The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells

 , and
, and
Abstract
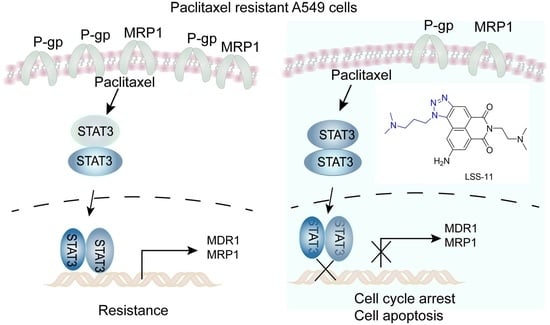
:
1. Introduction
2. Results
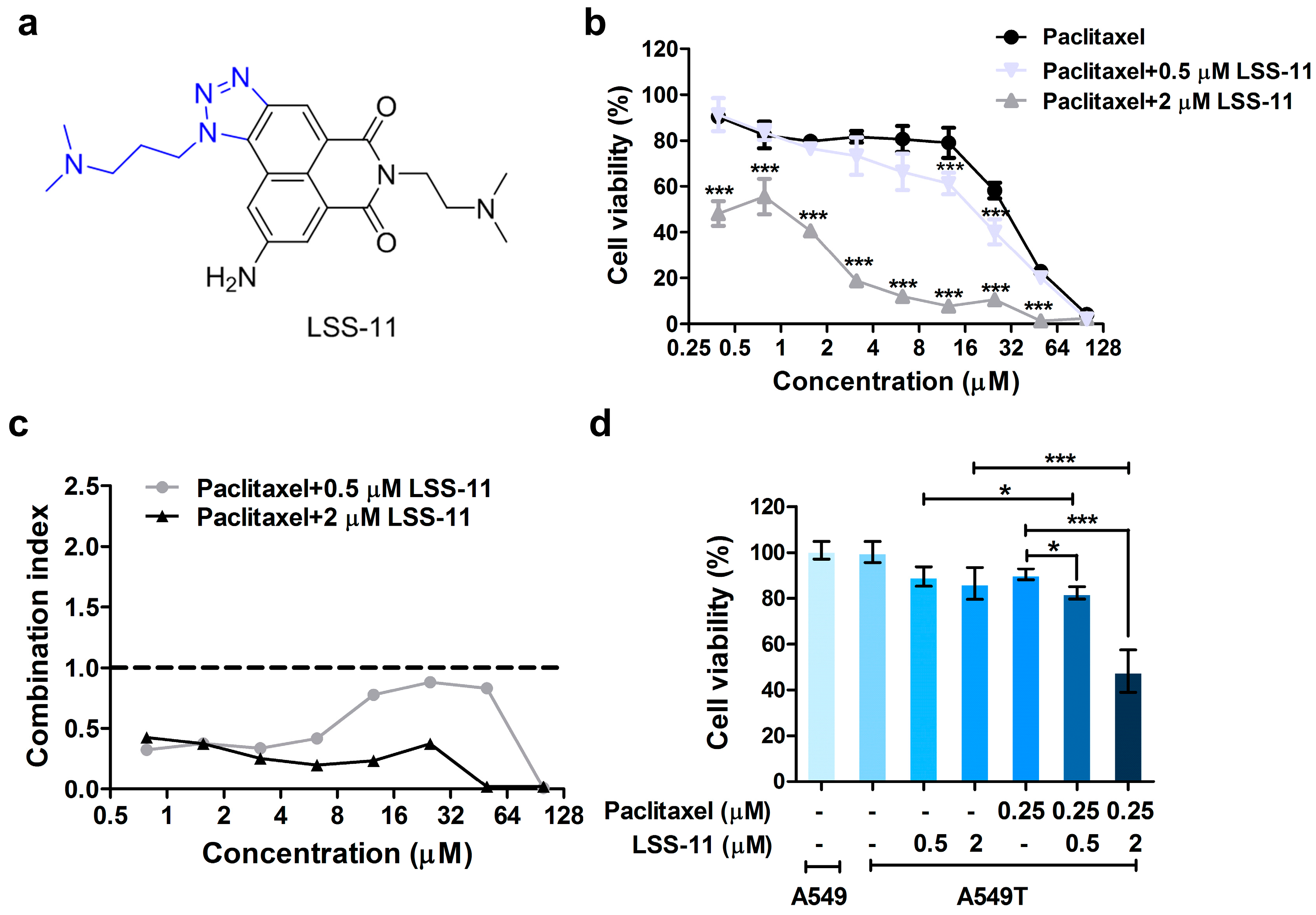
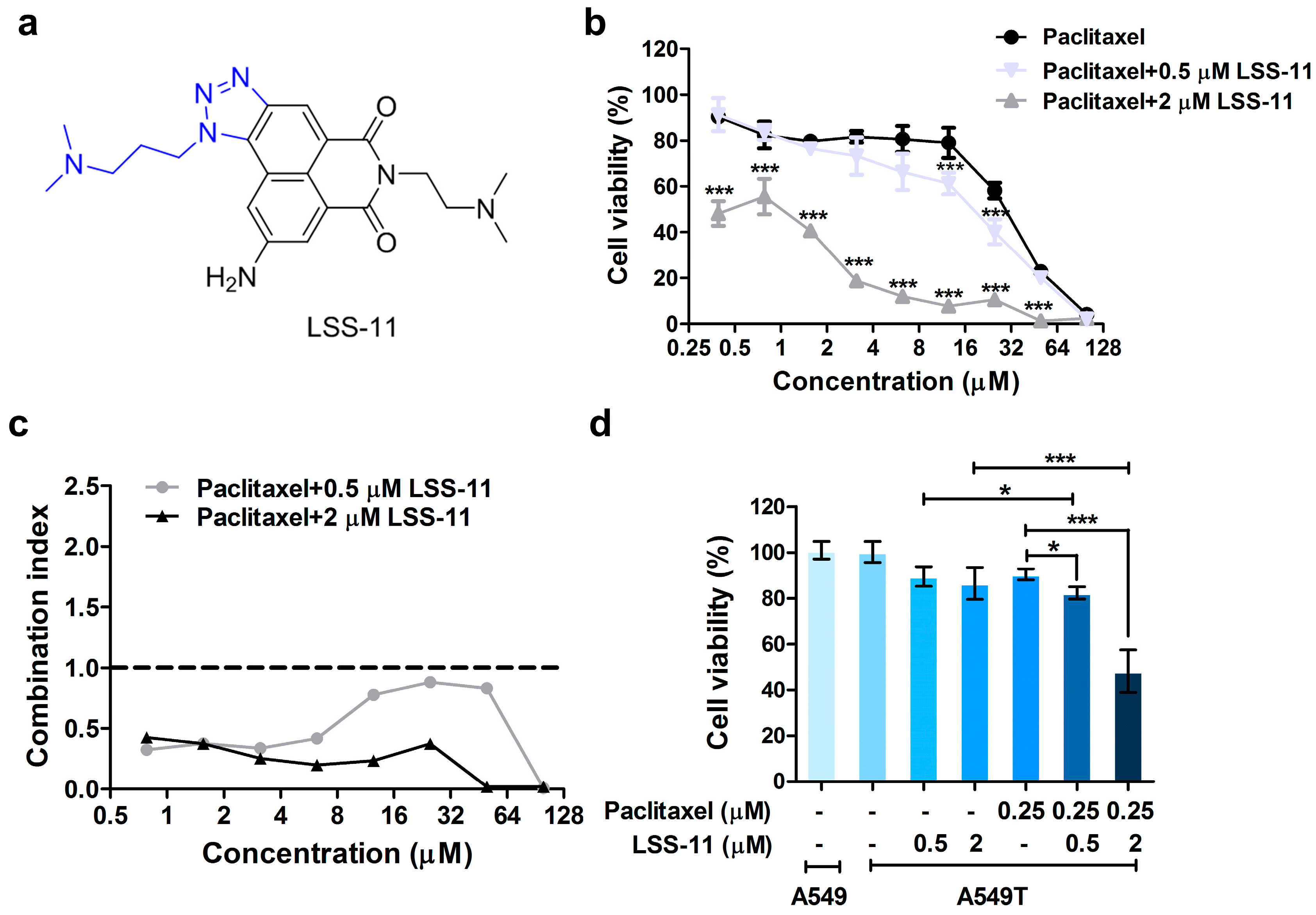
2.1. LSS-11 Reverses Paclitaxel Resistance in Lung Cancer Cells
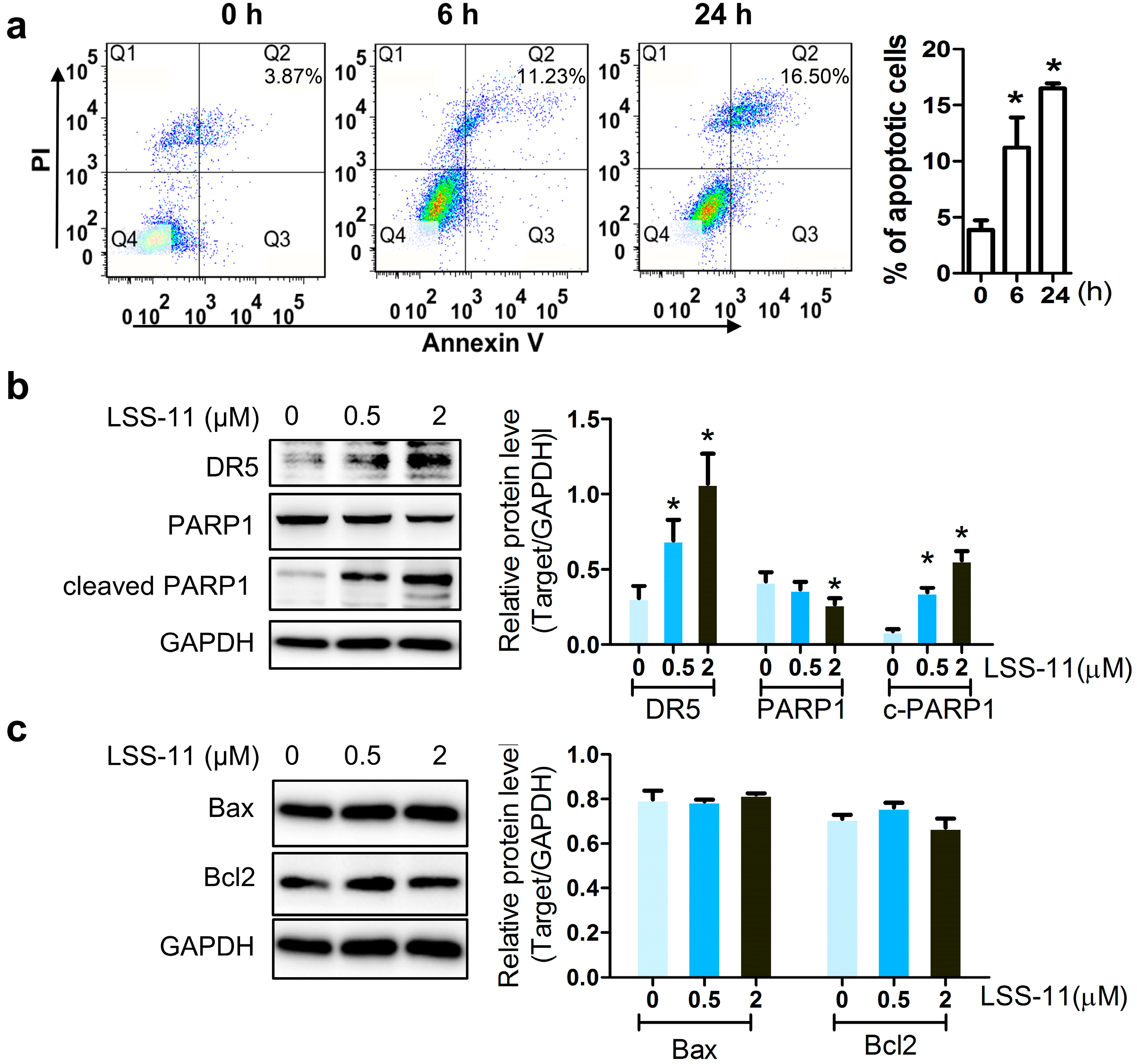
2.2. LSS-11 Augments the Pro-Apoptotic Effect in Paclitaxel-Resistant Lung Cancer Cells
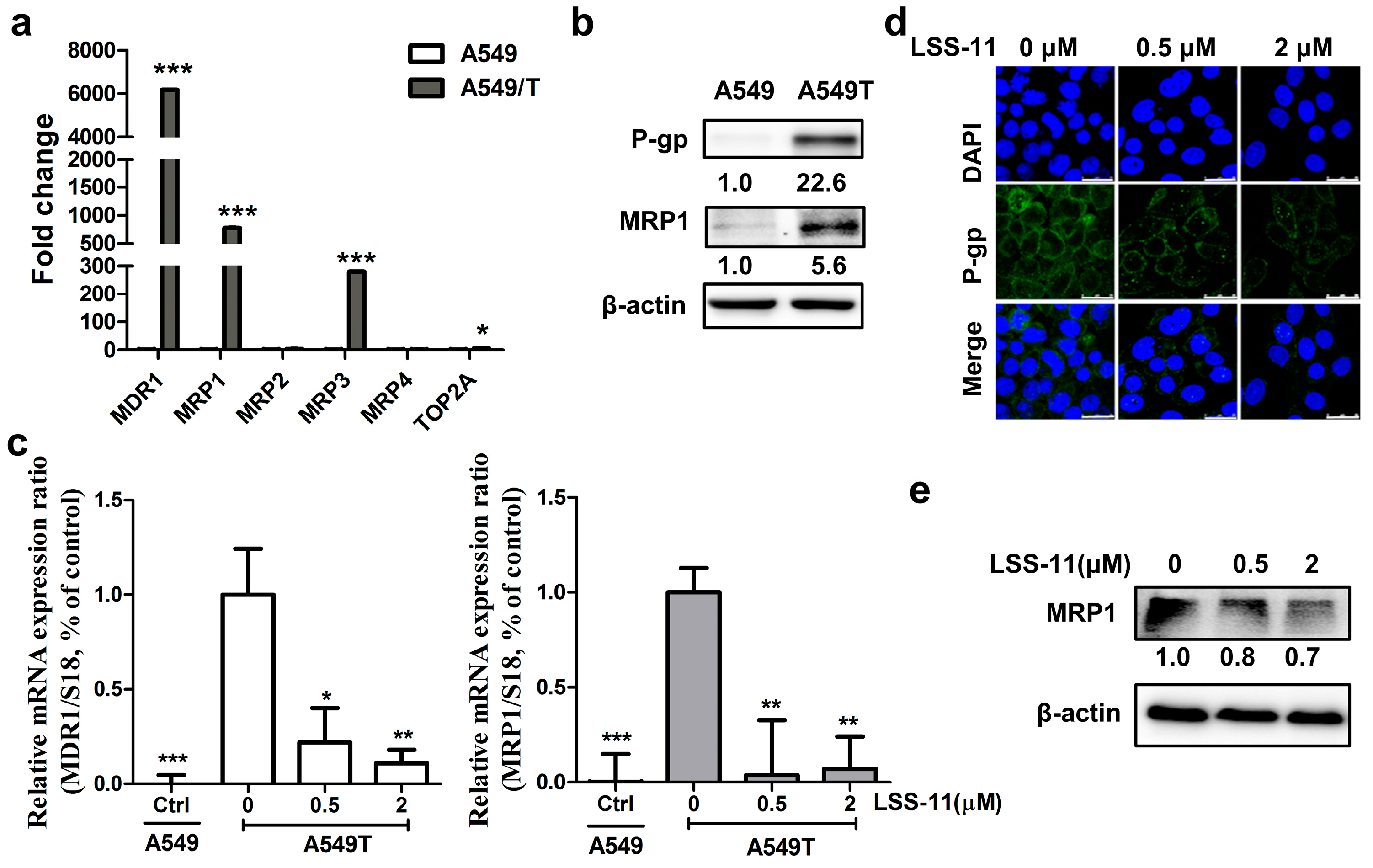
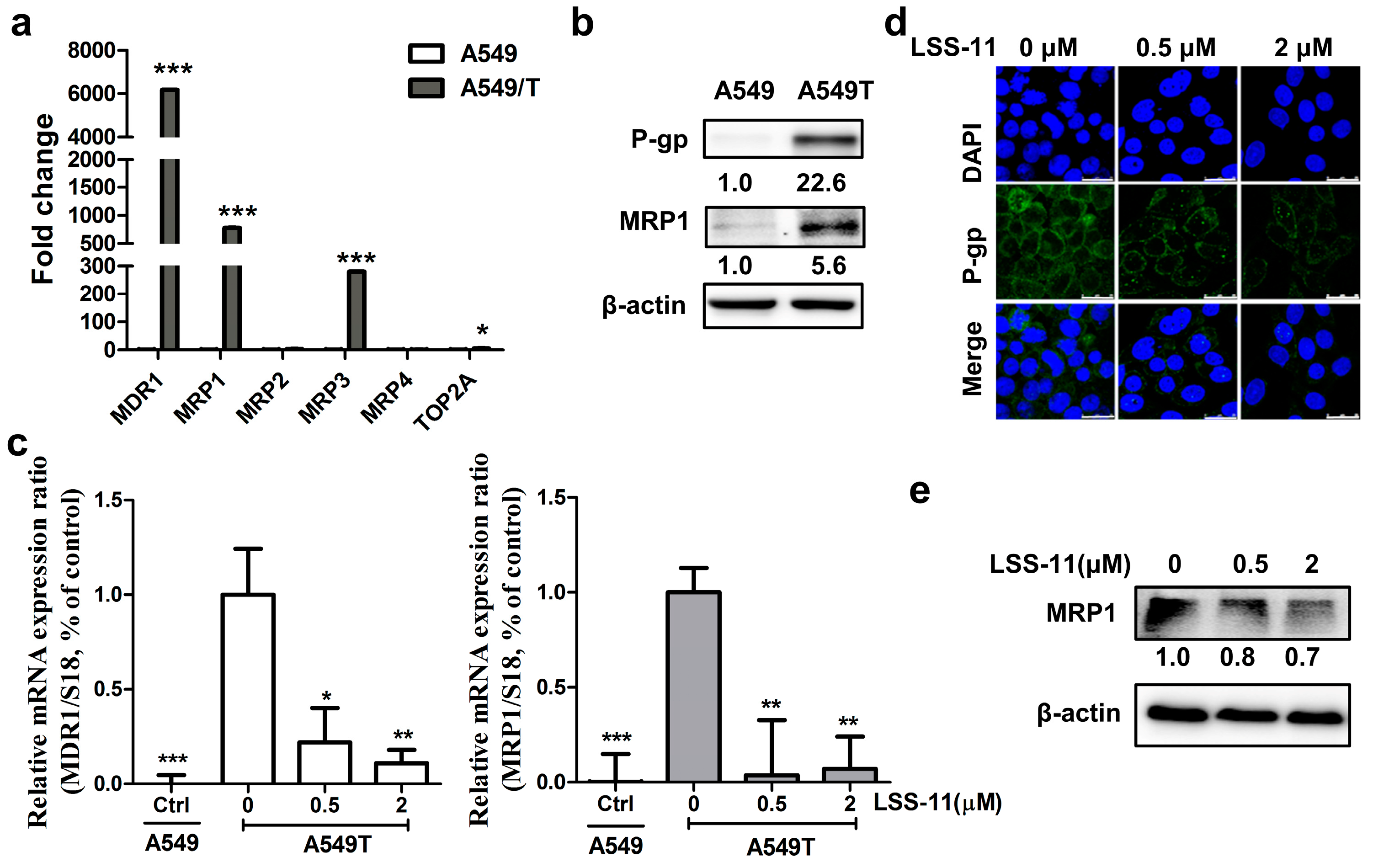
2.3. LSS-11 Has No Effect on P-gp Efflux Function in Drug-Insensitive A549/T Cells
2.4. LSS-11 Suppresses mRNA Levels of Drug Resistance Genes in A549/T Cells
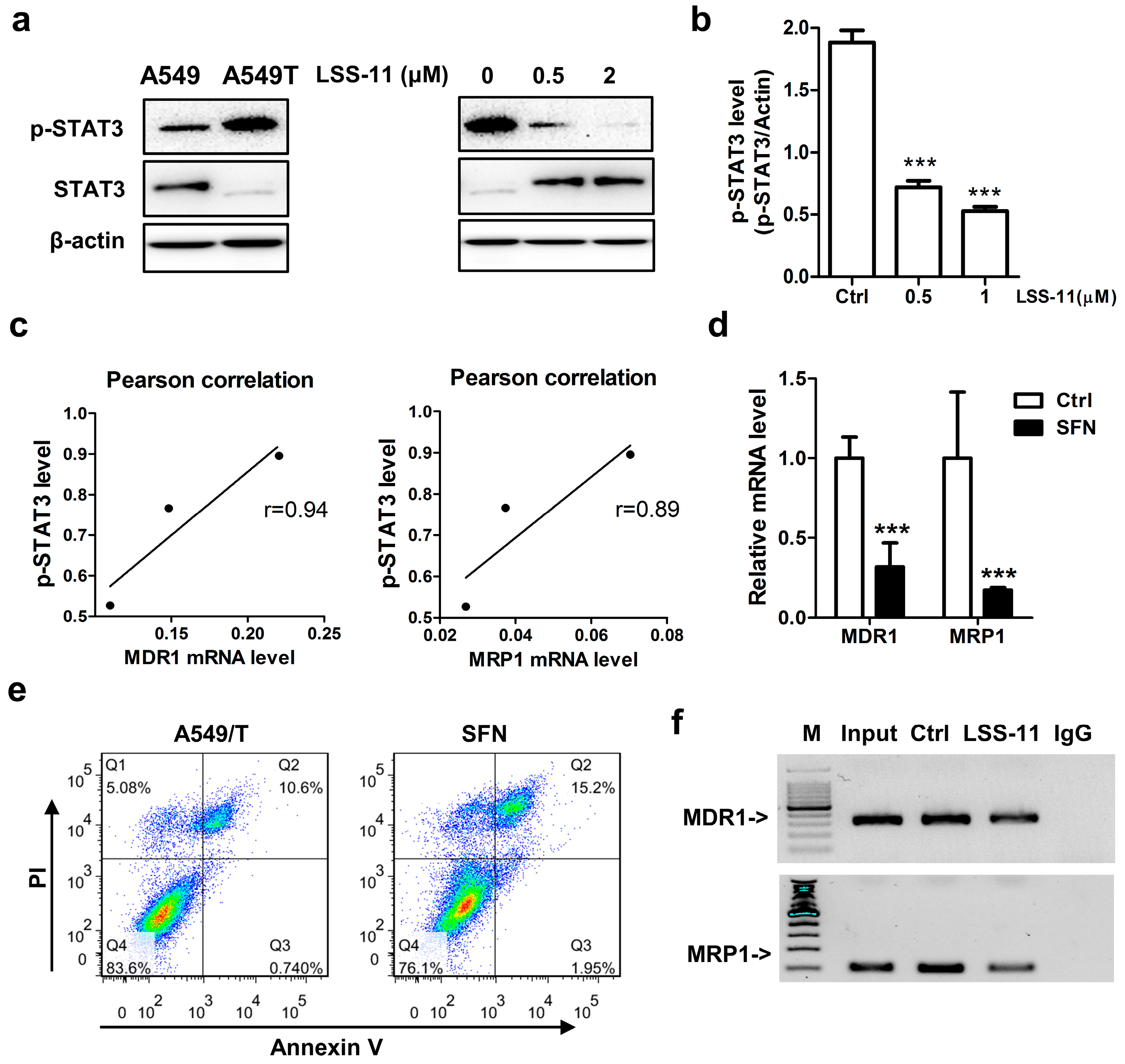
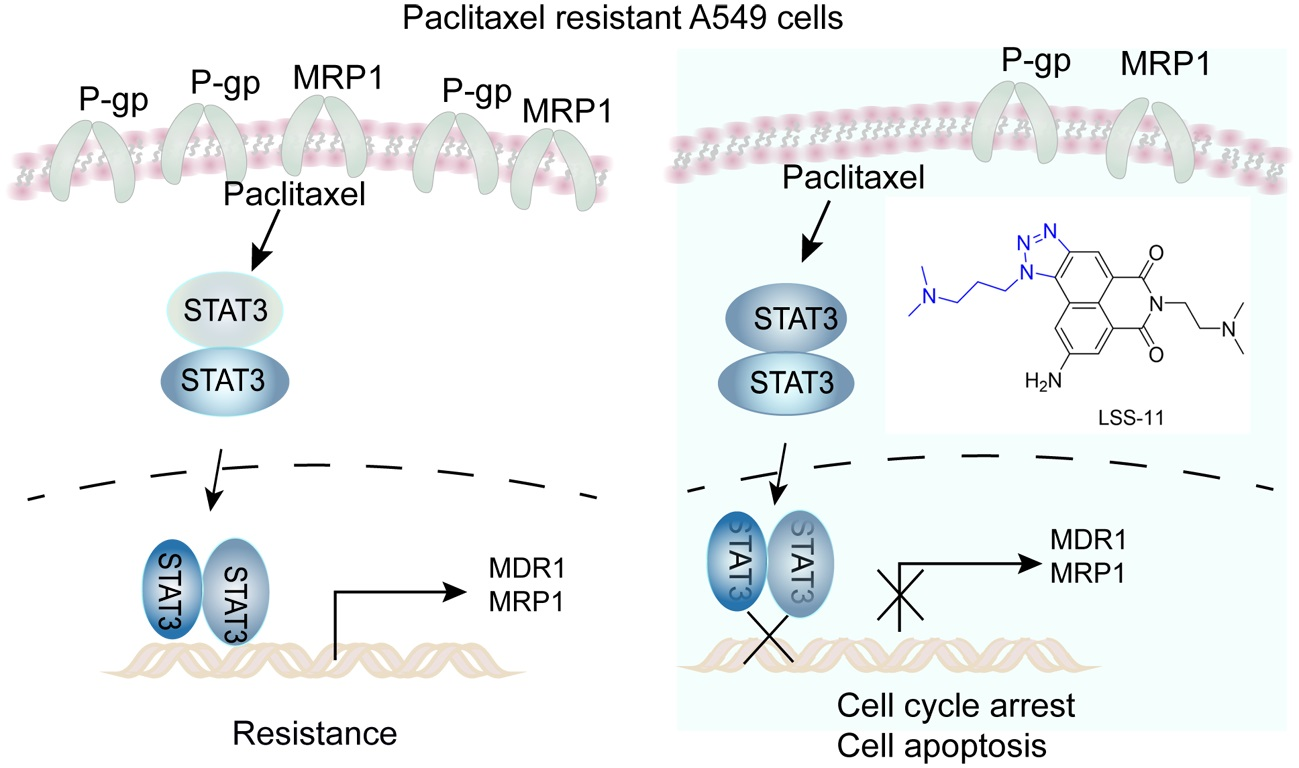
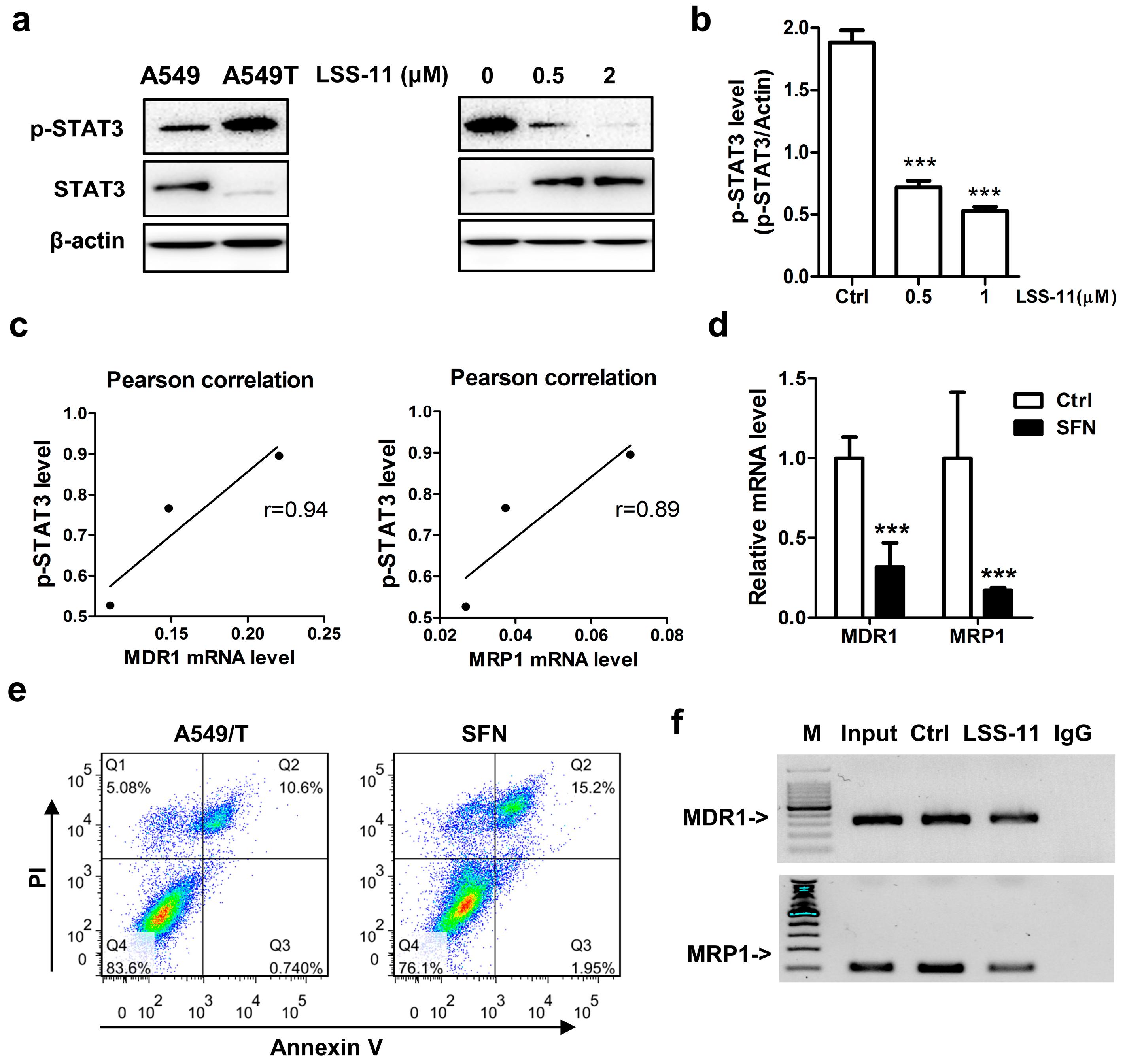
2.5. LSS-11 Downregulates MDR1 and MRP1 via Repression of STAT3 in A549/T Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. Drug Combination
4.5. Real Time PCR
4.6. Flow Cytometry
4.7. Immunofluorescence
4.8. Immunoblotting Assay
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA A Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Gong, W.; Sun, P.; Mu, Z.; Liu, J.; Yu, C.; Liu, A. Efficacy and Safety of Nab-Paclitaxel as Second-line Chemotherapy for Locally Advanced and Metastatic Non-small Cell Lung Cancer. Anticancer Res. 2017, 37, 4687–4691. [Google Scholar] [PubMed]
