Glypre: In Silico Prediction of Protein Glycation Sites by Fusing Multiple Features and Support Vector Machine

Abstract
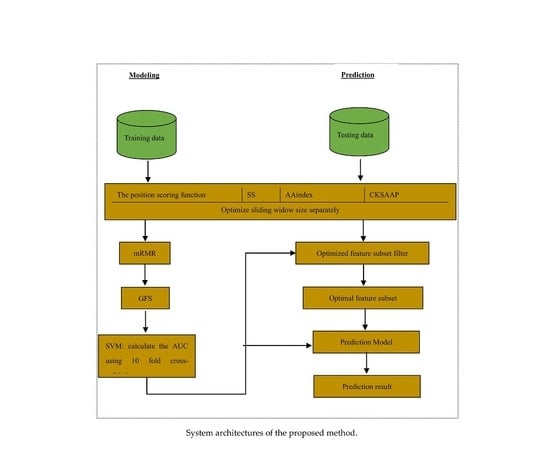
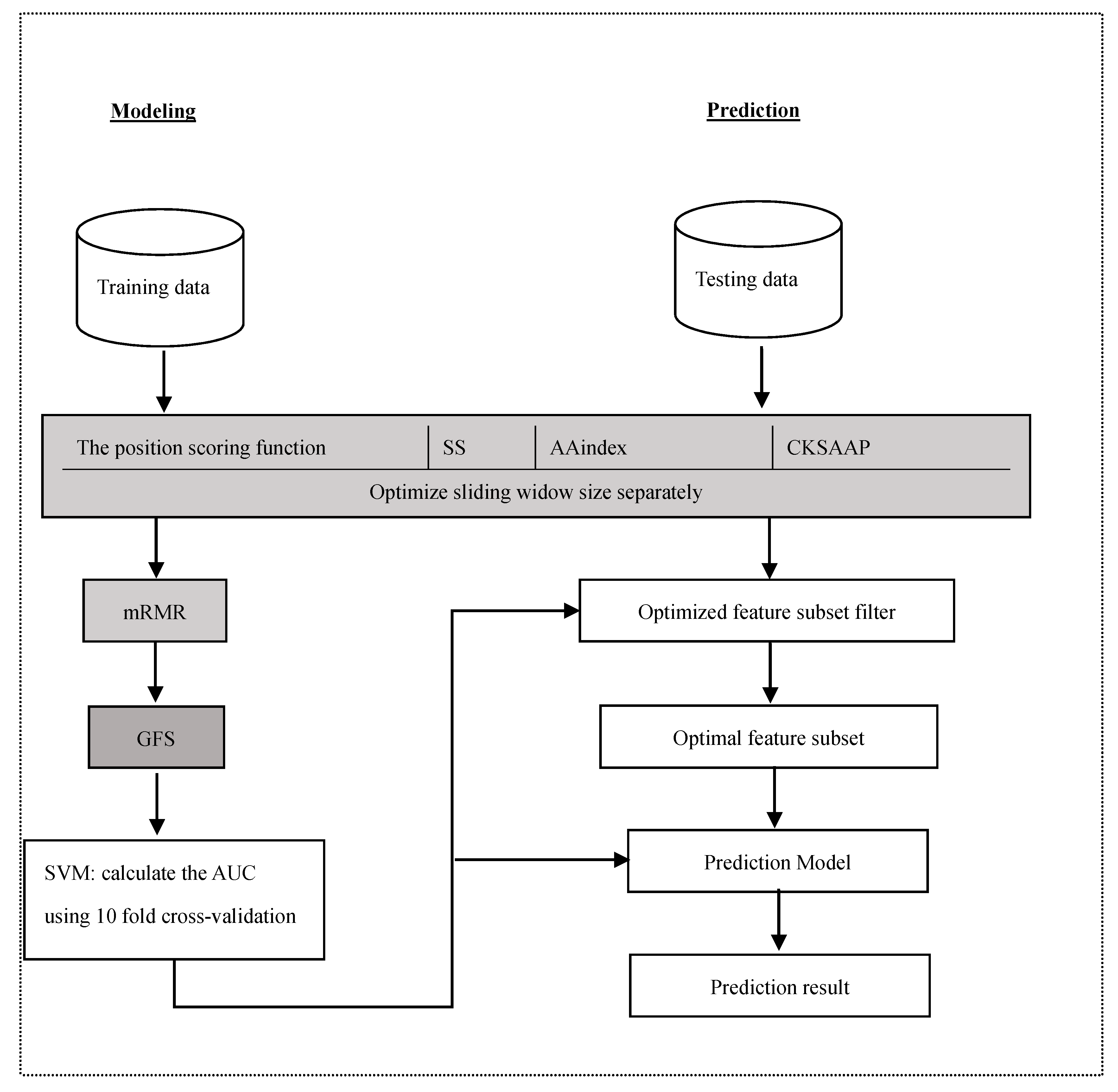
:
1. Introduction
2. Results and Discussion
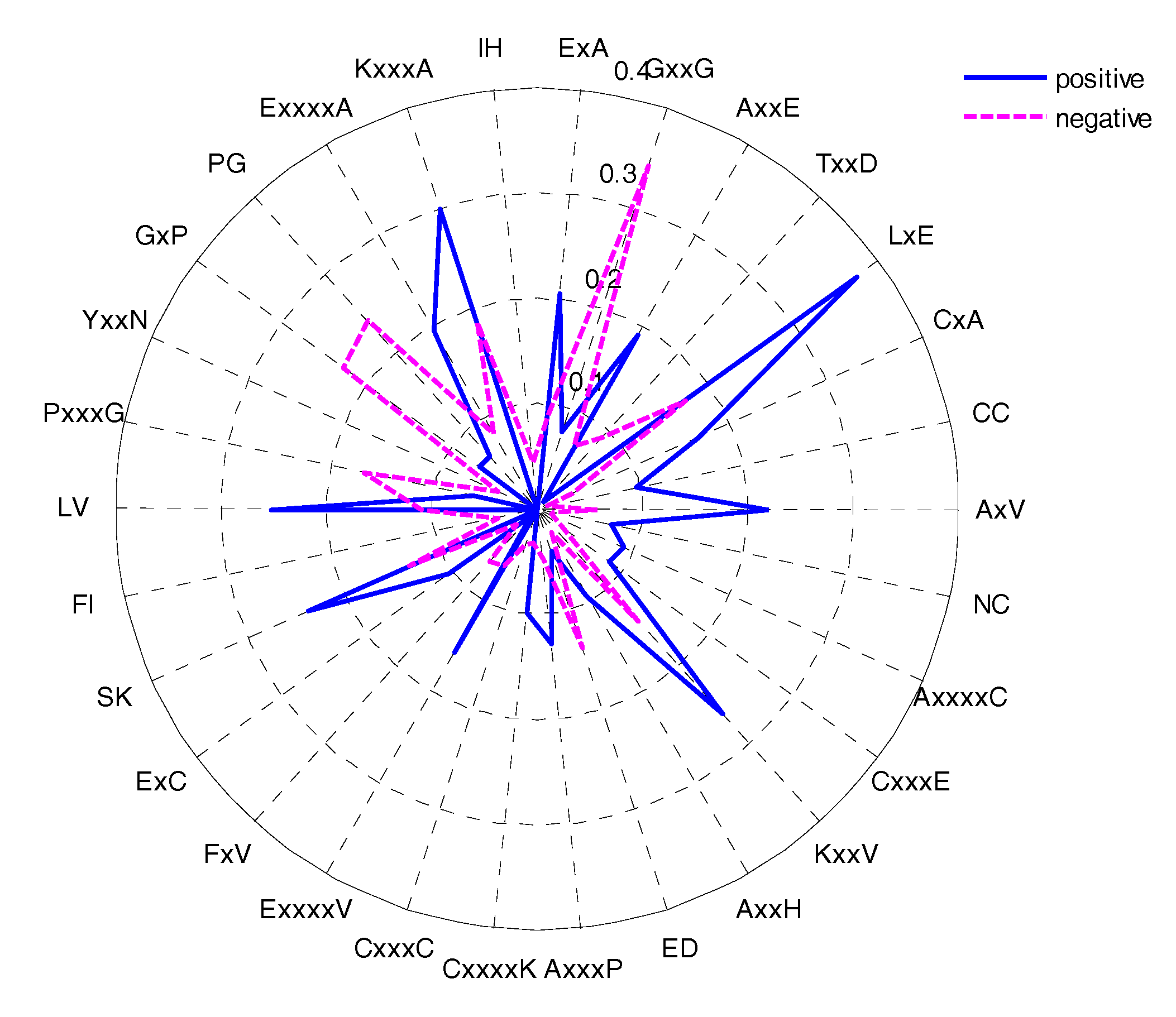
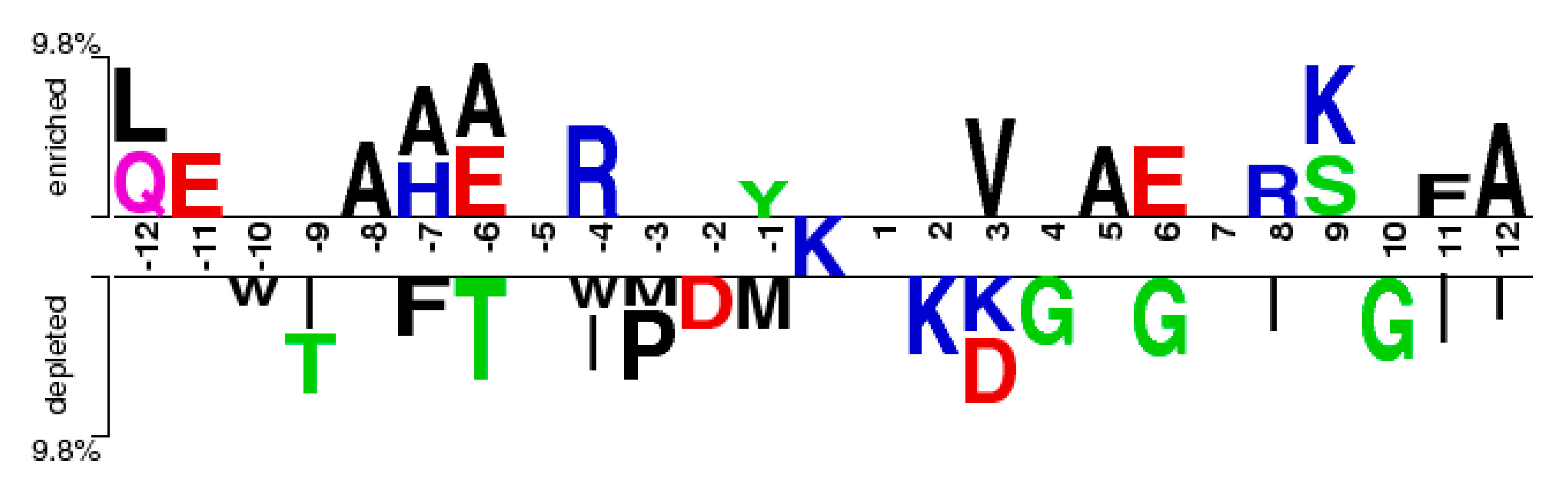
2.1. Investigation of Different Features
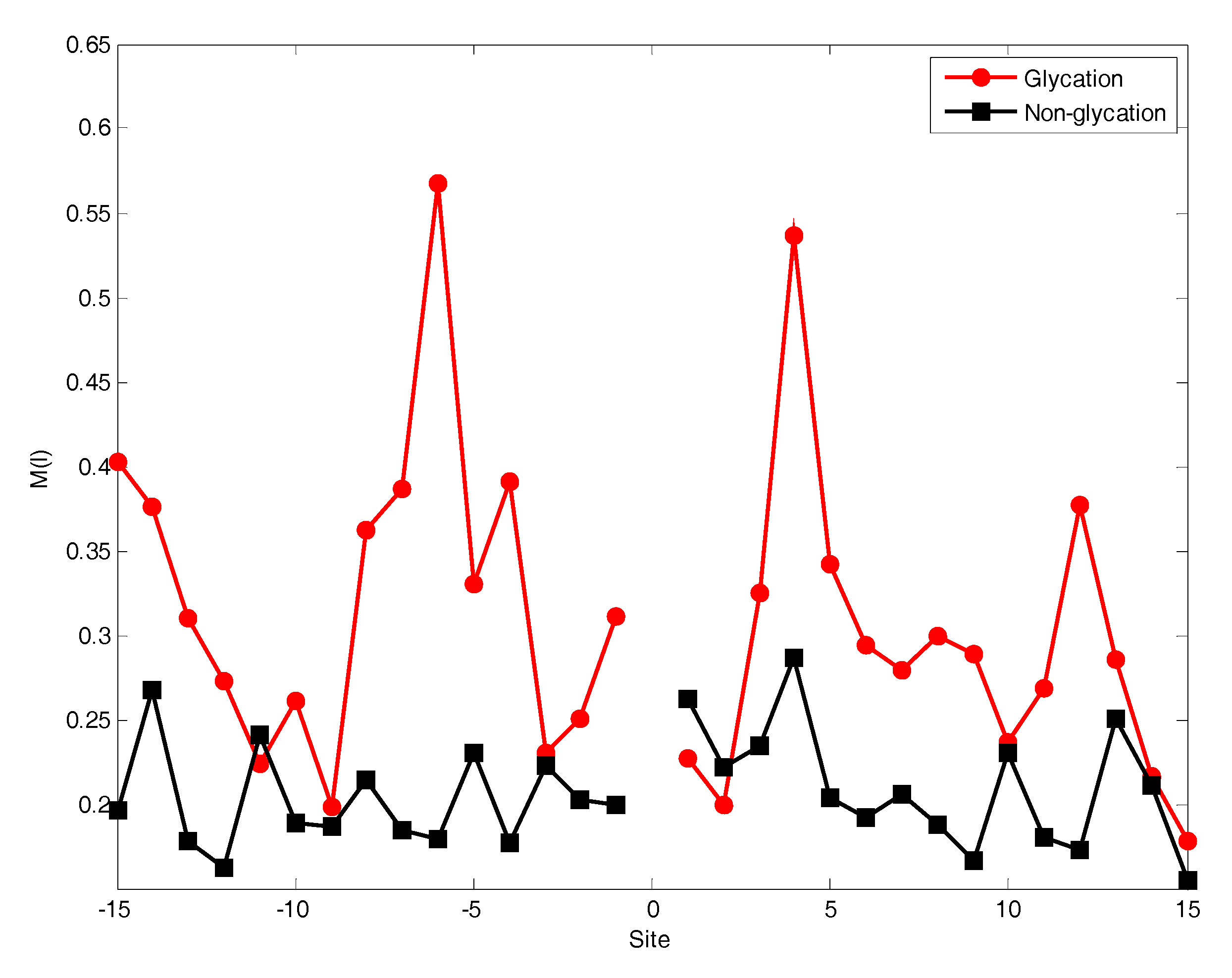
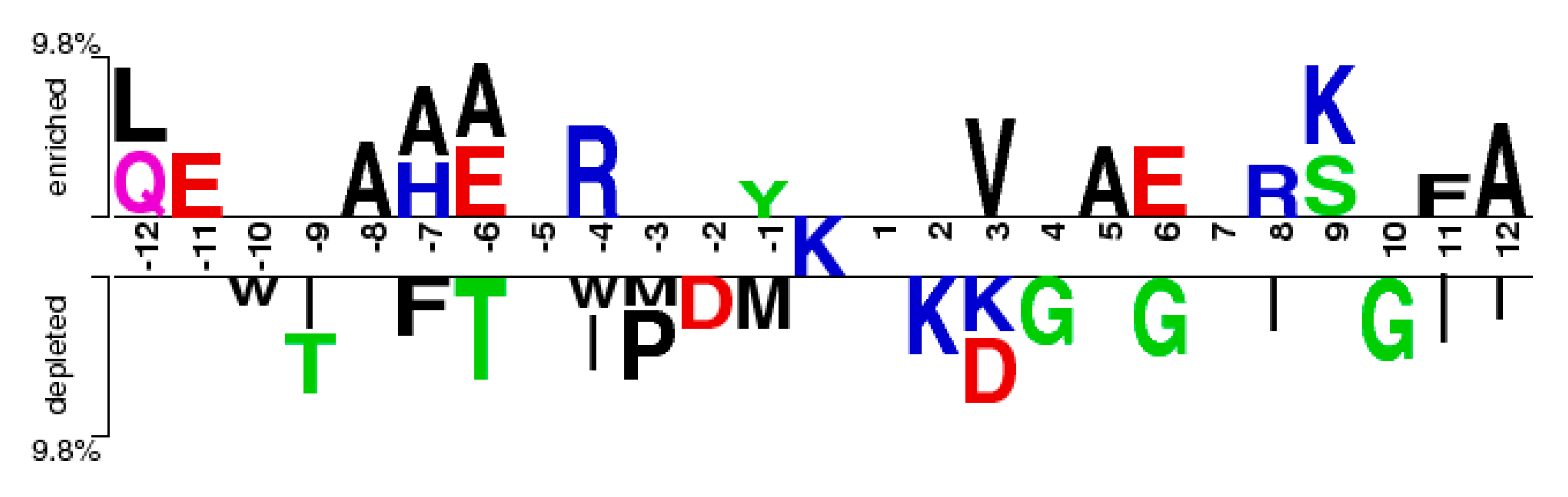
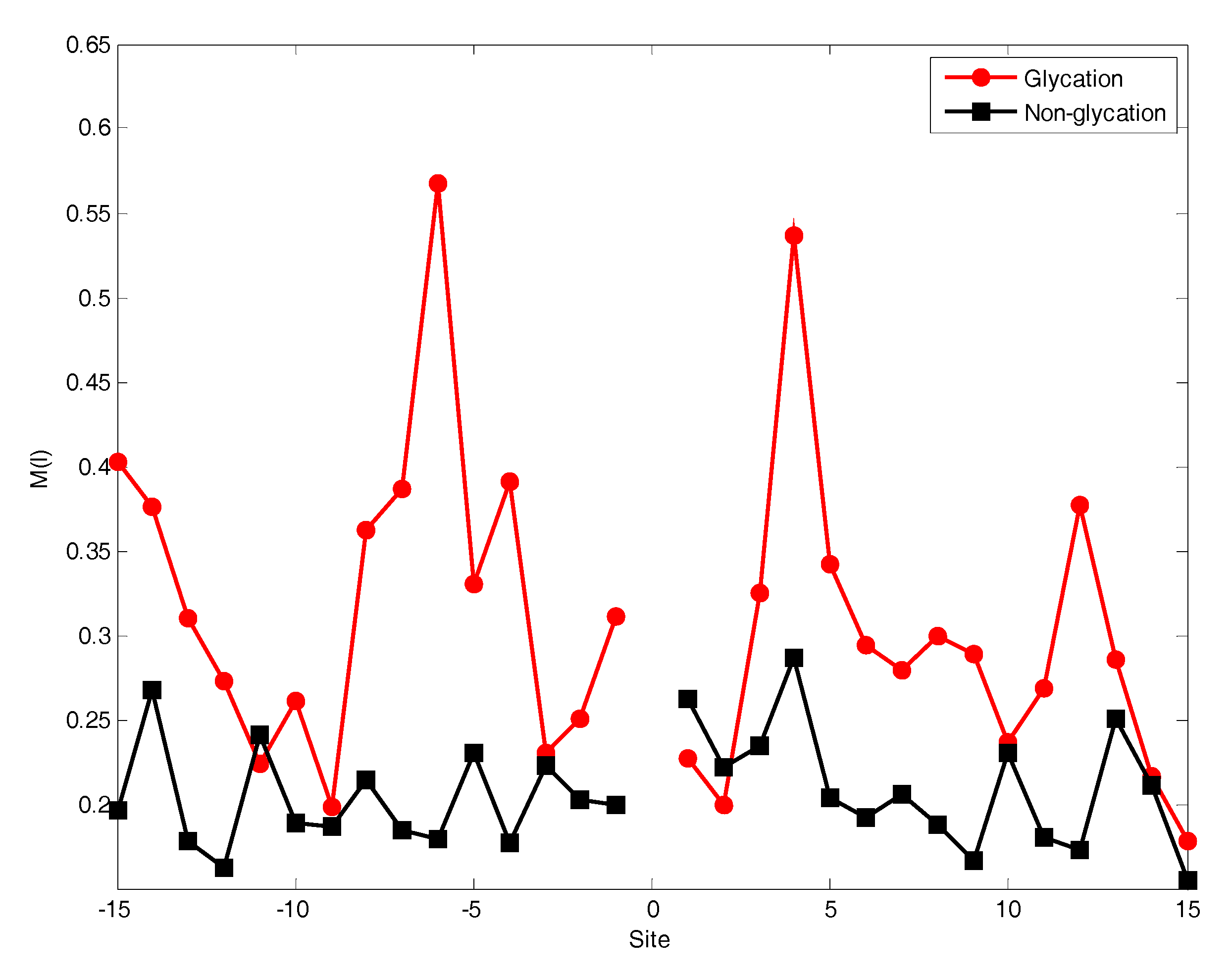
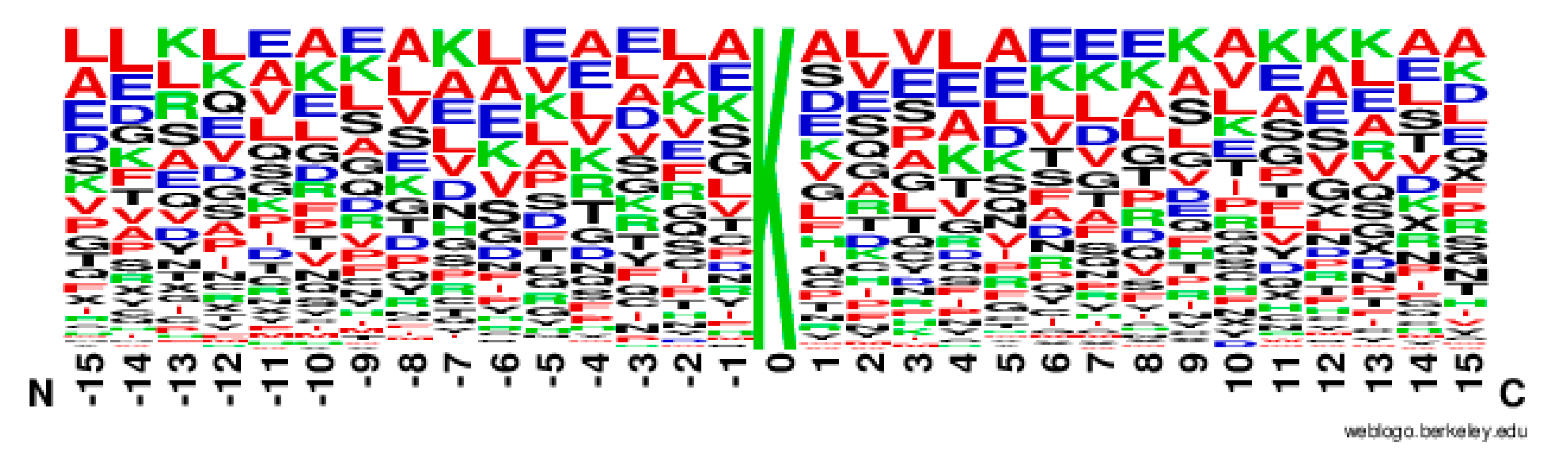
2.2. The Position Conservation Features Analysis
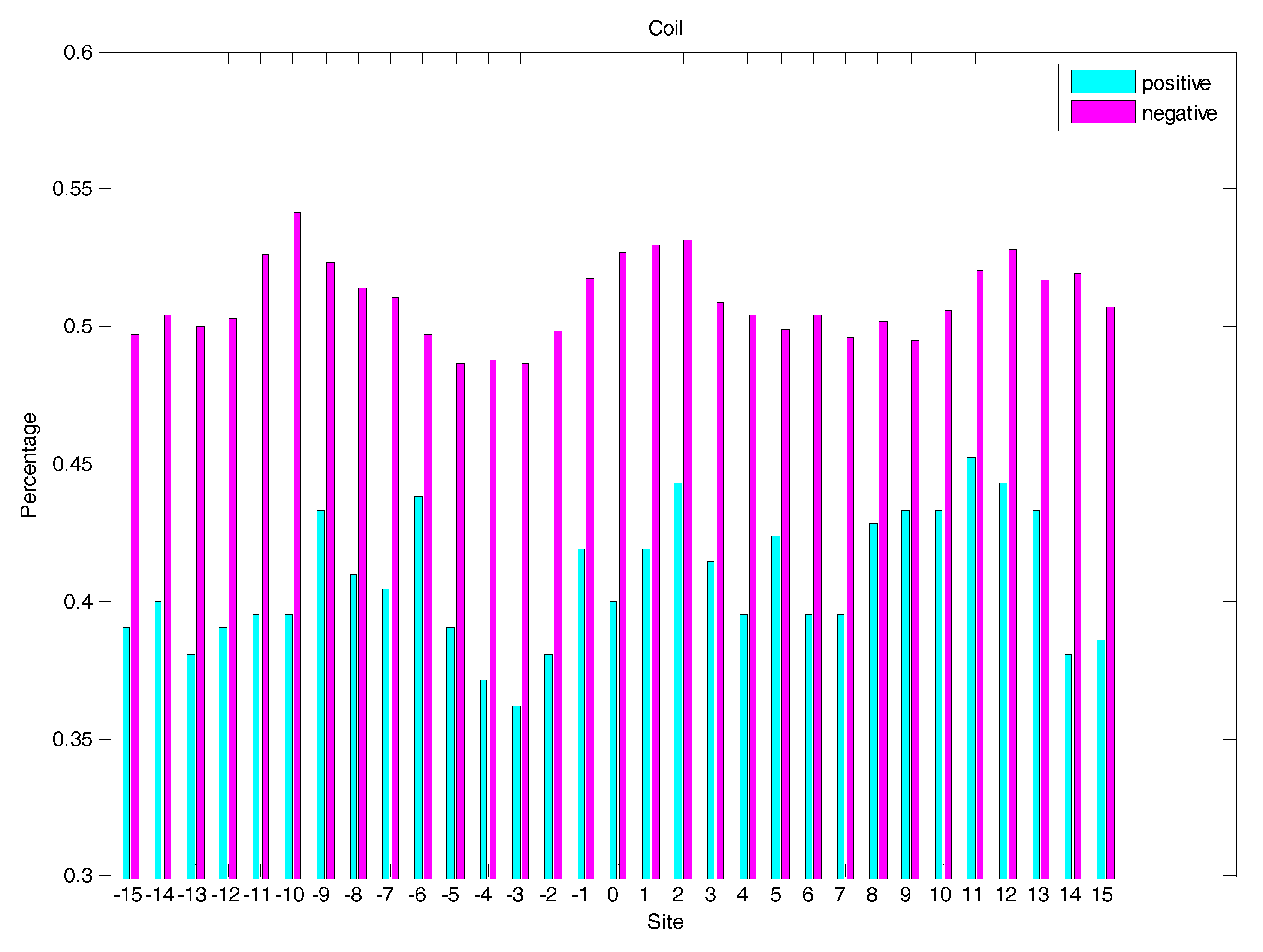
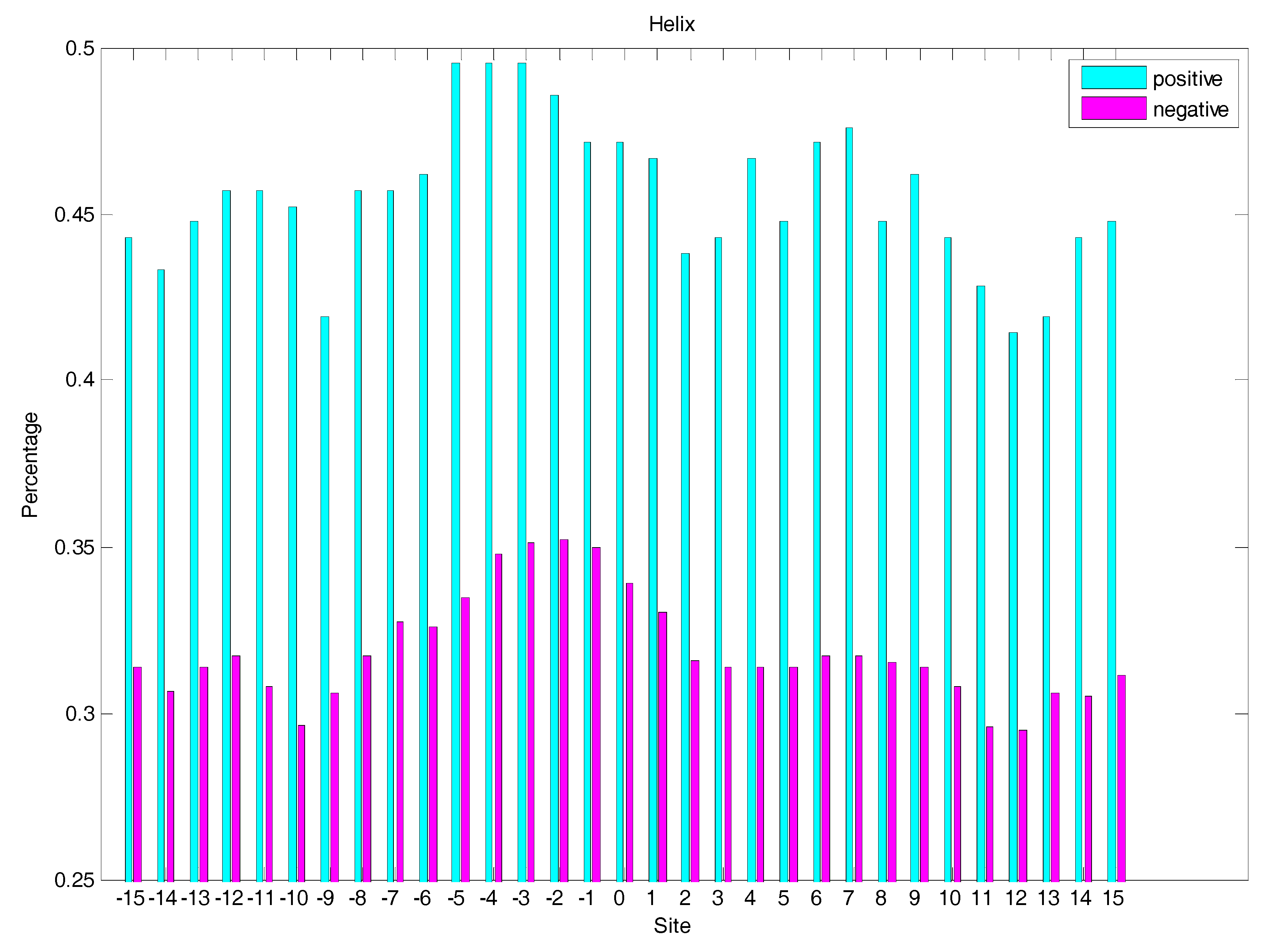
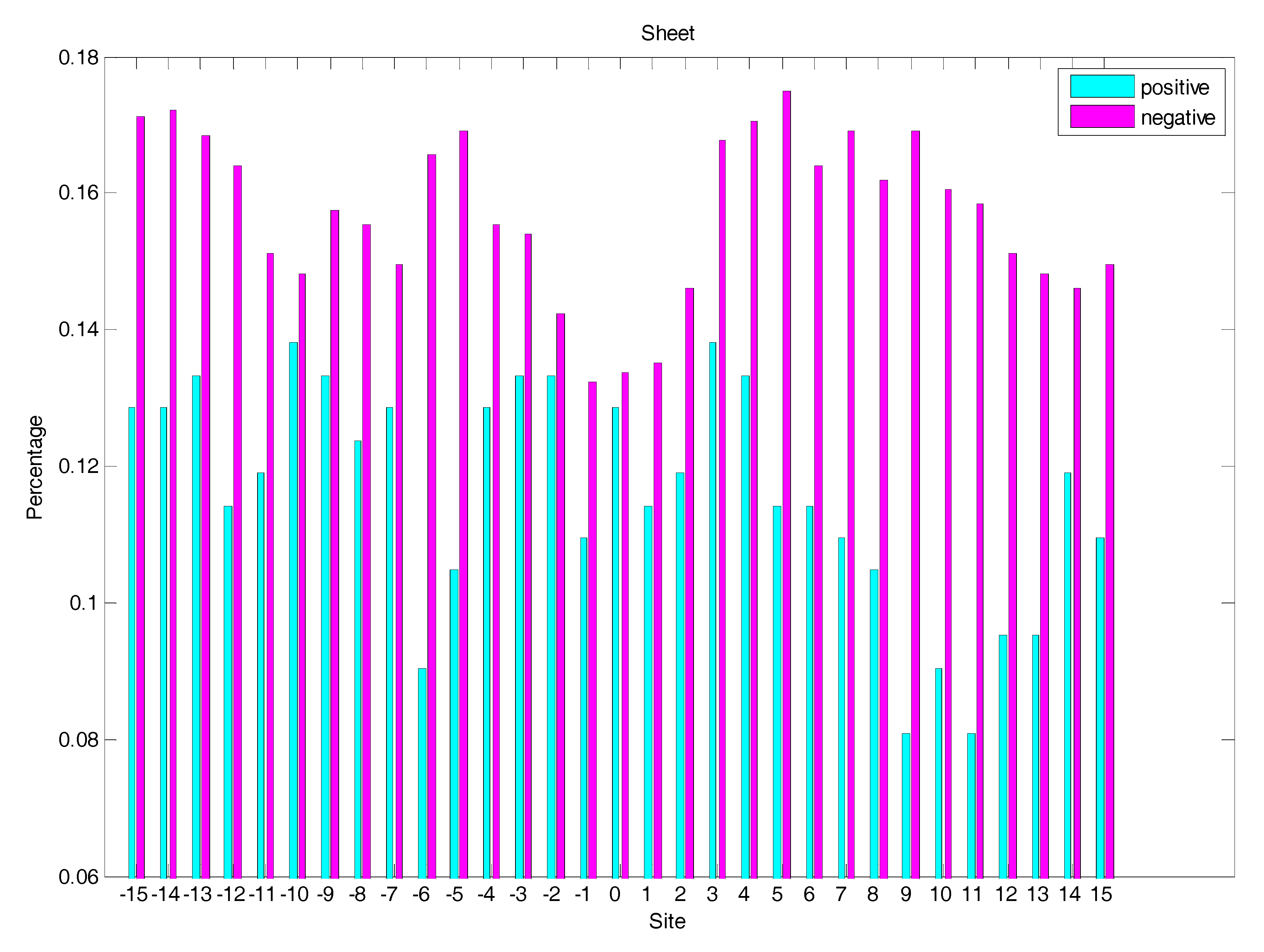
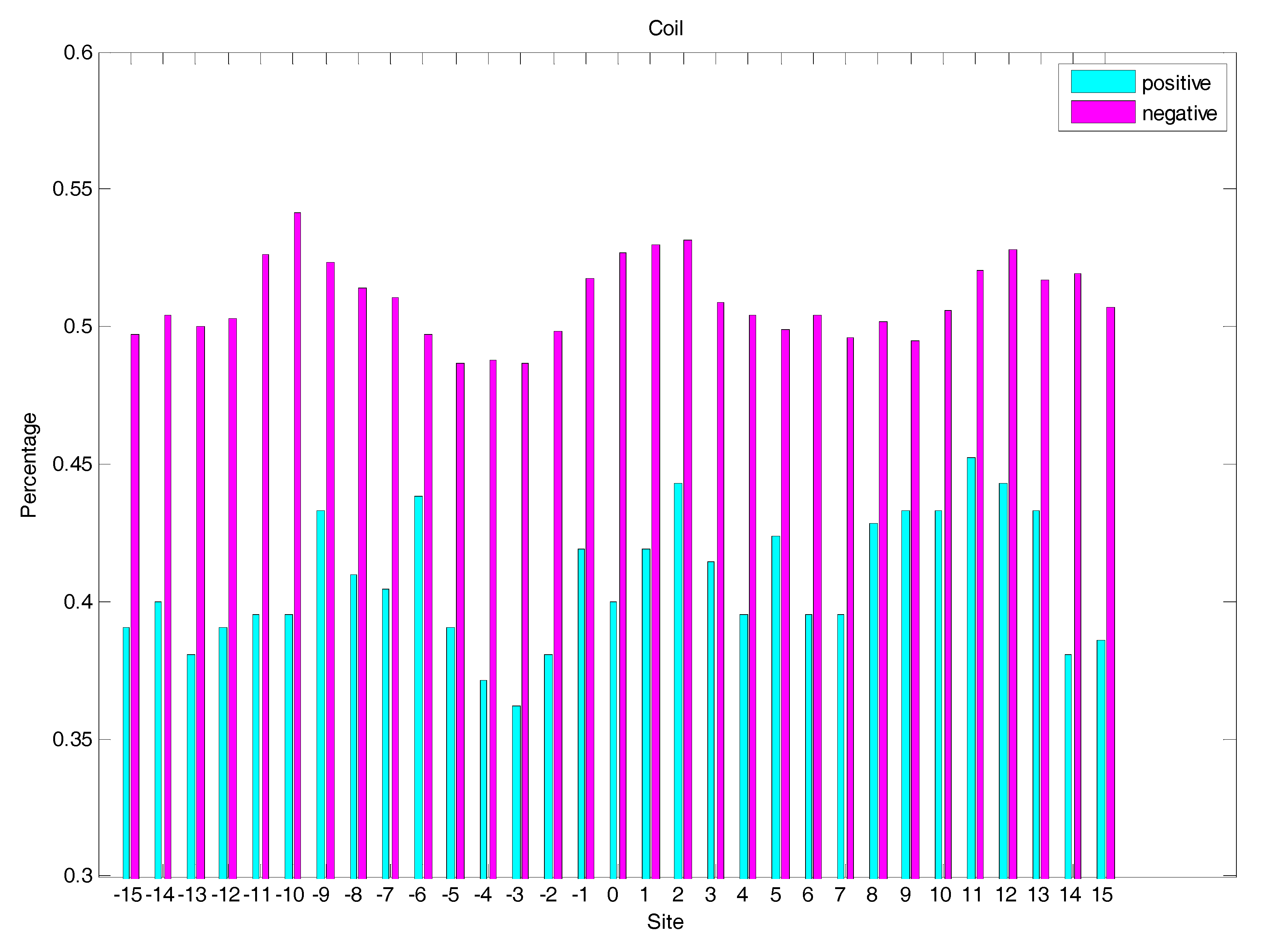
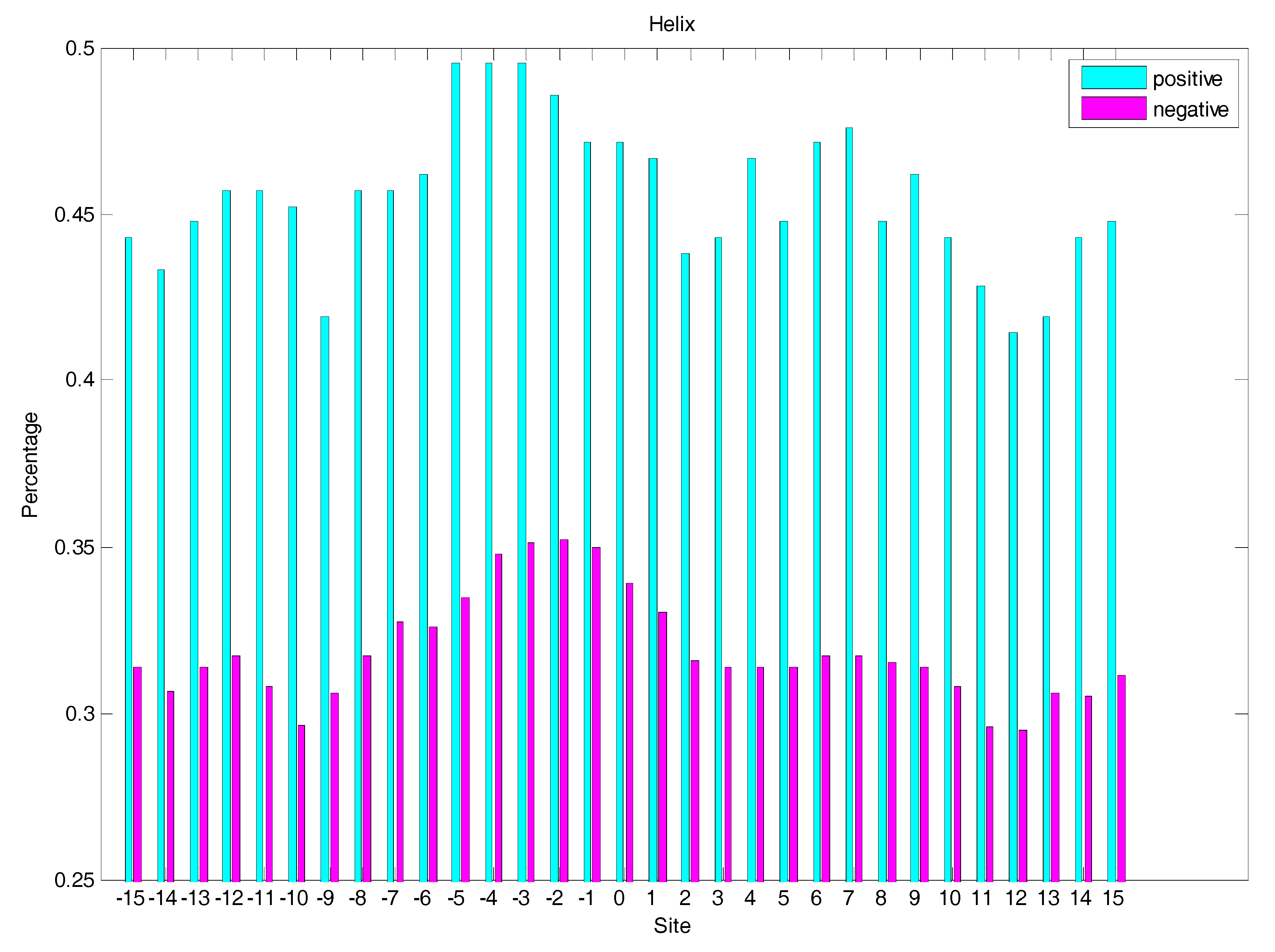
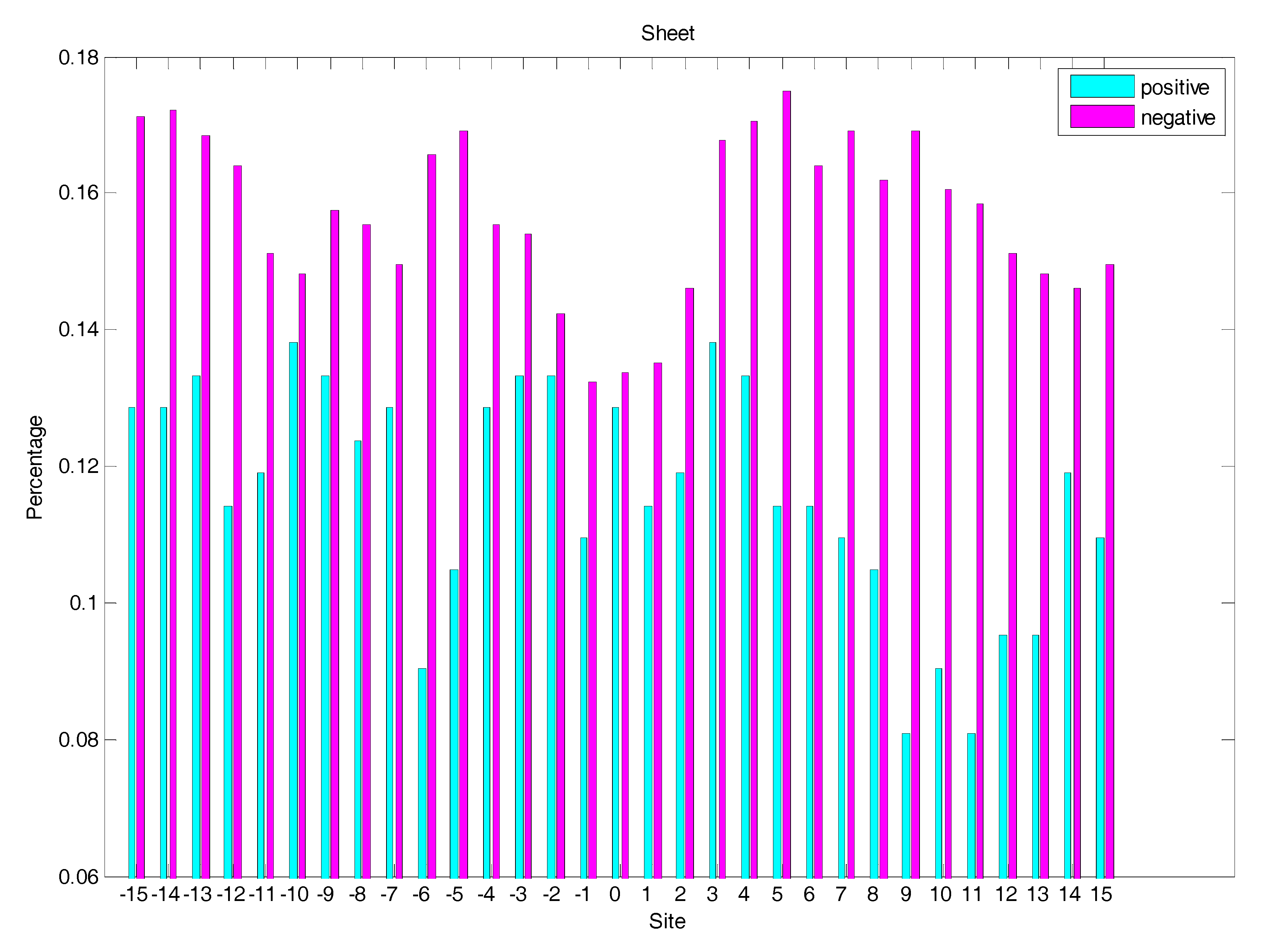
2.3. Secondary Structure Features Analysis
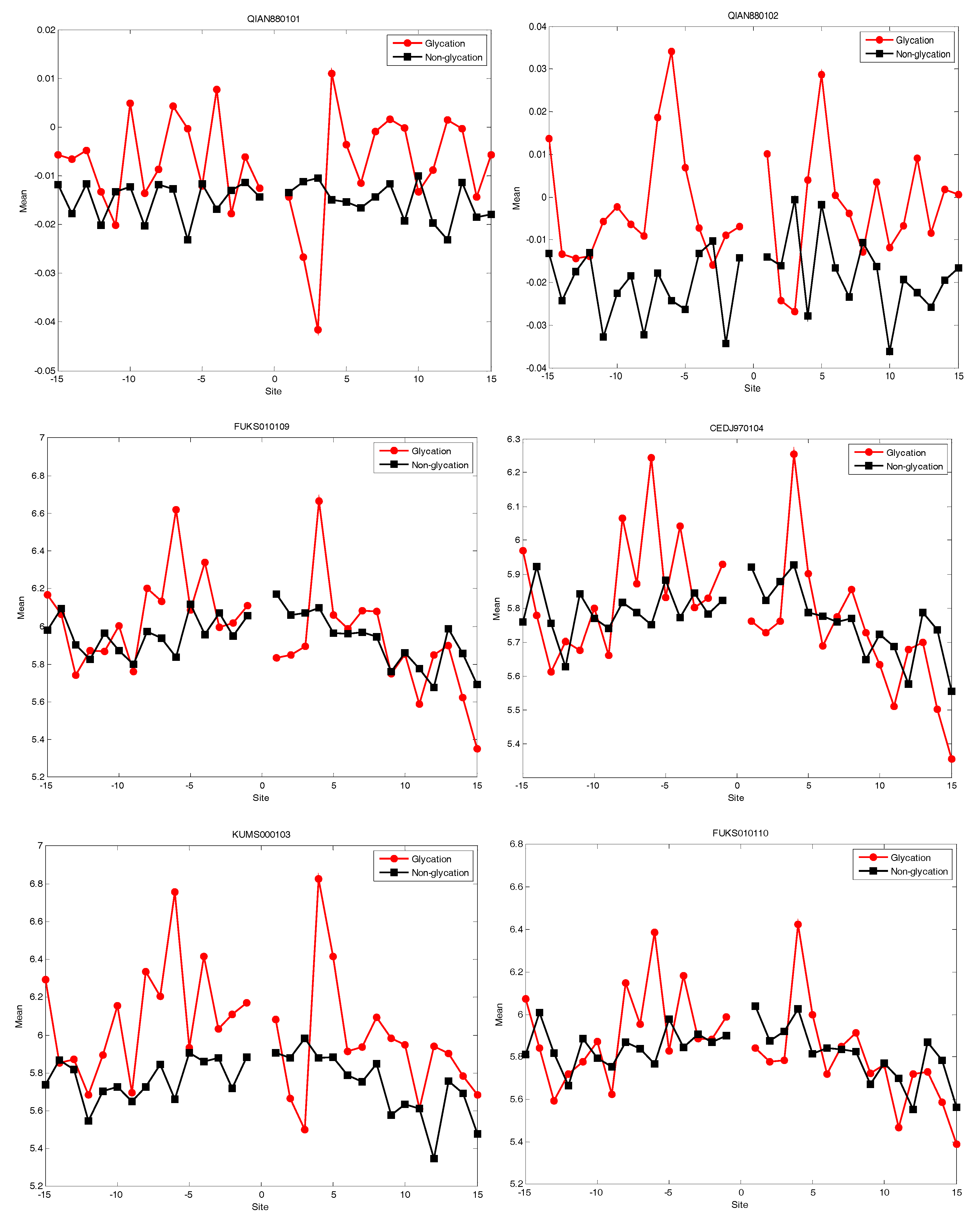
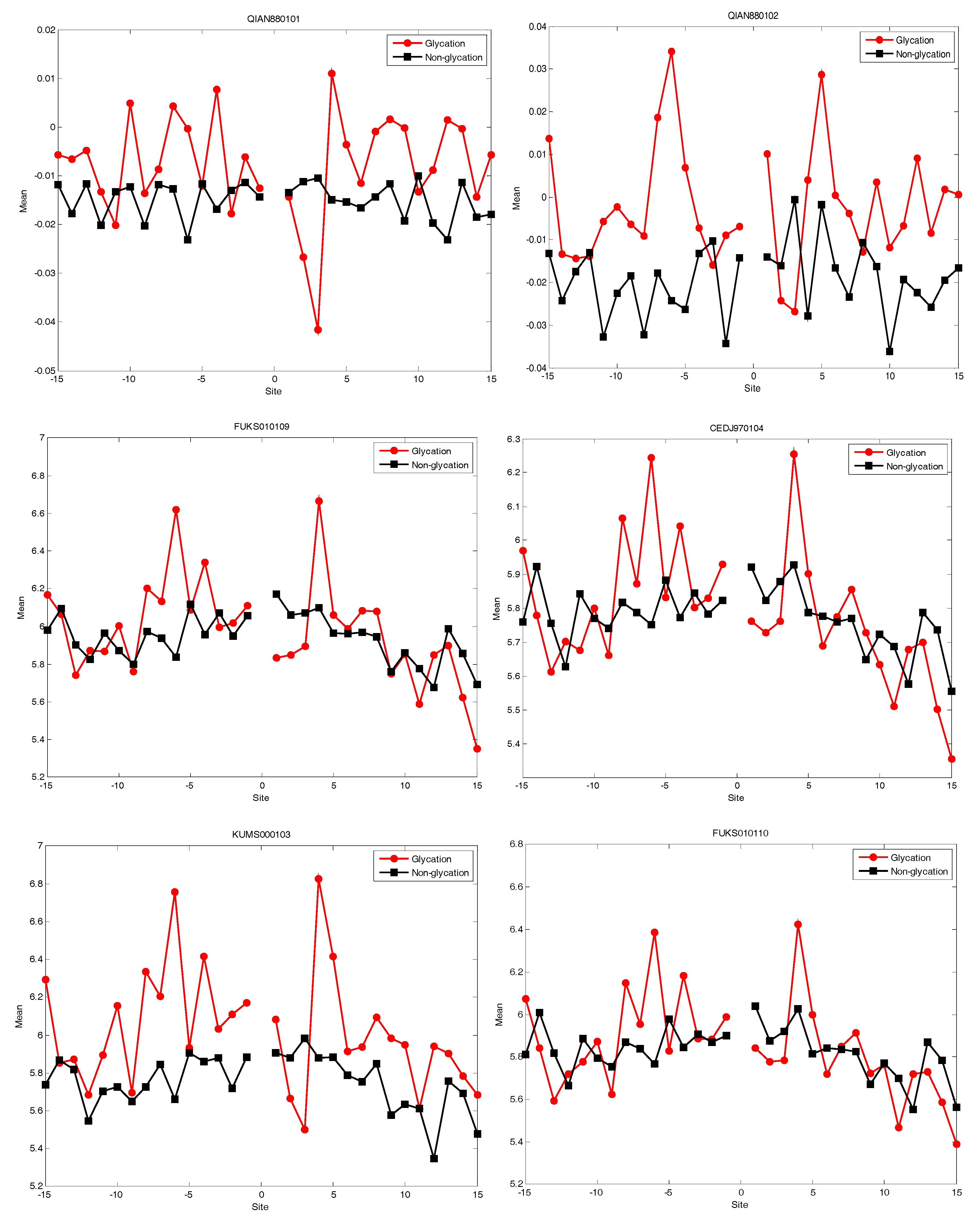
2.4. AAindex Features Analysis
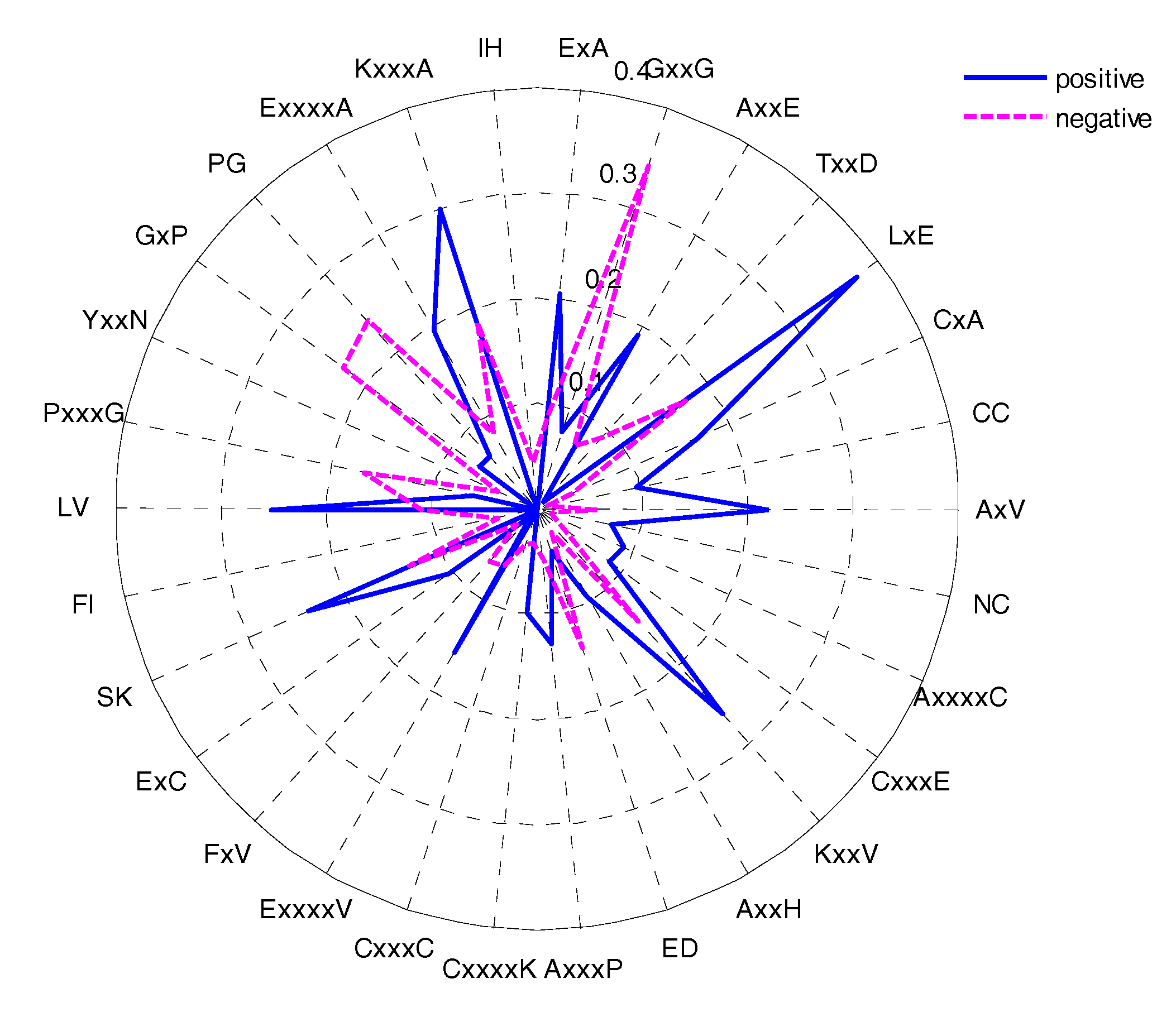
2.5. CKSAAP Feature Analysis
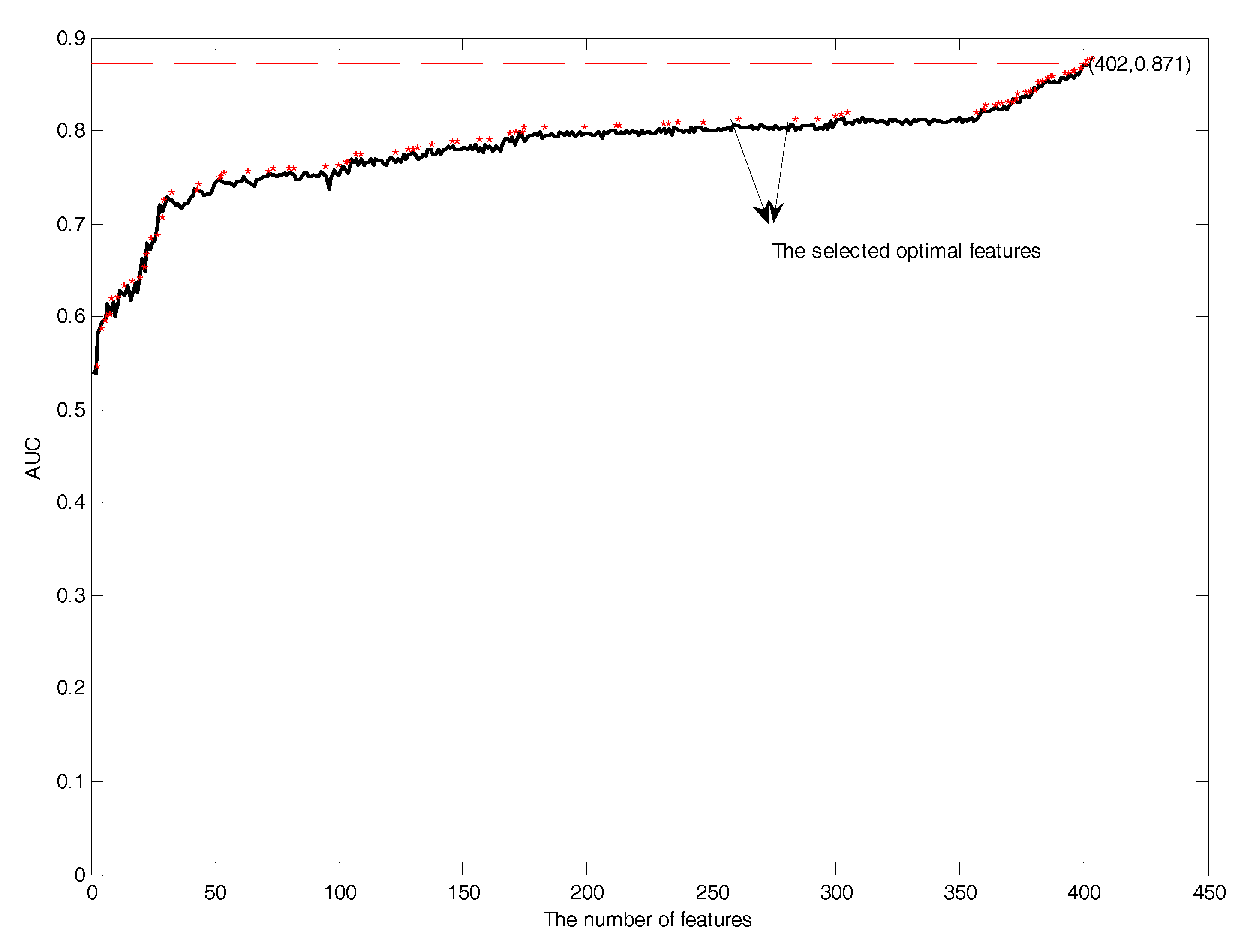
2.6. The Performance of the Proposed Predictor
2.7. Comparison with Existing Methods
2.8. Comparison with Other Predictors on the Independent Test Dataset
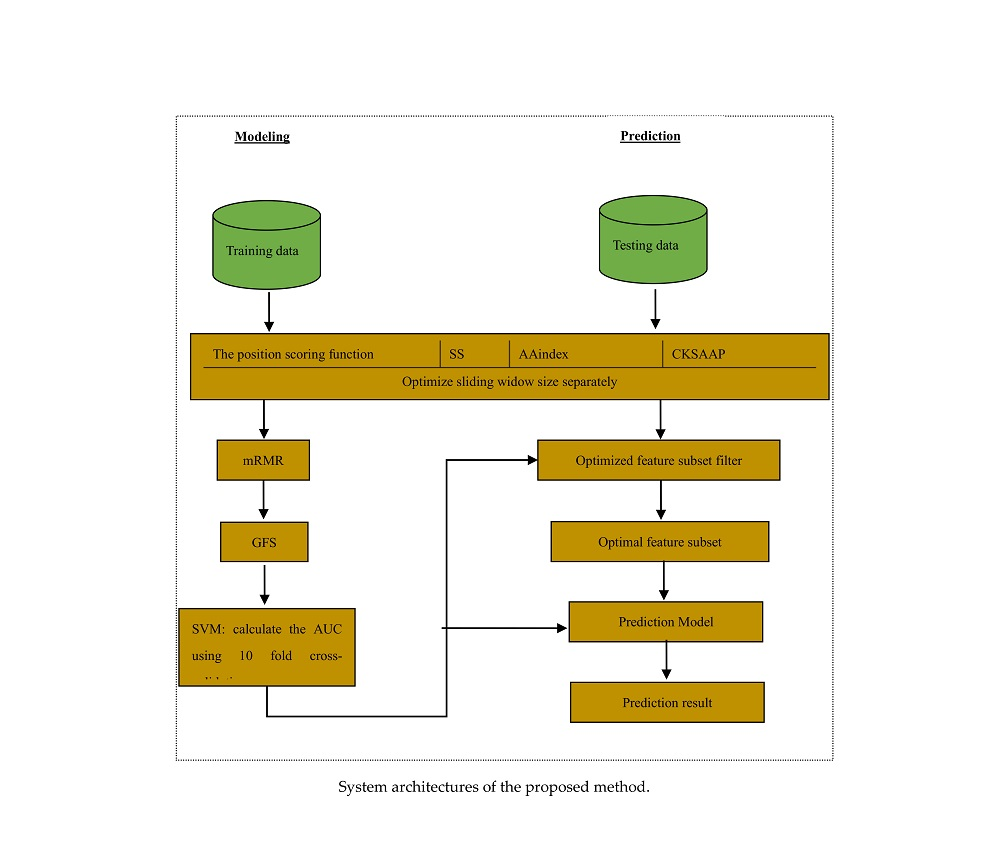
