An Optimized Synthesis, Molecular Structure and Characterization of Benzylic Derivatives of 1,2,4-Triazin-3,5(2H,4H)-dione

Abstract
:1. Introduction
2. Results and Discussion
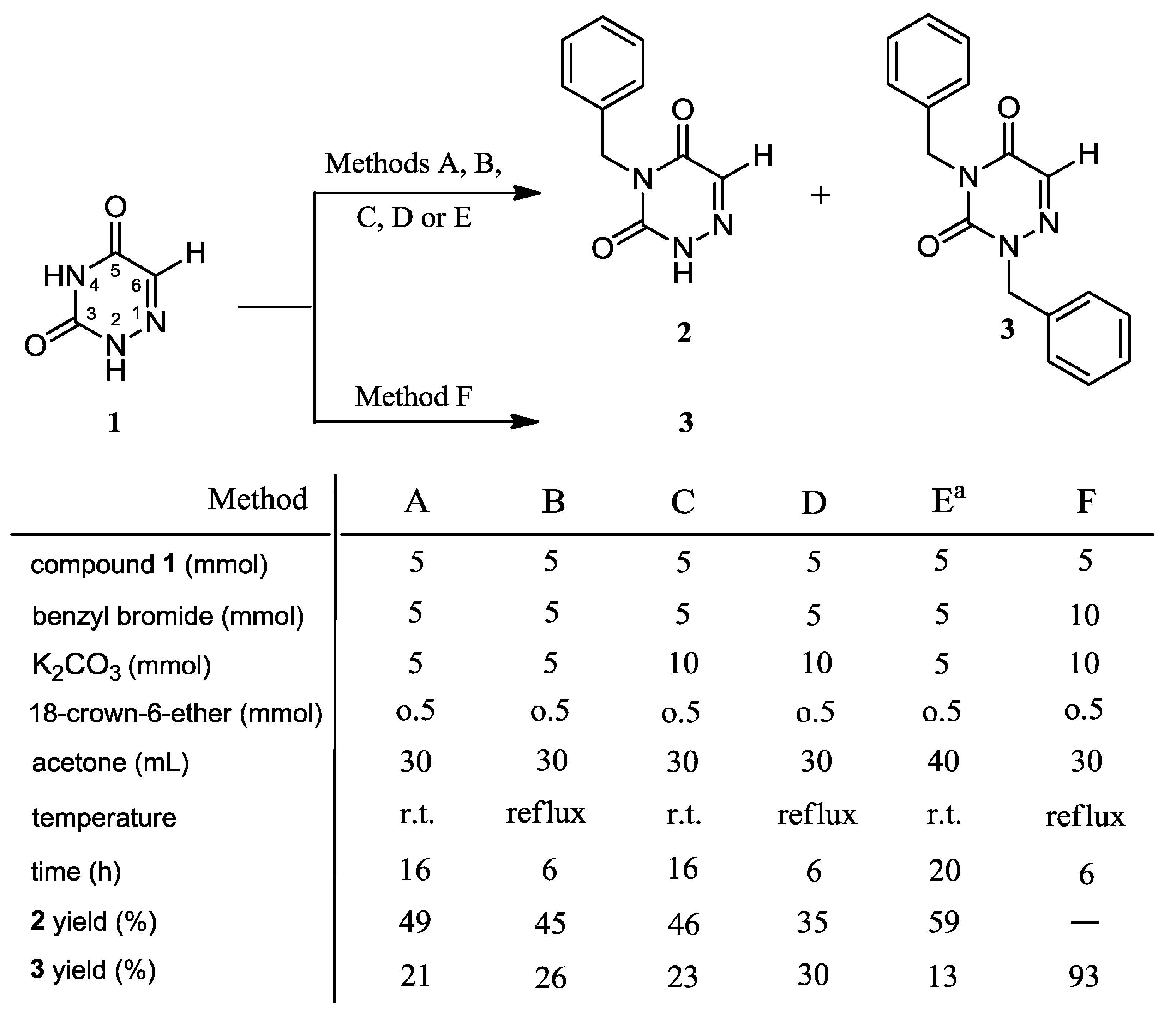
2.1. Chemistry
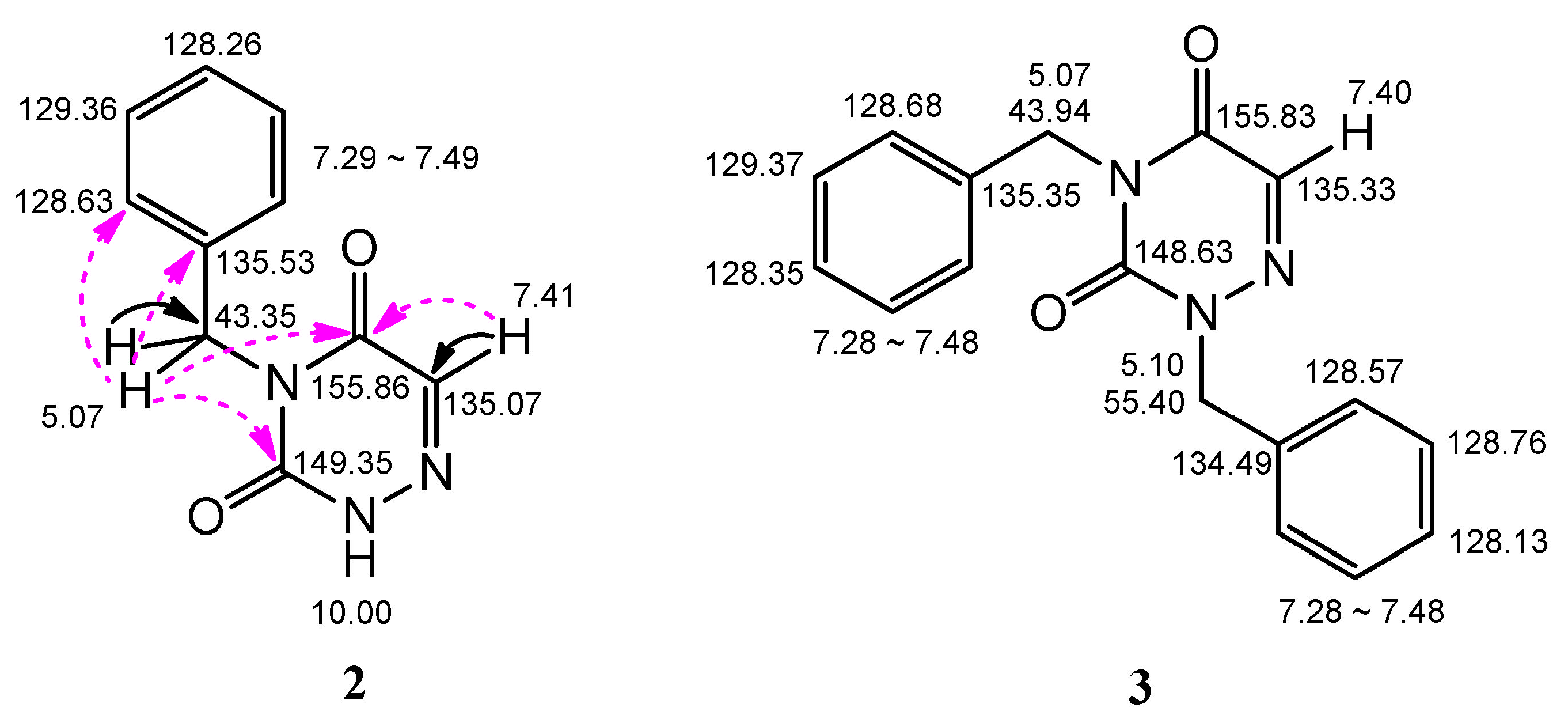
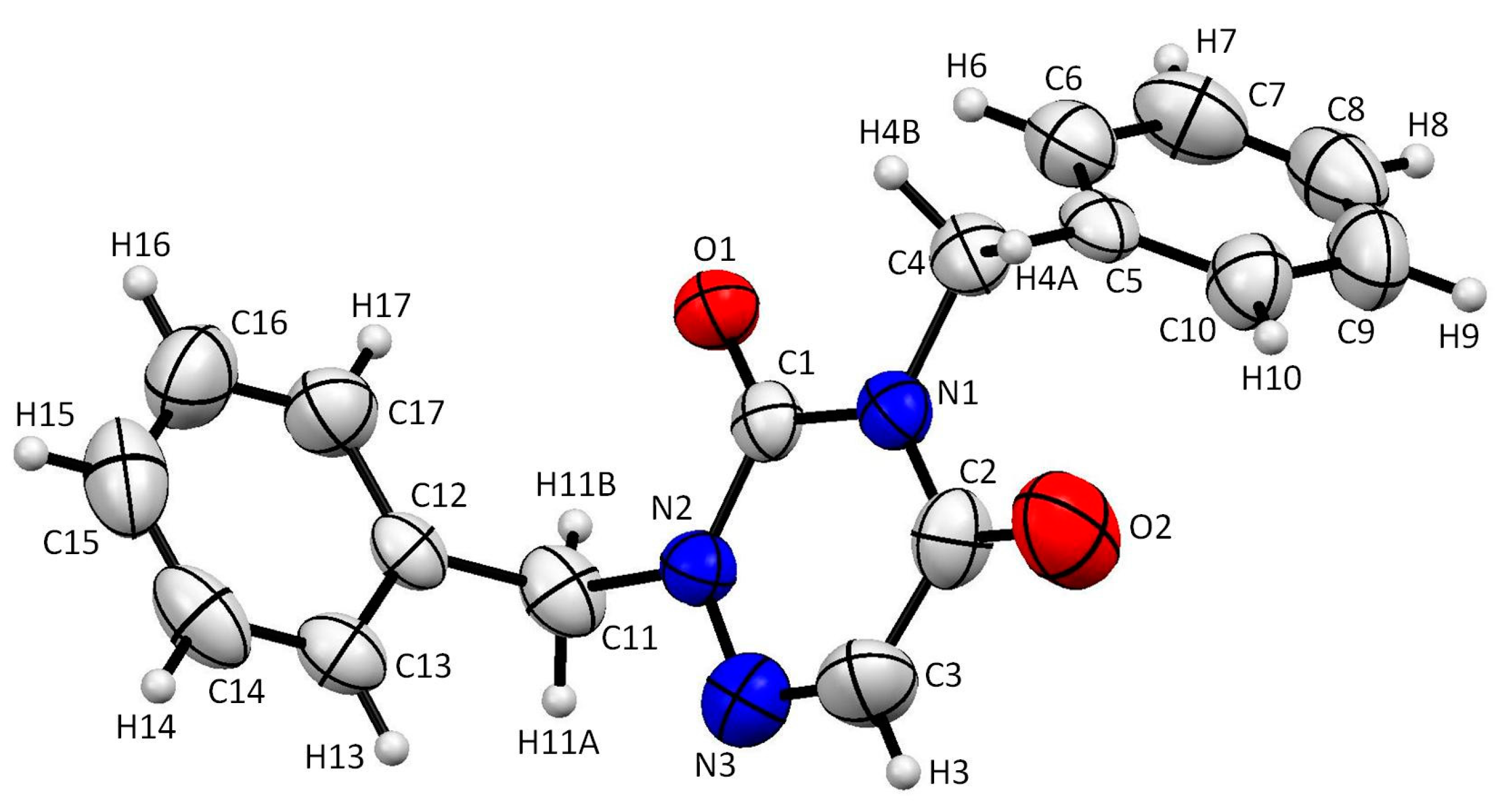
2.2. Analysis of the X-ray Crystallographic Structure
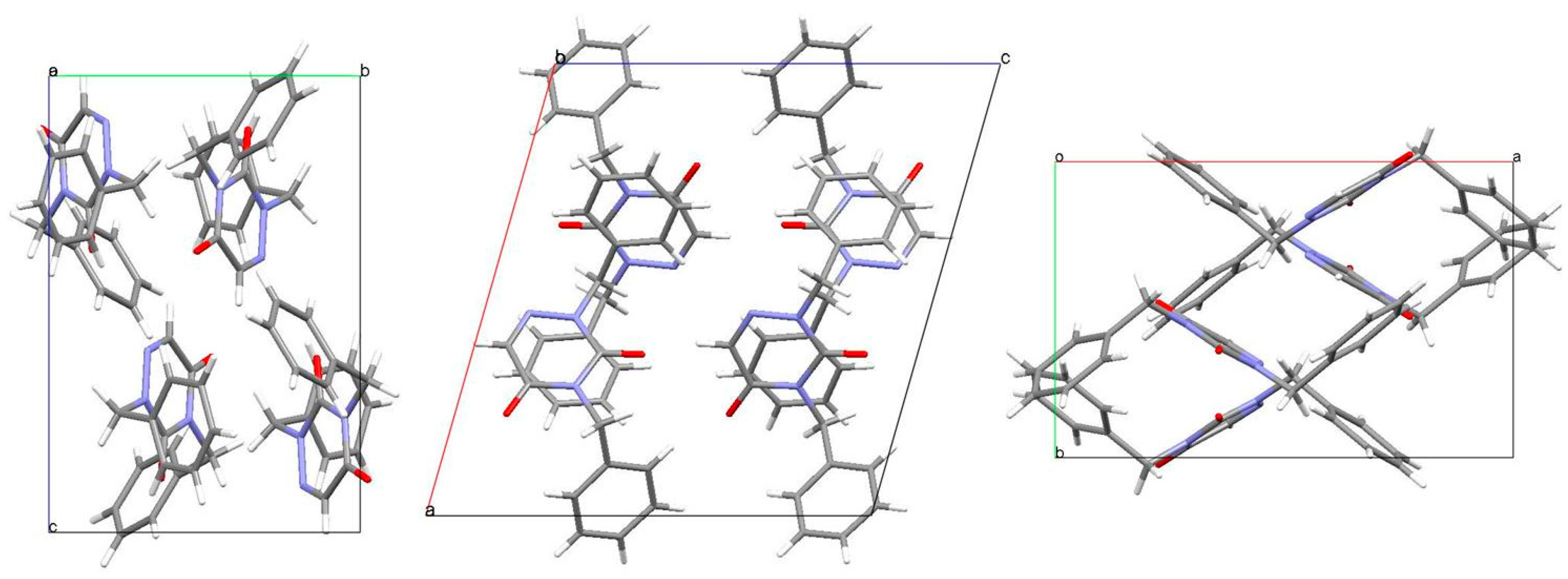
2.3. Analysis of the Molecular Packing
3. Materials and Methods
3.1. General Techniques
3.2. Syntheses
3.2.1. Syntheses of 4-Benzyl-1,2,4-triazin-3,5(2H,4H)-dione (2) and 2,4-Dibenzyl-1,2,4-triazin-3,5(2H,4H)-dione (3)
3.2.2. 2,4-Dibenzyl-1,2,4-triazin-3,5(2H,4H)-dione (3)
3.3. X-ray Diffraction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neunhoeffer, H.; Wiley, P.F. Chemistry of 1,2,3-Triazines and 1,2,4-Triazines, Tetrazines and Pentazines; Weissberger, A., Taylor, E.C., Eds.; John Wiley and Sons: New York, NY, USA, 1978; pp. 1001–1004. [Google Scholar]
- Szárics, E.; Riedl, Z.; Nyikos, L.; Hajós, G.; Kardos, J. Interaction of novel condensed triazine derivatives with central and peripheral type benzodiazepine receptors: Synthesis, in vitro pharmacology and modelling. Eur. J. Med. Chem. 2006, 41, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Pasternak, K.; Rajtar, B.; Sztanke, M.; Majek, M.; Polz-Dacewicz, M. Identification of antibacterial and antiviral activities of novel fused 1,2,4-triazine esters. Bioorg. Med. Chem. 2007, 15, 5480–5486. [Google Scholar] [CrossRef] [PubMed]
- Salimon, J.; Salih, N. Synthesis, characterization and biological activity of some new 1,2,4-triazine derivatives. Int. J. PharmTech Res. 2010, 2, 1041–1045. [Google Scholar]
