Glucopyranosylidene-Spiro-Thiazolinones: Synthetic Studies and Determination of Absolute Configuration by TDDFT-ECD Calculations



Abstract
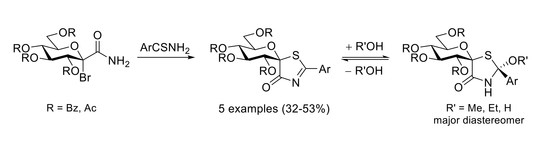
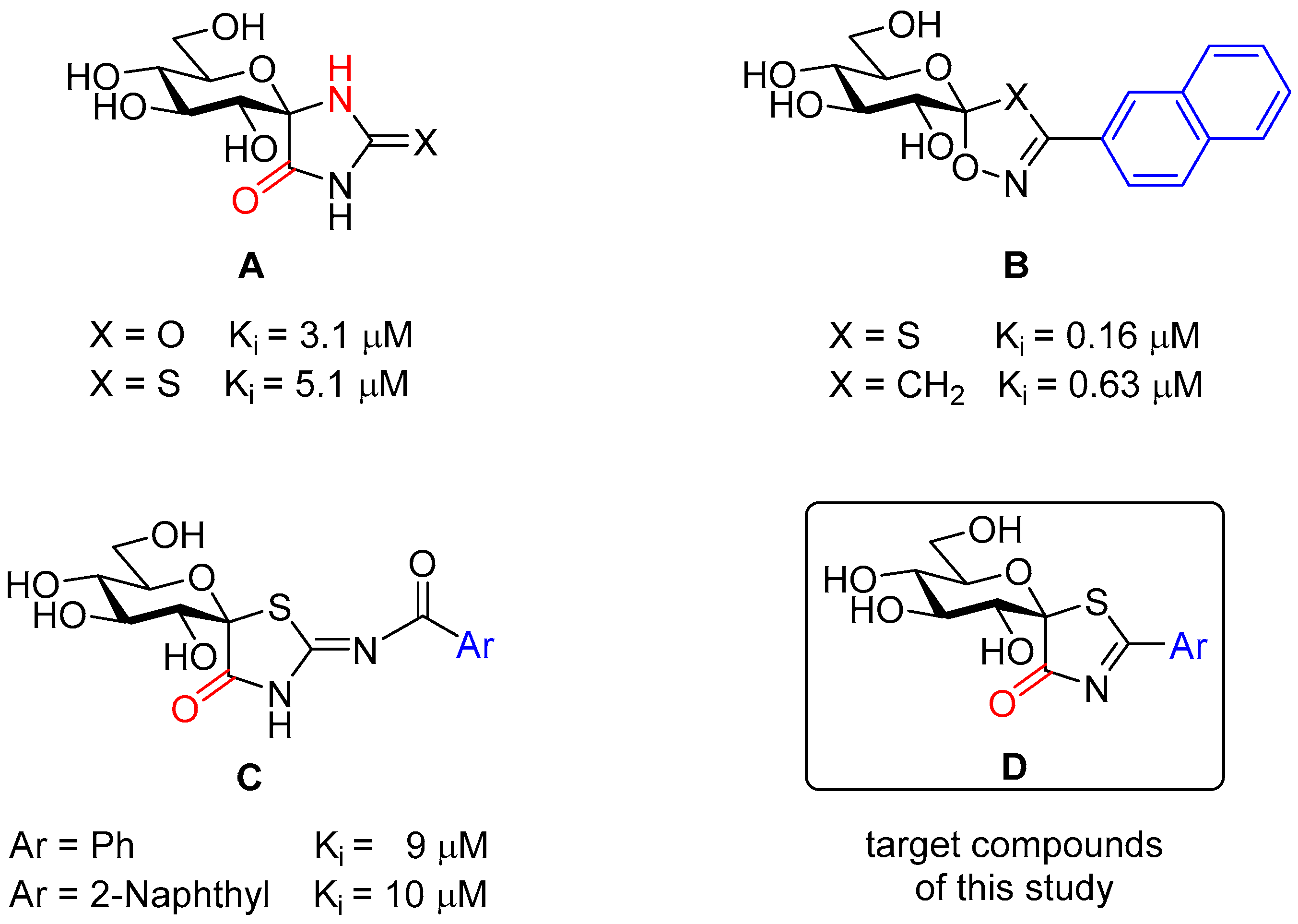
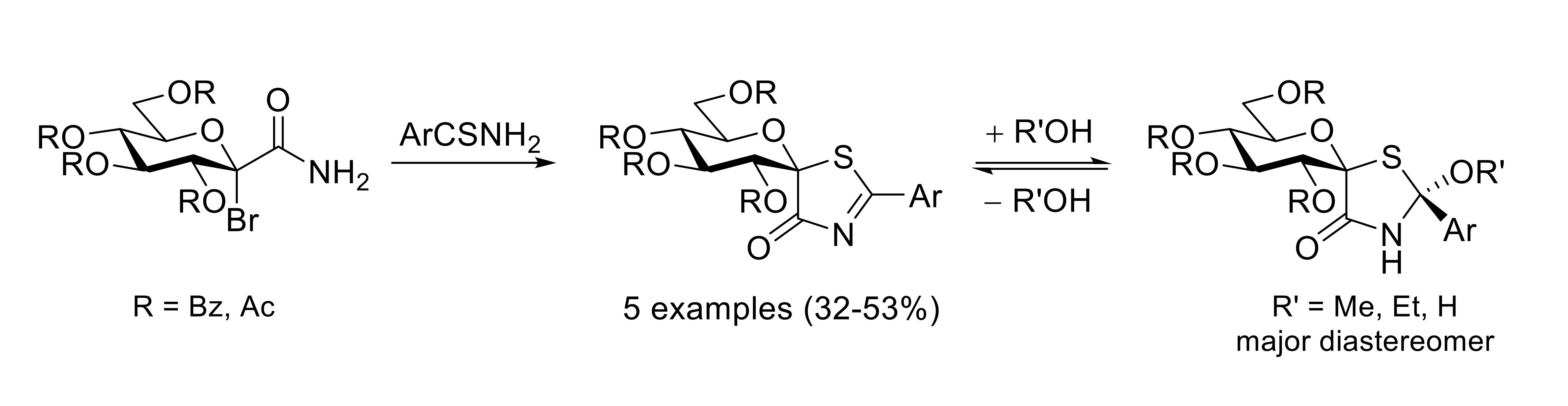
:
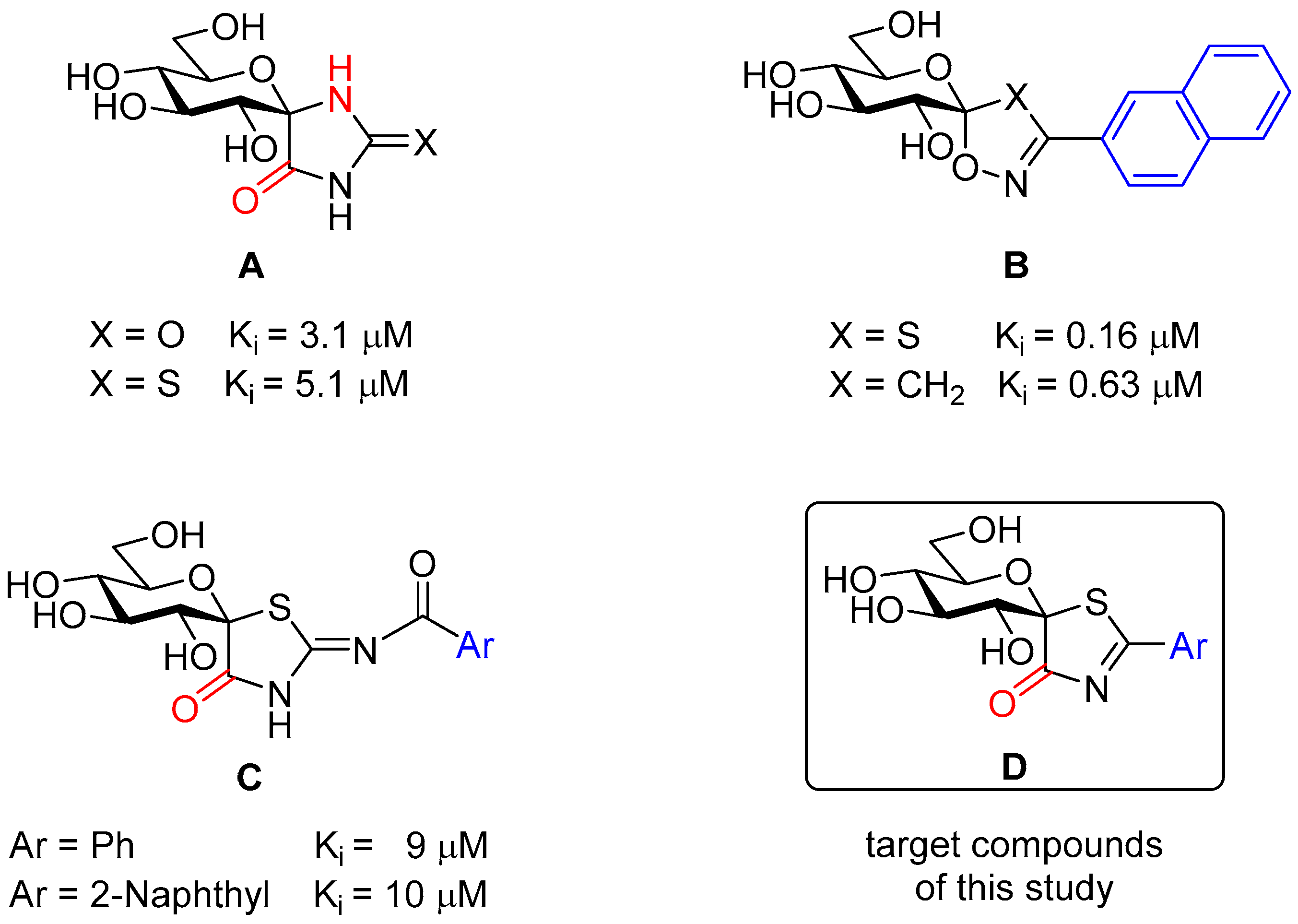
1. Introduction
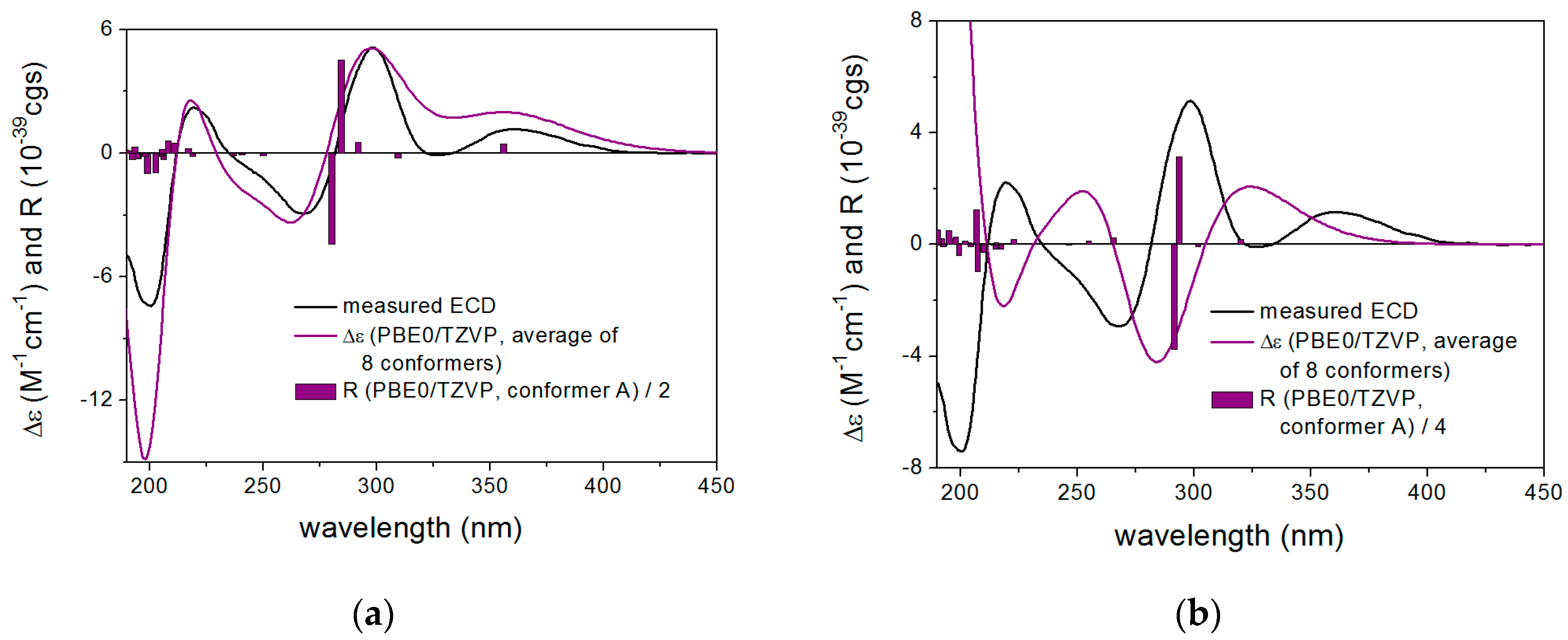
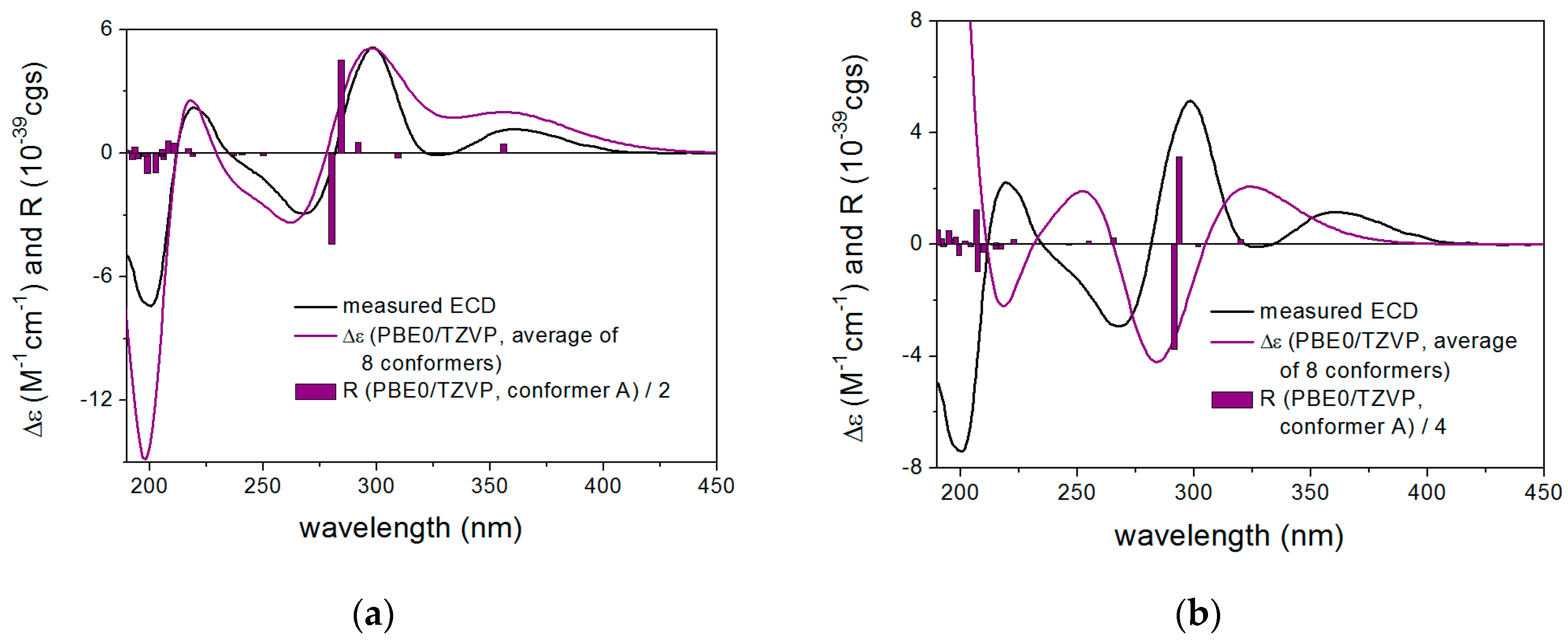
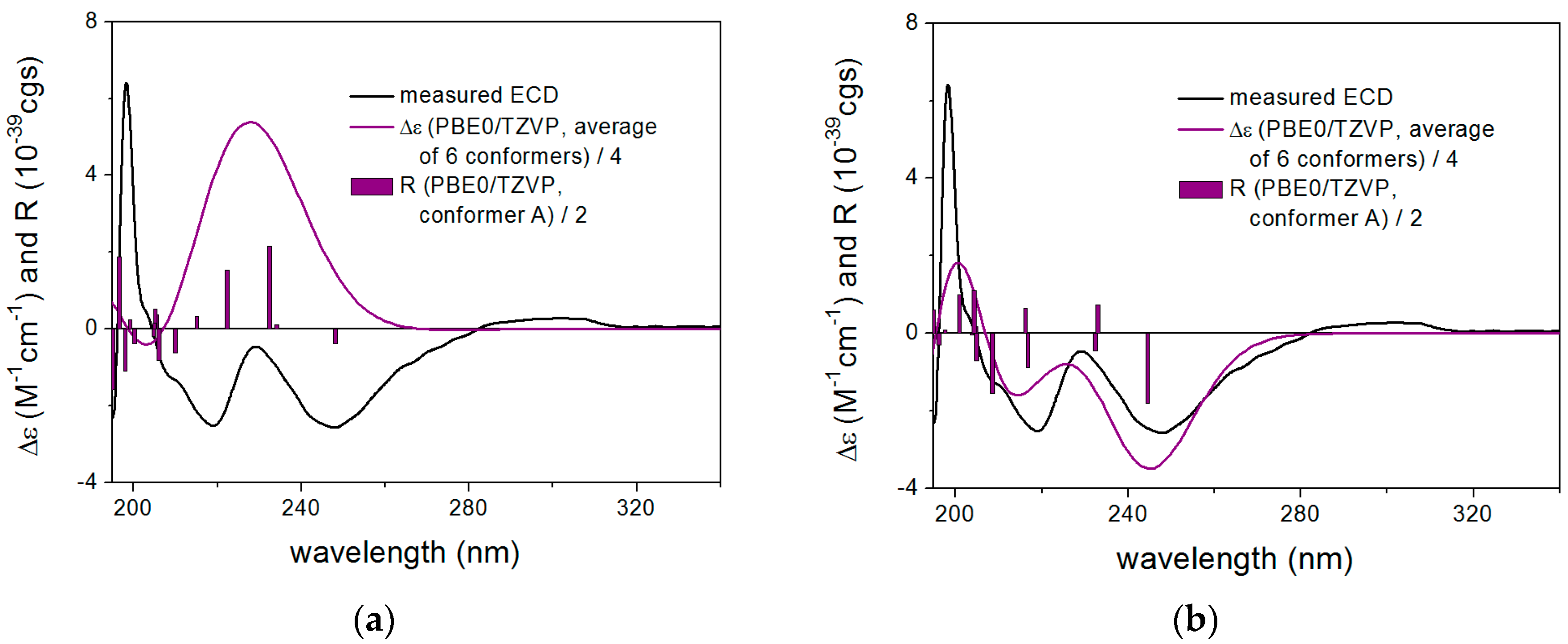
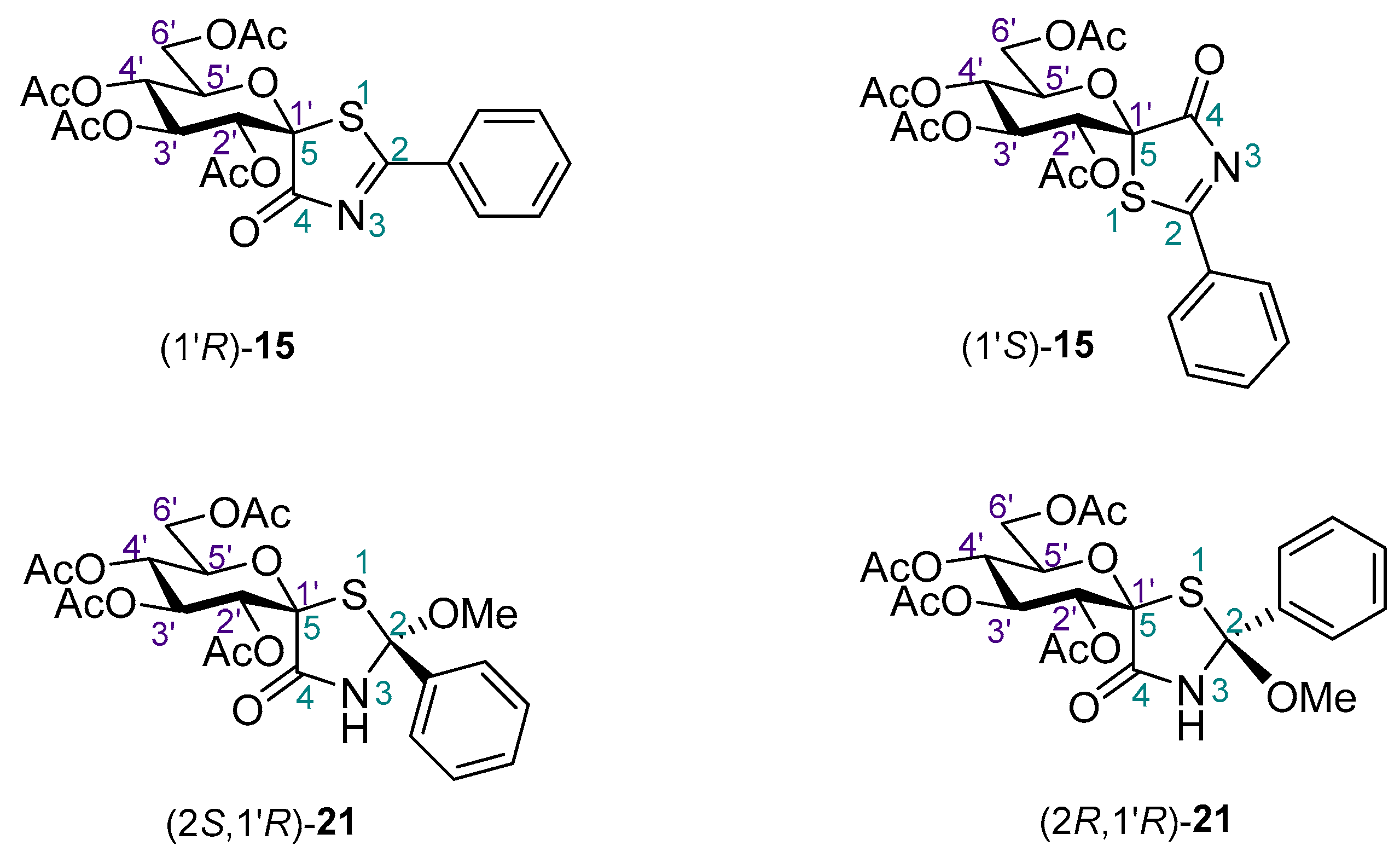
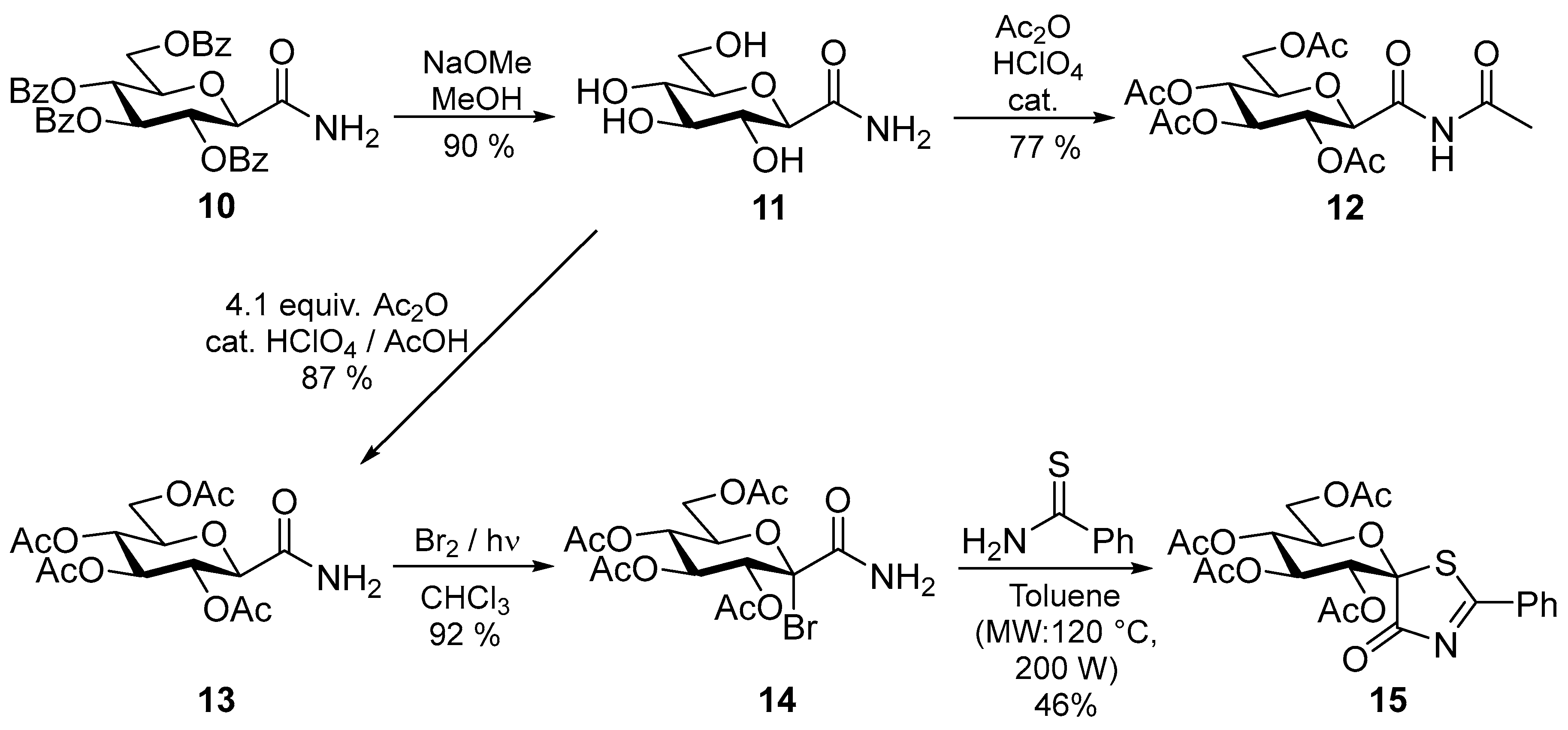
2. Results and Discussion
3. Conclusions
4. Experimental
4.1. General Methods
4.2. Characterization of the Compounds
4.3. Computational Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. Compt. Rend. Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef]
- Tite, T.; Tomas, L.; Docsa, T.; Gergely, P.; Kovensky, J.; Gueyrard, D.; Wadouachi, A. Synthesis of N-aryl spiro-sulfamides as potential glycogen phosphorylase inhibitors. Tetrahedron Lett. 2012, 53, 959–961. [Google Scholar] [CrossRef]
- Czifrák, K.; Páhi, A.; Deák, S.; Kiss-Szikszai, A.; Kövér, K.E.; Docsa, T.; Gergely, P.; Alexacou, K.-M.; Papakonstantinou, M.; Leonidas, D.D.; et al. Glucopyranosylidene-spiro-iminothiazolidinone, a New Bicyclic Ring System: Synthesis, Derivatization, and Evaluation for Inhibition of Glycogen Phosphorylase by Enzyme Kinetic and Crystallographic Methods. Bioorg. Med. Chem. 2014, 22, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Docsa, T.; Marics, B.; Németh, J.; Hüse, C.; Somsák, L.; Gergely, P.; Peitl, B. Insulin