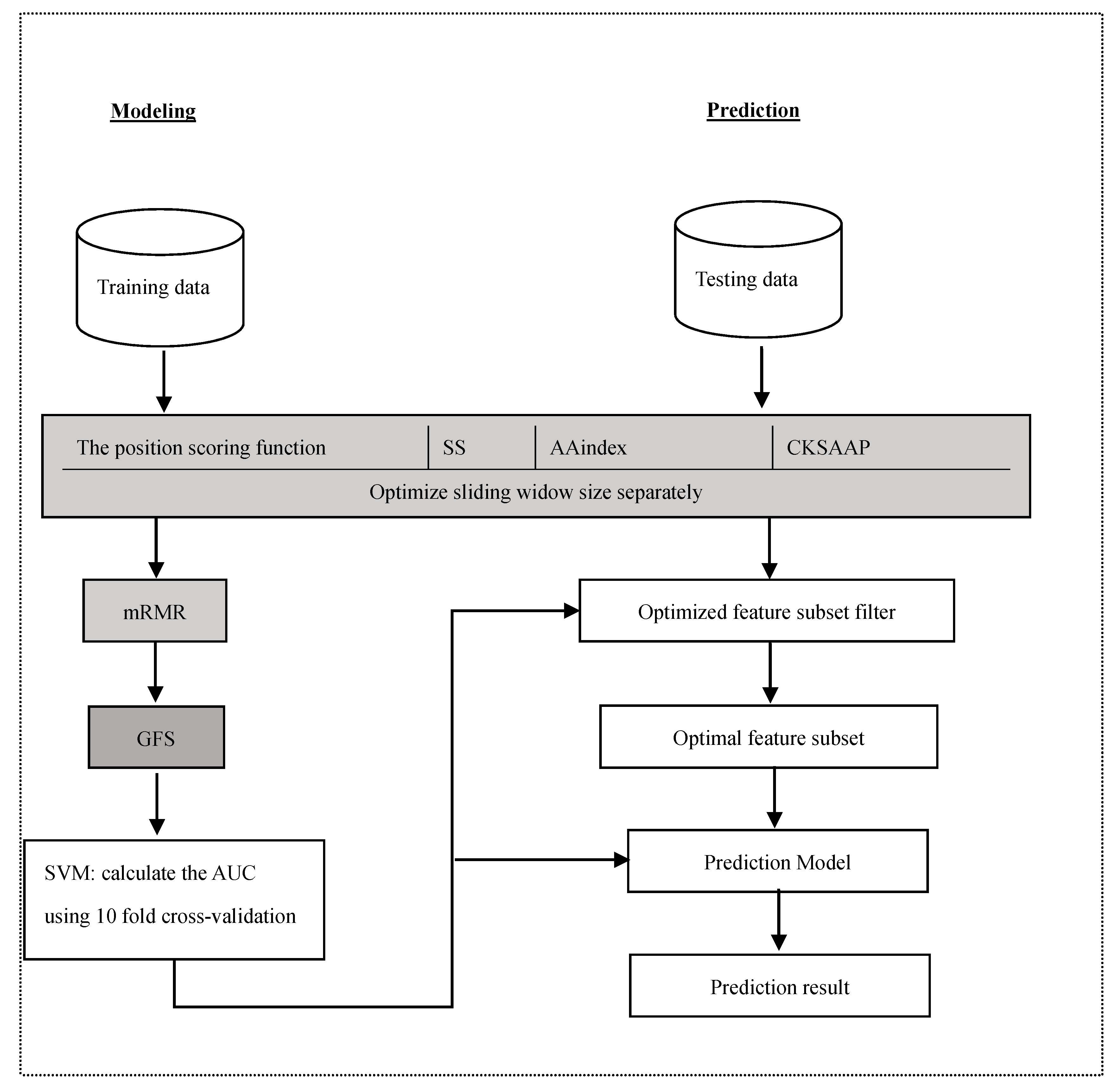
3. Materials and Methods
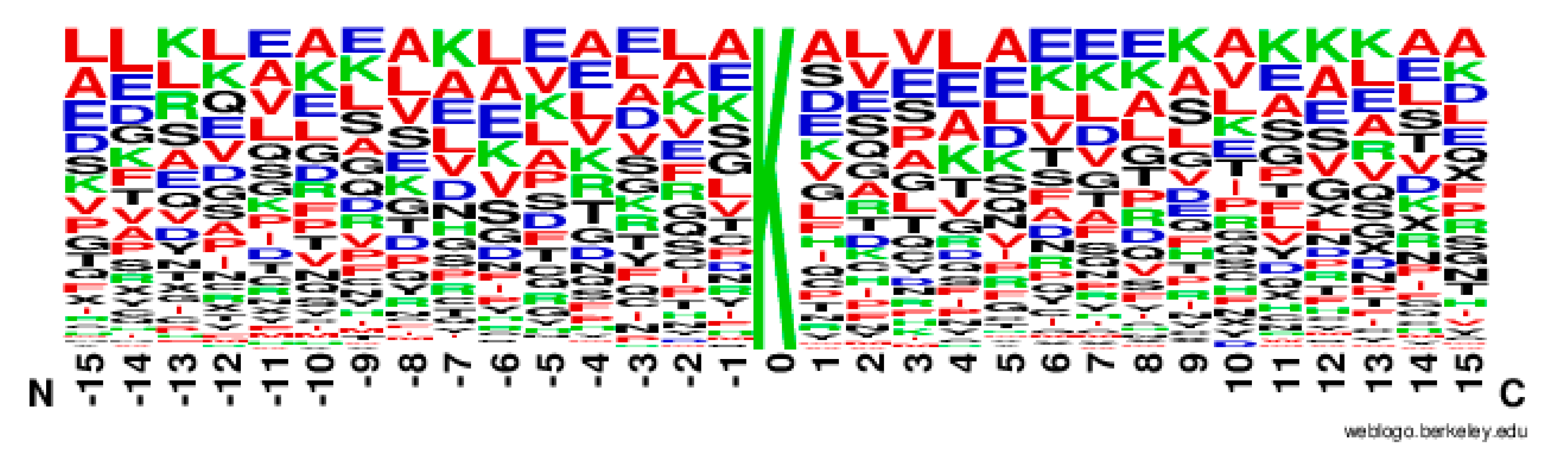
3.1. Datasets
3.2. Feature Construction
3.2.1. The Position Scoring Function
3.2.2. The Secondary Structure (SS)
3.2.3. Amino Acid Indices
3.2.4. The Composition of k-Spaced Amino Acid Pairs (CKSAAP)
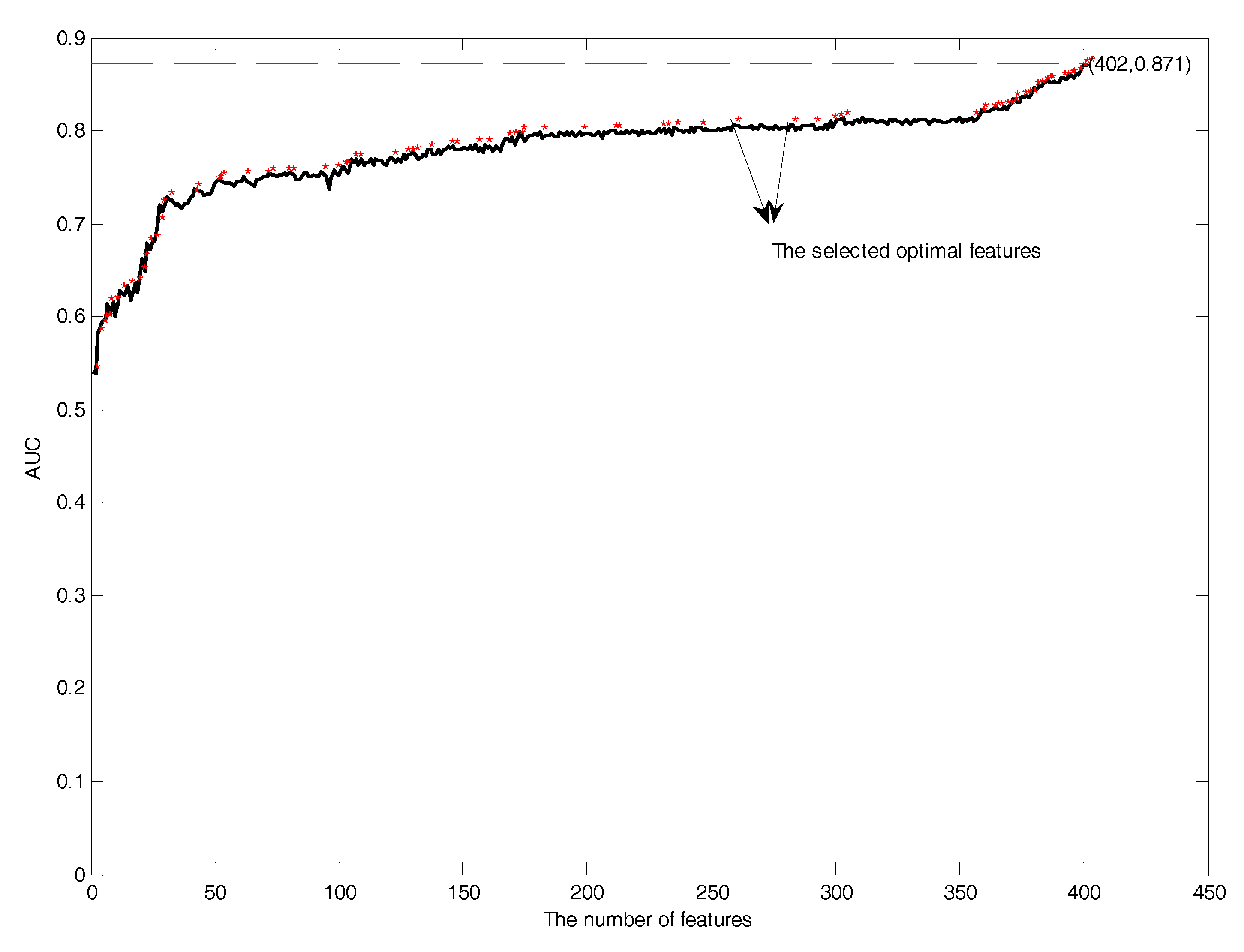
3.3. Feature Selection
3.4. Support Vector Machine
3.5. Performance Assessment
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nicolls, M.R. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Gerlach, M.; Sian, J.; Wong, A.; Riederer, P. Advanced glycation end products in neurodegeneration: More than early markers of oxidative stress? Ann. Neurol. 1998, 44, 85–88. [Google Scholar] [CrossRef]
- Lapolla, A.; Fedele, D.; Martano, L.; Arico, N.C.; Garbeglio, M.; Traldi, P.; Seraglia, R.; Favretto, D. Advanced glycation end products: A highly complex set of biologically relevant compounds detected by mass spectrometry. J. Mass Spectrom. 2001, 36, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Roman, G.; Yeboah, F.; Konishi, Y. The road to advanced glycation end products: A mechanistic perspective. Curr. Med. Chem. 2007, 14, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Guedes, S.; Rui, V.; Domingues, M.R.; Amado, F.; Domingues, P. Glycation