- d’Amato, T.A.; Landreneau, R.J.; McKenna, R.J.; Santos, R.S.; Parker, R.J. Prevalence of in vitro extreme chemotherapy resistance in resected nonsmall-cell lung cancer. Ann. Thorac. Surg. 2006, 81, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunaga, N.; Imai, H.; Kamide, Y.; Koga, Y.; Ono, A.; Kuwako, T.; Masuda, T.; Hisada, T.; Ishizuka, T.; et al. Phase I dose escalation study of amrubicin plus paclitaxel in previously treated advanced non-small cell lung cancer. Int. J. Clin. Oncol. 2016, 21, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Shanker, M.; Willcutts, D.; Roth, J.A.; Ramesh, R. Drug resistance in lung cancer. Lung Cancer (Auckl.) 2010, 1, 23–36. [Google Scholar] [PubMed]
- Yang, X.; Shen, J.; Gao, Y.; Feng, Y.; Guan, Y.; Zhang, Z.; Mankin, H.; Hornicek, F.J.; Duan, Z. Nsc23925 prevents the development of paclitaxel resistance by inhibiting the introduction of P-glycoprotein and enhancing apoptosis. Int. J. Cancer 2015, 137, 2029–2039. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Wang, L.L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–672. [Google Scholar] [CrossRef] [PubMed]
- Scotto, K.W. Transcriptional regulation of ABC drug transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Xiao, W.H.; Wang, L.H.; Tian, Z.J.; Zhang, J. Deactivation of Signal Transducer and Activator of Transcription 3 Reverses Chemotherapeutics Resistance of Leukemia Cells via Down-Regulating P-gp. PLoS ONE 