sensitivity is modified by a glycogen phosphorylase inhibitor: glucopyranosylidene-spiro-thiohydantoin in streptozotocin-induced diabetic rats. Curr. Top. Med. Chem. 2015, 15, 2390–2394. [Google Scholar] [CrossRef] [PubMed]
- Goyard, D.; Kónya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Patents 2016, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Tracey, W.; Treadway, J.; Magee, W.; McPherson, R.; Levy, C.; Wilder, D.; Li, Y.; Yue, C.; Zavadoski, W.; Gibbs, E.; et al. A novel glycogen phosphorylase inhibitor, CP-368296, reduces myocardial ischemic injury. Diabetes 2003, 52, A135. [Google Scholar]
- Tracey, W.R.; Treadway, J.L.; Magee, W.P.; Sutt, J.C.; McPherson, R.K.; Levy, C.B.; Wilder, D.E.; Yu, L.J.; Chen, Y.; Shanker, R.M.; Mutchler, A.K.; et al. Cardioprotective effects of ingliforib, a novel glycogen phosphorylase inhibitor. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1177–H1184. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini Rev. Med. Chem. 2010, 10, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Qian, Y.S.; Tang, X.Z.; Huang, M.H.; Huang, L.F.; Li, Y.M.; Sun, H.B. Maslinic Acid, a Natural Inhibitor of Glycogen Phosphorylase, Reduces Cerebral Ischemic Injury in Hyperglycemic Rats by GLT-1 Up-Regulation. J. Neurosci. Res. 2011, 89, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Schnier, J.B.; Nishi, K.; Monks, A.; Gorin, F.A.; Bradbury, E.M. Inhibition of glycogen phosphorylase (GP) by CP-91,149 induces growth inhibition correlating with brain GP expression. Biochem. Biophys. Res. Commun. 2003, 309, 126–134. [Google Scholar] [CrossRef]