and oxidation of histones H2B and H1: In vitro study and characterization by mass spectrometry. Anal. Bioanal. Chem. 2011, 399, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.R.; Chen, Y.Z.; Canchaya, C.A.; Zhang, Z. GANNPhos: A new phosphorylation site predictor based on a genetic algorithm integrated neural network. Protein Eng. Des. Sel. 2007, 20, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE. Trans. Nanobioscience 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; He, W.; Zou, Q. DephosSitePred: A High Accuracy Predictor for Protein Dephosphorylation Sites. Comb. Chem. High. Throughout. Screen. 2017, 20, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.W.; Lai, H.Y.; Tang, H.; Chen, W.; Lin, H. Prediction of phosphothreonine sites in human proteins by fusing different features. Sci. Rep. 2016, 6, 34817. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analysis and prediction of mammalian protein glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W.; Zhang, W.; Wang, J. Predict and Analyze Protein Glycation Sites with the mRMR and IFS Methods. Biomed. Res. Int. 2015, 2015, 561547. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, L.; Ding, J.; Wu, L.-Y.; Mai, G.; Zhou, F. Gly-PseAAC: Identifying protein lysine glycation through sequences. Gene 2016, 602, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W.; Watkins, N.G.; Fisher, C.I.; Hull, C.J.; Patrick, J.S.; Ahmed, M.U.; Dunn, J.A.; Thorpe, S.R. The Amadori product on protein: Structure and reactions. Prog. Clin. Biol. Res. 1989, 304, 43–67. [Google Scholar] [PubMed]
- Venkatraman, J.; Aggarwal, K.; Balaram, P. Helical peptide models for protein glycation: Proximity effects in catalysis of the Amadori rearrangement. Chem. Biol. 2001, 8, 611–625. [Google Scholar] [CrossRef]
- Zhao, X.; Ning, Q.; Ai, M.; Chai, H.; Yin, M. PGluS: Prediction of protein S-glutathionylation sites with multiple features and analysis. J. Theor. Boil. 2015, 380, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, X.; Yin, M.; Luo, N.; Zhang, J.; Wang, J. Prediction of methylation sites using the composition of K-spaced amino acid pairs. Protein Pept. Lett. 2013, 20, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Gao, T.; Pan, Z.; Cheng, H.; Yang, Q.; Cheng, Z.; Guo, A.; Ren, J.; Xue, Y. CPLM: A database of protein lysine modifications. Nucleic Acids Res. 2014, 42, D531–D536. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genomics. 