2011, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liu, Z.; Tang, L.; Liu, J.; Zhou, M.; Xie, F.; Wang, Z.; Wang, Y.; Shen, S.; Hu, L.; et al. Reversal of P-gp and MRP1-mediated multidrug resistance by H6, a gypenoside aglycon from Gynostemma pentaphyllum, in vincristine-resistant human oral cancer (KB/VCR) cells. Eur. J. Pharm. 2012, 696, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.; Cheng, F.Y.; Shieh, D.B.; Yeh, C.S.; Su, W.C. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int. J. Nanomed. 2012, 7, 4269–4283. [Google Scholar] [CrossRef] [PubMed]
- Tsyganov, M.M.; Rodionov, E.O.; Miller, S.V.; Litvyakov, N.V. Substantiation of Expressive Markers Use to Personalize Lung Cancer Chemotherapy. Antibiot. Khimioter. 2015, 60, 38–45. [Google Scholar] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, A.C.; Van Ark-Otte, J.; Span, S.; Scagliotti, G.V.; Van der Valk, P.; Postmus, P.E.; Giaccone, G. Topoisomerase IIalpha and other drug resistance markers in advanced non-small cell lung cancer. Lung Cancer 2001, 32, 117–128. [Google Scholar] [CrossRef]
- Hariri, G.; Edwards, A.D.; Merrill, T.B.; Greenbaum, J.M.; Van der Ende, A.E.; Harth, E. Sequential targeted delivery of paclitaxel and camptothecin using a cross-linked “nanosponge” network for lung cancer chemotherapy. Mol. Pharm. 2014, 11, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharm. Exp. Ther. 2001, 298, 1236–1242. [Google Scholar]