- Geschwind, J.-F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticanc. Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; Go, V.L.W.; Lee, W.-N.P. Glycogen phosphorylase inhibitor CP-320626 inhibits pancreatic cancer cell proliferation via inhibiting pentose cycle metabolism and glycolysis. Pancreas 2003, 27, 368–420. [Google Scholar]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.P.; Snell, C.; Steers, G.; Turley, H.; Li, J.-L.; Günther, U.L.; et al. Glucose Utilization via Glycogen Phosphorylase Sustains Proliferation and Prevents Premature Senescence in Cancer Cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen metabolism in cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Bichard, C.J.F.; Mitchell, E.P.; Wormald, M.R.; Watson, K.A.; Johnson, L.N.; Zographos, S.E.; Koutra, D.D.; Oikonomakos, N.G.; Fleet, G.W.J. Potent Inhibition of Glycogen Phosphorylase by a Spirohydantoin of Glucopyranose: First Pyranose Analogues of Hydantocidin. Tetrahedron Lett. 1995, 36, 2145–2148. [Google Scholar] [CrossRef]
- Ősz, E.; Somsák, L.; Szilágyi, L.; Kovács, L.; Docsa, T.; Tóth, B.; Gergely, P. Efficient inhibition of muscle and liver glycogen phosphorylases by a new glucopyranosylidene-spiro-thiohydantoin. Bioorg. Med. Chem. Lett. 1999, 9, 1385–1390. [Google Scholar] [CrossRef]