- Guerrini, G.; Ciciani, G.; Cambi, G.; Bruni, F.; Selleri, S.; Melani, F.; Montali, M.; Martini, C.; Ghelardini, C.; Norcini, M.; et al. Novel 3-aroylpyrazolo[5,1-c][1,2,4]benzotriazine 5-oxides 8-substituted, ligands at GABAA/benzodiazepine receptor complex: Synthesis, pharmacological and molecular modeling studies. Bioorg. Med. Chem. 2008, 16, 4471–4489. [Google Scholar] [CrossRef] [PubMed]
- Mullick, P.; Khan, S.A.; Begum, T.; Verma, S.; Kaushik, D.; Alam, O. Synthesis of 1,2,4-triazine derivatives as potential anti-anxiety and anti-inflammatory agents. Acta Pol. Pharm. Drug Res. 2009, 66, 379–385. [Google Scholar]
- Irannejad, H.; Amini, M.; Khodagholi, F.; Ansari, N.; Tusi, S.K.; Sharifzadeh, M.; Shafiee, A. Synthesis and in vitro evaluation of novel 1,2,4-triazine derivatives as neuroprotective agents. Bioorg. Med. Chem. 2010, 18, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Sangshetti, J.N.; Shinde, D.B. One pot synthesis and SAR of some novel 3-substituted 5,6-diphenyl-1,2,4-triazines as antifungal agents. Bioorg. Med. Chem. Lett. 2010, 20, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lin, J.; Wrobleski, S.T.; Lin, S.; Hynes, J.; Wu, H.; Dyckman, A.J.; Li, T.; Wityak, J.; Gillooly, K.M.; et al. Discovery of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38α MAP kinase inhibitor for the treatment of inflammatory diseases. J. Med. Chem. 2010, 53, 6629–6639. [Google Scholar] [CrossRef] [PubMed]