- Mamidipudi, V.; Shi, T.; Brady, H.; Surapaneni, S.; Chopra, R.; Aukerman, S.L.; Heise, C.; Sung, V. Increased cellular accumulation and distribution of amrubicin contribute to its activity in anthracycline-resistant cancer cells. Cancer Chemother. Pharm. 2012, 69, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Xu, H. Overview of naphthalimide analogs as anticancer agents. Curr. Med. Chem. 2009, 16, 4797–4813. [Google Scholar] [CrossRef] [PubMed]
- Sargent, J.M.; Williamson, C.J.; Yardley, C.; Taylor, C.G.; Hellmann, K. Dexrazoxane significantly impairs the induction of doxorubicin resistance in the human leukaemia line, K562. Br. J. Cancer 2001, 84, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Cheshire, P.J.; Hallman, J.C., 3rd; Gross, J.L.; McRipley, R.J.; Sun, J.H.; Behrens, C.H.; Dexter, D.L.; Houghton, J.A. Evaluation of a novel bis-naphthalimide anticancer agent, DMP 840, against human xenografts derived from adult, juvenile, and pediatric cancers. Cancer Chemother. Pharm. 1994, 33, 265–272. [Google Scholar] [CrossRef]
- Burcu, M.; O’Loughlin, K.L.; Ford, L.A.; Baer, M.R. Amonafide L-malate is not a substrate for multidrug resistance proteins in secondary acute myeloid leukemia. Leukemia 2008, 22, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, Z.; Li, S.; Zhong, W. 9-Substituted Triazole Para-Naphthalimide Derivative, Preparation Method Thereof and Application. Patent CN2012143814, 23 February 2012. [Google Scholar]