- Somsák, L.; Kovács, L.; Tóth, M.; Ősz, E.; Szilágyi, L.; Györgydeák, Z.; Dinya, Z.; Docsa, T.; Tóth, B.; Gergely, P. Synthesis of and a Comparative Study on the Inhibition of Muscle and Liver Glycogen Phosphorylases by Epimeric Pairs of d-Gluco- and d-Xylopyranosylidene-spiro-(thio)hydantoins and N-(d-Glucopyranosyl) Amides. J. Med. Chem. 2001, 44, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Somsák, L.; Nagy, V.; Vidal, S.; Czifrák, K.; Berzsényi, E.; Praly, J.-P. Novel design principle validated: glucopyranosylidene-spiro-oxathiazole as new nanomolar inhibitor of glycogen phosphorylase, potential antidiabetic agent. Bioorg. Med. Chem. Lett. 2008, 18, 5680–5683. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Benltifa, M.; Vidal, S.; Berzsényi, E.; Teilhet, C.; Czifrák, K.; Batta, G.; Docsa, T.; Gergely, P.; Somsák, L.; et al. Glucose-based spiro-heterocycles as potent inhibitors of glycogen phosphorylase. Bioorg. Med. Chem. 2009, 17, 5696–5707. [Google Scholar] [CrossRef] [PubMed]
- Benltifa, M.; Hayes, J.M.; Vidal, S.; Gueyrard, D.; Goekjian, P.G.; Praly, J.-P.; Kizilis, G.; Tiraidis, C.; Alexacou, K.-M.; Chrysina, E.D.; et al. Glucose-based Spiro-isoxazolines: A New Family of Potent Glycogen Phosphorylase Inhibitors. Bioorg. Med. Chem. 2009, 17, 7368–7380. [Google Scholar] [CrossRef] [PubMed]
- Gregoriou, M.; Noble, M.E.M.; Watson, K.A.; Garman, E.F.; Krülle, T.M.; Fuente, C.; Fleet, G.W.J.; Oikonomakos, N.G.; Johnson, L.N. The structure of a glycogen phosphorylase glucopyranose spirohydantoin complex at 1.8 A resolution and 100 K: The role of