- Kusch, P.; Deininger, S.; Specht, S.; Maniako, R.; Haubrich, S.; Pommerening, T.; Lin, P.K.T.; Hoerauf, A.; Kaiser, A. In vitro and in vivo antimalarial activity assays of seeds from Balanites aegyptiaca: Compounds of the extract show growth inhibition and activity against Plasmodial Aminopeptidase. J. Parasitol. Res. 2011, 2011. [Google Scholar] [CrossRef]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Mohamed, A.A. The reaction of cyanoacetylhydrazine with ω-bromo(4-methyl)acetophenone: Synthesis of heterocyclic derivatives with antitumor activity. Molecules 2010, 15, 3602–3617. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.H.; Moustafa, A.H. Synthesis and anticancer activity of some new S-glycosyl and S-alkyl 1,2,4-triazinone derivatives. Molecules 2011, 16, 5682–5700. [Google Scholar] [CrossRef] [PubMed]
- Zołnowska, B.; Sławinski, J.; Belka, M.; Baczek, T.; Kawiak, A.; Chojnacki, J.; Pogorzelska, A.; Szafranski, K. Synthesis, molecular structure, metabolic stability and QSAR studies of a novel series of anticancer N-acylbenzenesulfonamides. Molecules 2015, 20, 19101–19129. [Google Scholar] [CrossRef] [PubMed]
- Zołnowska, B.; Sławinski, J.; Pogorzelska, A.; Szafranski, K.; Kawiak, A.; Stasilojc, G.; Belka, M.; Ulenberg, S.; Baczek, T.; Chojnacki, J. Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability. Molecules 2016, 21, 808. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Welch, A.D. Microbial studies of 6-azauracil, an antagonist of uracil. Cancer Res. 1956, 16, 965–969. [Google Scholar] [PubMed]
- Falke, D.; Rada, B. 6-Azauridine as an inhibitor of the synthesis of Herpesvirus hominis. Acta Virol. 1970, 14, 115–123. [Google Scholar] [PubMed]
- Sidwell, R.W.; Dixon, G.J.; Sellers, S.M.; Schabel, F.M., Jr. In vivo antiviral properties of biologically active compounds. II. Studies with influenza and vaccinia viruses. Appl. Microbiol. 1968, 16, 370–392. [Google Scholar] [PubMed]