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Shi, S.P.; Qiu, J.D.; Sun, X.-Y.; Suo, S.-B.; Liang, R.-P. PredSulSite: Prediction of protein tyrosine sulfation sites with multiple features and analysis. Anal. Biochem. 2012, 428, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mcguffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino Acid Index Database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, X.; Sun, P.; Ma, Z. PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC. Int. J. Mol. Sci. 2014, 15, 11204–11219. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Tang, Y.-R.; Sheng, Z.-Y.; Zhang, Z.D. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinform. 2008, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. A Comparative Study on Feature Selection Methods for Drug Discovery. J. Chem. Inf. Comput. Sci. 2004, 44, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, L.; Zhang, H. Ensemble gene selection by grouping for microarray data classification. J. Biomed. Inform. 2010, 43, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhang, B.; Kong, J.; Wang, J. An improved locality sensitive discriminant analysis approach for feature extraction. Multimed. Tools. Appl. 2015, 74, 85–104. [Google Scholar] [CrossRef]
- Yi, Y.; Shi, Y.; Zhang, H.; Kong, J. Label propagation based semi-supervised non-negative matrix factorization for feature extraction. Neurocomputing 2015, 149, 1021–1037. [Google Scholar] [CrossRef]
- Liu, H.; Liu, L.; Zhang, H. Boosting feature selection using information metric for classification. Neurocomputing 2009, 73, 295–303. [Google Scholar] [CrossRef]
- Shi, Y.; Yi, Y.; Yan, H. Region contrast and supervised locality-preserving projection-based saliency detection. Visual. Comput. 2015, 31, 1191–1205. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 389–396. [Google Scholar] [CrossRef]
- Zhao, X.; Ning, Q.; Ai, M.; Chai, H.; Yang, G. Identification of S-glutathionylation sites in species-specific proteins by incorporating five sequence-derived features into the general pseudo-amino acid composition. J. Theor. Biol. 2016, 398, 96–102. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | |||
|---|---|---|---|
| QIAN880101 | FUKS010109 | RACS820107 | QIAN880118 |
| FUKS010102 | CEDJ970104 | NAKH920106 | GEIM800107 |
| FUKS010101 | KUMS000103 | KARP850103 | PARS000102 |
| QIAN880102 | CHAM830102 | FUKS010104 | FUKS010110 |
| PALJ810108 | RACS820104 | QIAN880104 | BURA740102 |
| Cross-Validation | Sen (%) | Spe (%) | Acc (%) | AUC | MCC |
|---|---|---|---|---|---|