- Ji, L.; Yang, S.; Li, S.; Liu, S.; Tang, S.; Liu, Z.; Meng, X.; Yu, S. A novel triazolonaphthalimide induces apoptosis and inhibits tumor growth by targeting DNA and DNA-associated processes. Oncotarget 2017, 8, 37394–37408. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, N.; Sakata, K.; Yamasaki, T.; Terashima, H.; Mio, T.; Miyazaki, Y.; Fujii, T.; Kitada, K. Gene amplification and expression in lung cancer cells with acquired paclitaxel resistance. Cancer Genet. Cytogenet. 2007, 173, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Guo, X.; Bian, H.; Zhou, Y.; Li, T.; Yang, J. Changed expression and function of P-gp in peripheral blood CD56 + cells predicting chemoresistance in non-Hodgkin lymphoma patients. Cancer Biomark. Sect. A Dis. Mark. 2015, 15, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Singh, S.V. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer Prev. Res. 2010, 3, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Locke, V.L.; Davey, R.A.; Davey, M.W. Modulation of drug and radiation resistance in small cell lung cancer cells by paclitaxel. Anti-Cancer Drugs 2003, 14, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Nemcova-Furstova, V.; Kopperova, D.; Balusikova, K.; Ehrlichova, M.; Brynychova, V.; Vaclavikova, R.; Daniel, P.; Soucek, P.; Kovar, J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol. Appl. Pharm. 