- Creasey, W.A.; Fink, M.E.; Handschurnacher, R.E.; Calabresi, P. Clinical and pharmacological studies with 2′,3′,5′-triacetyl-6-azauridine. Cancer Res. 1963, 23, 444–453. [Google Scholar] [PubMed]
- Walters, T.R.; Aur, R.J.A.; Hernandez, K.; Vietti, T.; Pinkel, D. 6-Azauridine in combination chemotherapy of childhood acute myelocytic leukemia. Cancer 1972, 29, 1057–1060. [Google Scholar] [CrossRef]
- Matolcsy, G. Aufnahme von Uracyl, Azauracyl und Maleinhydrazid durch Pflanzen in Abhängigkeit von der Konzentration un dihre gegenseitige Beeinflussung. Acta Phytopathol. 1966, 1, 245–250. [Google Scholar]
- Pasternak, C.A.; Handschumacher, R.E. The biochemical activity of 6-azauridine: Interference with pyrimidine metabolism in transplantable mouse tumors. J. Biol. Chem. 1959, 234, 2992–2997. [Google Scholar] [PubMed]
- Handschumacher, R.E. Orotidylic acid decarboxylase: Inhibition studies with azauridine 5′-phosphate. J. Biol. Chem. 1959, 235, 2917–2919. [Google Scholar]
- Exinger, F.; Lacroute, F. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr. Genet. 1992, 22, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Mylari, B.L.; Miller, M.W.; Howes, H.L., Jr.; Figdor, S.K.; Lynch, J.E.; Koch, R.C. Anticoccidial derivatives of 6-azauracil. 1. Enhancement of activity by benzylation of nitrogen-1. Observations on the design of nucleotide analogues in chemotherapy. J. Med. Chem. 1977, 20, 475–483. [Google Scholar] [PubMed]
- Sarges, R.; Schnur, R.C.; Belletire, J.L.; Peterson, M.J. Spiro hydantoin aldose reductase inhibitors. J. Med. Chem. 1988, 31, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Hwang, L.C.; Wei, D.C.; Cheng, M.C.; Wang, Y.; Tzeng, C.C. Synthesis and structure assignment of 1-(2-acetoxyethoxy)methyl derivatives of 5-chloro-6-azauracil and 5-bromo-6-azaisocytosine. Nucleosides Nucleotides 1994, 13, 2185–2193. [Google Scholar] [CrossRef]