| 10-fold | 57.47 (1.31) | 90.78 (0.56) | 79.68 (0.57) | 0.8629 (0.0035) | 0.5232 (0.0140) |
| 8-fold | 57.10 (1.41) | 90.95 (0.65) | 79.67 (0.71) | 0.8629 (0.0050) | 0.5227 (0.0175) |
| 6-fold | 56.30 (1.74) | 91.06 (0.74) | 79.47 (0.88) | 0.8600 (0.0059) | 0.5175 (0.0218) |
| LOO | 57.62 | 90.24 | 79.37 | 0.8693 | 0.5162 |
| Predictor | Sen (%) | Spe (%) | Acc (%) | AUC | MCC |
|---|---|---|---|---|---|
| Glypre a | 85.11 | 93.06 | 89.77 | 0.9557 | 0.7884 |
| Glypre b | 80.96 | 91.55 | 87.16 | 94.20 | 0.7344 |
| Gly-PseAAC | 56.06 | 80.17 | 68.12 | 0.7705 | 0.38 |
| PreGly a | 71.06 | 95.85 | 85.51 | - | 0.70 |
| GlyNN b | 78.65 | 80.15 | 79.50 | 0.77 | 0.58 |
| Protein | Glycation | Glypre | Gly-PseAAC |
|---|---|---|---|
| P62760 | 7, 18 | 0.3278, 0.5980 | <0.35, 0.3835 |
| Q9Y5I3 | 677 | 0.8407 | 0.5878 |
| Q9Y6P5 | 55 | 0.2178 | <0.35 |
| A6NE02 | 302 | 0.0722 | <0.35 |
| Q9NPC3 | 119 | 0.7346 | 0.3831 |
| P29122 | 573 | 0.4627 | <0.35 |
| O96005 | 207, 209 | 0.3228, 0.3646 | 0.5519, 0.5515 |
| P47869 | 231, 247 | 0.7250, 0.2822 | <0.35, <0.35 |
| Q8TC59 | 770 | 0.2745 | <0.35 |
| Q8IUR6 | 216, 493 | 0.1153, 0.2307 | <0.35, <0.35 |
| Q9Y587 | 53 | 0.0524 | <0.35 |
