 6.35 | |
| 8 | 97.5 | 6.93 | 100 | 10.8 | |
| Parishin E | 306 | 96.3 | 2.33 | 99.8 | 1.87 |
| 61.2 | 98.5 | 2.14 | 101 | 1.56 | |
| 12.2 | 99.2 | 3.54 | 100 | 2.65 | |
| Compounds | Parameter | Unit | Low Dose | Medium Dose | High Dose |
|---|---|---|---|---|---|
| Gastrodin | AUC(0–t) | ng/mL·h | 4890 ± 2060 | 8830 ± 1860 | (17.0 ± 4.24) × 103 |
| AUC(0–∞) | ng/mL·h | 5080 ± 2110 | 9170 ± 1790 | (17.6 ± 4.71) × 103 | |
| MRT(0-t) | h | 2.98 ± 1.01 | 3.46 ± 0.29 | 3.54 ± 0.69 | |
| MRT(0–∞) | h | 3.33 ±1.09 | 3.80 ± 0.28 | 3.93 ± 0.99 | |
| t1/2 | h | 1.86 ± 0.67 | 1.90 ± 0.26 | 2.09 ± 0.68 | |
| CL | L/h/kg | 0.21 ± 0.13 | 0.20 ± 0.04 | 0.21 ± 0.05 | |
| Tmax | h | 1.10 ± 0.52 | 1.75 ± 0.90 | 2.00 ± 0.71 | |
| Cmax | ng/mL | 1200 ± 120 | 2050 ± 495 | 3760 ± 778 | |
| Parishin | AUC(0–t) | ng/mL·h | 868 ± 314 | 1500 ± 836 | 3270 ± 1370 |
| AUC(0–∞) | ng/mL·h | 899 ± 299 | 1520 ± 832 | 3280± 1370 | |
| MRT(0–t) | h | 1.80 ± 0.42 | 2.49 ± 0.40 | 2.48 ± 0.36 | |
| MRT(0–∞) | h | 2.00 ± 0.30 | 2.65 ± 0.44 | 2.50 ± 0.35 | |
| t1/2 | h | 1.15 ± 0.18 | 1.38 ± 0.34 | 1.05 ± 0.16 | |
| CL | L/h/kg | 2.69 ± 1.01 | 3.56 ± 1.75 | 3.18 ± 1.65 | |
| Tmax | h | 1.00 ± 0.56 | 1.55 ± 0.62 | 1.45 ± 0.76 | |
| Cmax | ng/mL | 388 ± 116 | 462 ± 230 | 969 ± 292 | |
| Parishin B | AUC(0–t) | ng/mL·h | 1550 ± 530 | 2700 ± 1130 | 4700 ± 2330 |
| AUC(0–∞) | ng/mL·h | 1570 ± 533 | 2720 ± 1140 | 4720 ± 2330 | |
| MRT(0–t) | h | 2.25 ± 0.26 | 2.78 ± 0.42 | 2.76 ± 0.44 | |
| MRT(0–∞) | h | 2.32 ± 0.25 | 2.84 ± 0.44 | 2.80 ± 0.42 | |
| t1/2 | h | 1.08 ± 0.11 | 1.18 ± 0.29 | 1.17 ± 0.22 | |
| CL | L/h/kg | 0.72 ± 0.39 | 0.78 ± 0.23 | 1.11 ± 0.84 | |
| Tmax | h | 1.25 ± 0.69 | 1.80 ± 0.45 | 2.00 ± 0.71 | |
| Cmax | ng/mL | 526 ± 143 | 746 ± 320 | 1220 ± 562 | |
| Parishin C | AUC(0–t) | ng/mL·h | 349 ± 100 | 567 ± 198 | 1090 ± 523 |
| AUC(0–∞) | ng/mL·h | 359 ± 99.3 | 578 ± 200 | 1100 ± 527 | |
| MRT(0–t) | h | 2.47 ± 0.28 | 3.14 ± 0.54 | 3.18 ± 0.69 | |
| MRT(0–∞) | h | 2.70 ± 0.28 | 3.31 ± 0.59 | 3.25 ± 0.72 | |
| t1/2 | h | 1.37 ± 0.18 | 1.56 ± 0.41 | 1.29 ± 0.36 | |
| CL | L/h/kg | 0.72 ± 0.23 | 0.91± 0.23 | 1.26 ± 1.05 | |
| Tmax | h | 1.35 ± 0.60 | 2.00 ± 0.71 | 2.40 ± 0.55 | |
| Cmax | ng/mL | 107 ± 29.4 | 144 ± 55.1 | 243 ± 99.6 | |
| Parishin E | AUC(0-t) | ng/mL·h | 545 ± 188 | 865 ± 257 | 1620 ± 76 |
| AUC(0-∞) | ng/mL·h | 553 ± 190 | 868 ± 254 | 1630 ± 759 | |
| MRT(0-t) | h | 3.86 ± 1.05 | 3.74 ± 0.66 | 4.36 ± 0.59 | |
| MRT(0-∞) | h | 4.19 ± 1.36 | 3.79 ± 0.60 | 4.52 ± 0.66 | |
| t1/2 | h | 3.09 ± 2.03 | 1.72 ± 0.13 | 3.00 ± 1.71 | |
| CL | L/h/kg | 1.33 ± 0.82 | 1.50 ± 0.35 | 2.01 ± 1.44 | |
| Tmax | h | 1.35 ± 0.60 | 2.00 ± 0.71 | 1.80 ± 0.45 | |
| Cmax | ng/mL | 136 ± 35.3 | 192 ± 57.0 | 312 ± 138 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Chen, S.; Cheng, J.; Zhang, J.; Wang, Y.; Liu, A. An Optimized and Sensitive Pharmacokinetic Quantitative Method of Investigating Gastrodin, Parishin, and Parishin B, C and E in Beagle Dog Plasma using LC-MS/MS after Intragastric Administration of Tall Gastrodia Capsules. Molecules 2017, 22, 1938. https://doi.org/10.3390/molecules22111938
Liu J, Chen S, Cheng J, Zhang J, Wang Y, Liu A. An Optimized and Sensitive Pharmacokinetic Quantitative Method of Investigating Gastrodin, Parishin, and Parishin B, C and E in Beagle Dog Plasma using LC-MS/MS after Intragastric Administration of Tall Gastrodia Capsules. Molecules. 2017; 22(11):1938. https://doi.org/10.3390/molecules22111938
Chicago/Turabian StyleLiu, Junqiu, Sha Chen, Jintang Cheng, Jun Zhang, Yuesheng Wang, and An Liu. 2017. "An Optimized and Sensitive Pharmacokinetic Quantitative Method of Investigating Gastrodin, Parishin, and Parishin B, C and E in Beagle Dog Plasma using LC-MS/MS after Intragastric Administration of Tall Gastrodia Capsules" Molecules 22, no. 11: 1938. https://doi.org/10.3390/molecules22111938