the water structure and its contribution to the binding. Protein Sci. 1998, 7, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Oikonomakos, N.G.; Skamnaki, V.T.; Ősz, E.; Szilágyi, L.; Somsák, L.; Docsa, T.; Tóth, B.; Gergely, P. Kinetic and crystallographic studies of glucopyranosylidene spirothiohydantoin binding to glycogen phosphorylase b. Bioorg. Med. Chem. 2002, 10, 261–268. [Google Scholar] [CrossRef]
- Nagy, V.; Czifrák, K.; Bényei, A.; Somsák, L. Synthesis of some O-, S-, and N-glycosides of hept-2-ulopyranosonamides. Carbohydr. Res. 2009, 344, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Páhi, A.; Czifrák, K.; Kövér, K.E.; Somsák, L. Anomeric Spirocycles by Solvent Incorporation: Reactions of O-Peracylated (Glyculopyranose and Glyculopyranosyl Bromide)onamide Derivatives with Ketones. Carbohydr. Res. 2015, 403, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kun, S.; Deák, S.; Czifrák, K.; Juhász, L.; Somsák, L.; Apelt, O. Preparation of 2,6-anhydro-hept-2-enonic acid derivatives and their 3-deoxy counterparts. In Carbohydrate Chemistry: Proven Synthetic Methods; Vogel, C., Murphy, P.V., Eds.; CRC Press: Boca Raton, FL, USA, 2017; Volume 4, pp. 79–90. [Google Scholar]
- Somsák, L.; Nagy, V. A new, scalable preparation of a glucopyranosylidene-spiro-thiohydantoin: one of the best inhibitors of glycogen phosphorylases. Tetrahedron Asymm. 2000, 11, 1719–1727. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Myers, R.W.; Lee, Y.C. Synthesis of diazomethyl β-d-galactopyranosyl and β-d-glucopyranosyl ketones. Potential affinity-labeling reagents for carbohydrate-binding proteins. Carbohydr. Res. 1986, 152, 143–158. [Google Scholar] [CrossRef]