- Hwang, L.C.; Wang, E.C.; Lee, K.H.; Tzeng, C.C. Synthetic and antiviral studies on certain acyclonucleosides of 5-substituted-6-azauracils. Chin. Pharm. J. 1995, 47, 1–11. [Google Scholar]
- Tzeng, C.C.; Hwang, L.C.; Chen, C.C.; Wei, D.C. Synthesis of racemic 5-substituted 1-(2,3-dihydroxypropyl)-5-substituted-6-azauracils and their isosterric isomers. Nucleosides Nucleotides 1995, 14, 1425–1435. [Google Scholar] [CrossRef]
- Niedballa, U.; Vorbrüggen, H. A general synthesis of pyrimidine nucleosides. Angew. Chem. Int. Ed. 1970, 9, 461–462. [Google Scholar] [CrossRef]
- Novacek, A.; Vondracek, B.; Gut, J.; Hesoun, D.; Luksik, L. 6-Azauracil. Czech Patent 108383, 15 Septemebr 1963. [Google Scholar]
- Hwang, L.C.; Jane, S.Y.; Lai, H.Y.; Tu, C.H.; Lee, G.H. Synthesis, molecular structure and characterization of allylic derivatives of 6-amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]triazin-8(7H)-one. Molecules 2006, 11, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R.; Frederikse, H.P.R. CRC Handbook of Chemistry and Physics, 80th ed.; CRC Press: New York, NY, USA, 1999; pp. 1–14. [Google Scholar]
- Sheldrick, G.M. SHELXS-97, Program for the Solution of Crystal Structure; University of Göttingen: Göttingen, Germany, 1990. [Google Scholar]
- Sheldrick, G.M. SHELXL-2014/7, Program for the Refinement of Crystal Structure; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C17H15N3O2 |
|---|---|
| Formula weight | 293.32 |