2016, 310, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wei, X.; Lu, Y. Chaetominine reduces MRP1-mediated drug resistance via inhibiting PI3K/Akt/Nrf2 signaling pathway in K562/Adr human leukemia cells. Biochem. Biophys. Res. Commun. 2016, 473, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Lacreusette, A.; Barbieux, I.; Nguyen, J.M.; Pandolfino, M.C.; Dreno, B.; Jacques, Y.; Godard, A.; Blanchard, F. Defective activations of STAT3 Ser727 and PKC isoforms lead to oncostatin M resistance in metastatic melanoma cells. J. Pathol. 2009, 217, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.; Ahn, K.S.; Jeong, S.J.; Lee, H.J.; Kim, M.; Lee, H.J.; Lee, E.O.; Kim, S.H. Reactive oxygen species involved in sulforaphane-induced STAT3 inactivation and apoptosis in DU145 prostate cancer cells. Chin. Sci. Bull 2010, 55, 3922–3928. [Google Scholar] [CrossRef]
- Duan, Z.; Foster, R.; Bell, D.A.; Mahoney, J.; Wolak, K.; Vaidya, A.; Hampel, C.; Lee, H.; Seiden, M.V. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5055–5063. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.Q.; Zhang, Y.H.; Li, Q.; Xu, F.H.; Miao, J.W.; Zhao, J.; Wang, C.J. 3-Nitro-naphthalimide and nitrogen mustard conjugate NNM-25 induces hepatocellular carcinoma apoptosis via PARP-1/p53 pathway. Apoptosis Int. J. Program. Cell Death 2012, 17, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Li, Y.; Wang, Z.; Xiao, M.; Yin, P.; Lu, Y.; Qian, X.; Xu, Y.; Liu, J. 7b, a novel naphthalimide derivative, exhibited anti-inflammatory effects via targeted-inhibiting TAK1 following down-regulation of ERK1/2- and p38 MAPK-mediated activation of NF-kappaB in LPS-stimulated RAW264.7 macrophages. Int. Immunopharmacol. 