| P28289 | 191, 214, 221, 228, 249, 255, 286, 297, 308, 314 | 0.4737, 0.1668, 0.3140, 0.4933, 0.1197, 0.4824, 0.1432, 0.1932, 0.0227, 0.2218 | <0.35, <0.35, 0.7305, 0.4924, <0.35, <0.35, <0.35, <0.35, 0.3583, <0.35 |
| O94919 | 252, 281, 300 | 0.0252, 0.4890, 0.6587 | <0.35, <0.35, 0.3520 |
| P01877 | 155 | 0.1260 | <0.35 |
| Q93034 | 137 | 0.2791 | <0.35 |
| Q13011 | 267, 276 | 0.6926, 0.8448 | <0.35, 0.4122 |
| Q6P6C2 | 274 | 0.0244 | <0.35 |
| Q8IZI9 | 70 | 0.5000 | <0.35 |
| Q15084 | 73, 245 | 0.6981, 0.0404 | <0.35, <0.35 |
| Q8IY21 | 1077 | 0.5758 | <0.35 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zhao, X.; Bao, L.; Zhang, Y.; Dai, J.; Yin, M. Glypre: In Silico Prediction of Protein Glycation Sites by Fusing Multiple Features and Support Vector Machine. Molecules 2017, 22, 1891. https://doi.org/10.3390/molecules22111891
Zhao X, Zhao X, Bao L, Zhang Y, Dai J, Yin M. Glypre: In Silico Prediction of Protein Glycation Sites by Fusing Multiple Features and Support Vector Machine. Molecules. 2017; 22(11):1891. https://doi.org/10.3390/molecules22111891
Chicago/Turabian StyleZhao, Xiaowei, Xiaosa Zhao, Lingling Bao, Yonggang Zhang, Jiangyan Dai, and Minghao Yin. 2017. "Glypre: In Silico Prediction of Protein Glycation Sites by Fusing Multiple Features and Support Vector Machine" Molecules 22, no. 11: 1891. https://doi.org/10.3390/molecules22111891