- Somsák, L.; Ferrier, R.J. Radical-mediated Brominations at Ring Positions of Carbohydrates. Adv. Carbohydr. Chem. Biochem. 1991, 49, 37–92. [Google Scholar] [CrossRef] [PubMed]
- Somsák, L.; Czifrák, K. Radical-mediated brominations at ring-positions of carbohydrates–35 years later. Carbohydr. Chem. 2013, 39, 1–37. [Google Scholar] [CrossRef]
- Rudnichenko, A.V.; Timoshenko, V.M.; Shermolovich, Y.G. Cycloaddition reactions of polyfluoroalkylthioncarboxylic acid derivatives with dimethyl acetylenedicarboxylate (DMAD). J. Fluorine Chem. 2004, 125, 439–444. [Google Scholar] [CrossRef]
- Rudnichenko, A.V.; Timoshenko, V.M.; Chernega, A.N.; Nesterenko, A.M.; Shermolovich, Y.G. Addition reactions of 2-polyfluoroalkyl substituted 1,3-thiazolin-4-one derivatives. J. Fluorine Chem. 2004, 125, 1351–1356. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xu, D.-X.; Mándi, A.; Kurtán, T.; Li, T.-J.; Schulz, B.; Zhang, W. Structure, Absolute Configuration, and Conformational Study of 12-Membered Macrolides from the Fungus Dendrodochium sp. Associated with the Sea Cucumber Holothuria nobilis Selenka. J. Org. Chem. 2013, 78, 7030–7047. [Google Scholar] [CrossRef] [PubMed]