2013, 17, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Stefanick, D.F.; Horton, J.K.; Kedar, P.S.; Wilson, S.H. PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair 2009, 8, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Giannakakou, P. BRCA1 regulates microtubule dynamics and taxane-induced apoptotic cell signaling. Oncogene 2014, 33, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Wu, C.H.; Choi, M.R.; Najafi, F.; Emami, A.; Safa, A.R. P-glycoprotein enhances TRAIL-triggered apoptosis in multidrug resistant cancer cells by interacting with the death receptor DR5. Biochem. Pharmacol. 2006, 72, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Sun, Y.; Zhou, T.; McDonald, J.; Chen, Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4750–4759. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound LSS-11 are available from Dr. Meng. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | A549 (μM) | A549/T (μM) | Resistance Index |
|---|---|---|---|
| Paclitaxel | 1.9 ± 0.36 | 36.32 ± 8.29 | 19 |
| LSS-11 | >10 | 6.87 ± 0.77 | ~0.6 |
| Gene Symbol | Accession Number 1 | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|
| TOP2A | NM_001067.3 | 5′-GACGCTTCGTTATGGGAAGATA-3′ | 5′- GGGCCAGTTGTGATGGATAA -3′ |
| MDR1 | NM_001348946.1 | 5′-CAGCTATTCGAAGAGTGGGC-3′ | 5′-CCTGACTCACCACACCAATG-3′ |
| MRP1 | NM_004996.3 | 5′-ACCAAGACGTATCAGGTGGC-3′ | 5′-CTGTCAGGTTCCAGCTCCTC-3′ |
| MRP2 | NM_000392.4 | 5′-GCAGCGATTTCTGAAACACA-3′ | 5′-CAACAGCCACAATGTTGGTC-3′ |
| MRP3 | NM_003786.3 | 5′-CGCACACCGGCTTAACACTATCATGG-3′ | 5′-AAACCAGGAAAGGCCAGGAGGAAATC-3′ |
| MRP4 | NM_001301829.1 | 5′-GAGTTGCAAGGGTTCTGGGA-3′ | 5′-AAAGTCAGCACCGTGGCATA-3′ |
| RPS18 | NM_022551.2 | 5′-GATATGCTCATGTGGTGTTG-3′ | 5′-AATCTTCTTCAGTCGCTCCA-3′ |
| MDR1-promoter | - | 5′-GCAAGCTTCTAGAGAGGTGCAAC-3′ | 5′-AAAAGCTTGCGGCCTCTG-3′ |
| MRP1-promoter | - | 5′-TCTGTGTGACTCAGCTTTGG-3′ | 5′-GTGCAGAGAGGTTGAGTGATT-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, L.; Liu, X.; Zhang, S.; Tang, S.; Yang, S.; Li, S.; Qi, X.; Yu, S.; Lu, L.; Meng, X.; et al. The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells. Molecules 2017, 22, 1822. https://doi.org/10.3390/molecules22111822
Ji L, Liu X, Zhang S, Tang S, Yang S, Li S, Qi X, Yu S, Lu L, Meng X, et al. The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells. Molecules. 2017; 22(11):1822. https://doi.org/10.3390/molecules22111822
Chicago/Turabian StyleJi, Liyan, Xi Liu, Shuwei Zhang, Shunan Tang, Simin Yang, Shasha Li, Xiaoxiao Qi, Siwang Yu, Linlin Lu, Xiangbao Meng, and et al. 2017. "The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells" Molecules 22, no. 11: 1822. https://doi.org/10.3390/molecules22111822