| Temperature | 295(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a = 13.7844(13) Å | |
| Unit cell dimensions | b = 8.5691(8) Å, β = 105.961(2)° |
| c = 13.0527(12) Å | |
| Volume | 1482.3(2) Å3 |
| Z | 4 |
| Density (calculated) | 1.314 Mg/m3 |
| Absorption coefficient | 0.089 mm−1 |
| F(000) | 616 |
| Theta range for data collection | 2.831 to 24.999° |
| Index ranges | ‒16 ≤ h ≤ 16, −9 ≤ k ≤ 10, −15 ≤ l ≤ 15 |
| Reflections collected | 8796 |
| Independent reflections | 2612 (Rint = 0.0509) |
| Completeness to θ = 24.999° | 99.90% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.756 and 0.659 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2612/0/199 |
| Goodness-of-fit on F2 | 1.085 |
| Final R indices (I > 2σ(I)) | R1 = 0.0851, wR2 = 0.1922 |
| R indices (all data) | R1 = 0.1281, wR2 = 0.2139 |
| Largest difference peak and hole | 0.566 and −0.189 eÅ−3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, L.-C.; Yang, S.-Y.; Chuang, C.-L.; Lee, G.-H. An Optimized Synthesis, Molecular Structure and Characterization of Benzylic Derivatives of 1,2,4-Triazin-3,5(2H,4H)-dione. Molecules 2017, 22, 1924. https://doi.org/10.3390/molecules22111924
Hwang L-C, Yang S-Y, Chuang C-L, Lee G-H. An Optimized Synthesis, Molecular Structure and Characterization of Benzylic Derivatives of 1,2,4-Triazin-3,5(2H,4H)-dione. Molecules. 2017; 22(11):1924. https://doi.org/10.3390/molecules22111924
Chicago/Turabian StyleHwang, Long-Chih, Shiun-Yau Yang, Chung-Lin Chuang, and Gene-Hsiang Lee. 2017. "An Optimized Synthesis, Molecular Structure and Characterization of Benzylic Derivatives of 1,2,4-Triazin-3,5(2H,4H)-dione" Molecules 22, no. 11: 1924. https://doi.org/10.3390/molecules22111924