- Nicu, V.P.; Mándi, A.; Kurtán, T.; Polavarapu, P.L. On the Complementarity of ECD and VCD Techniques. Chirality 2014, 26, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. MacroModel 10.8.011; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA; Oxfordshire, UK, 2013. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Varetto, U. MOLEKEL; 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| The observed by-products: |  |  |  |  | |
| 6 [25] | 7 [26] | 8 [27] | 9 [28] | ||
| Entry | Ar | Solvent | Reaction Time | Product | Yield 1 (%) |
| 1. | Ph | Ethanol | 18 h | 6 | 65 |
| 2. | Ph | Pentan-3-one | 3 days | 7 | 33 |
| 3. | Ph | Pyridine | 0.5 h | 8 | 22 |
| 4. | Ph | DMF | 2 h | 9 | 75 |
| 5. | Ph | Toluene | 1.5 days | 2 | 34 |
| 6. | Ph | m-Xylene | 4 h | 2 | 33 |
| 7. | Ph | Dibutyl ether | 1 days | 2 | 26 |
| 8. | Ph | Anisole 2 | 1 days | 2 | 27 |
| 9. | Ph | Dioxane | 1.5 days | 2 | 28 |
| 10. | Ph | Nitromethane | 1.5 days | 2 | 33 |
| 11. | Ph | m-Xylene/Ar atm. | 4 h | 2 | 27 |
| 12. | Ph | m-Xylene 3 | 2.5 h | 2 | 30 |
| 13. | Ph | m-Xylene (MW: 120 °C, 200 W) | 1 h | 2 | 40 |
| 14. | Ph | m-Xylene (MW: 140 °C, 200 W) | 1.5 h | 2 | 53 |
| 15. | Ph | m-Xylene (MW: 140 °C, 200 W) 4 | 1.5 h | 2 | 13 |
| 16. | 1-Napth | m-Xylene (MW: 140 °C, 200 W) | 1.5 h | 3 | 32 |
| 17. | 2-Napth | m-Xylene (MW: 140 °C, 200 W) | 1.5 h | 4 | 40 |
| 18. | 4-Me-Ph | m-Xylene (MW: 140 °C, 200 W) | 1.5 h | 5 | 53 |
 | ||
|---|---|---|
| Entry | Conditions | Observations |
| 1. | cat. NaOMe/MeOH, 1 day | Complex reaction mixture |
| 2. | K2CO3, CHCl3: MeOH = 6:1, 1 day | Decomposition |
| 3. | 1 equiv. KCN/MeOH, r.t., 4 weeks | Complex reaction mixture |
| 4. | 50% NH3/MeOH, 2 h | Complex reaction mixture |
| 5. | NaOH, Bu4NHSO4/DCM, r.t., 1 day | Decomposition |
| 6. | 4 equiv. DBU/Toluene, 60 °C, Ar atm. | Complex reaction mixture |
| 7. | 4 equiv. LiOH/MeOH, 0 °C, 1 h | 16 (17%) + 17 (17%) |
| 8. | 0.5 equiv. LiOH/MeOH, 0 °C, 7 h | 16 (32%) + 17 (32%) |
| 9. | cat. AcCl/MeOH, r.t., 1 week | Complex reaction mixture |
| 10. | 4 Å Molecular sieves/MeOH, r.t., 4 weeks | No reaction |
| 11. | KHSO4/MeOH, r.t., 2 weeks | 18 (25%) |
 | |||
|---|---|---|---|
| Entry | Starting Compound | R’ | Product 1 |
| 1. | 2 | Me | 19 |
| 2. | Et | 20 | |
| 3. | 15 | Me | 21 |
| 4. | Et | 22 | |
| 5. | H | 23 2 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, K.E.; Kun, S.; Mándi, A.; Kurtán, T.; Somsák, L. Glucopyranosylidene-Spiro-Thiazolinones: Synthetic Studies and Determination of Absolute Configuration by TDDFT-ECD Calculations. Molecules 2017, 22, 1760. https://doi.org/10.3390/molecules22101760
Szabó KE, Kun S, Mándi A, Kurtán T, Somsák L. Glucopyranosylidene-Spiro-Thiazolinones: Synthetic Studies and Determination of Absolute Configuration by TDDFT-ECD Calculations. Molecules. 2017; 22(10):1760. https://doi.org/10.3390/molecules22101760
Chicago/Turabian StyleSzabó, Katalin E., Sándor Kun, Attila Mándi, Tibor Kurtán, and László Somsák. 2017. "Glucopyranosylidene-Spiro-Thiazolinones: Synthetic Studies and Determination of Absolute Configuration by TDDFT-ECD Calculations" Molecules 22, no. 10: 1760. https://doi.org/10.3390/molecules22101760