Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin

 , and
, and
Abstract
: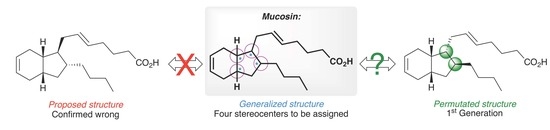
1. Introduction
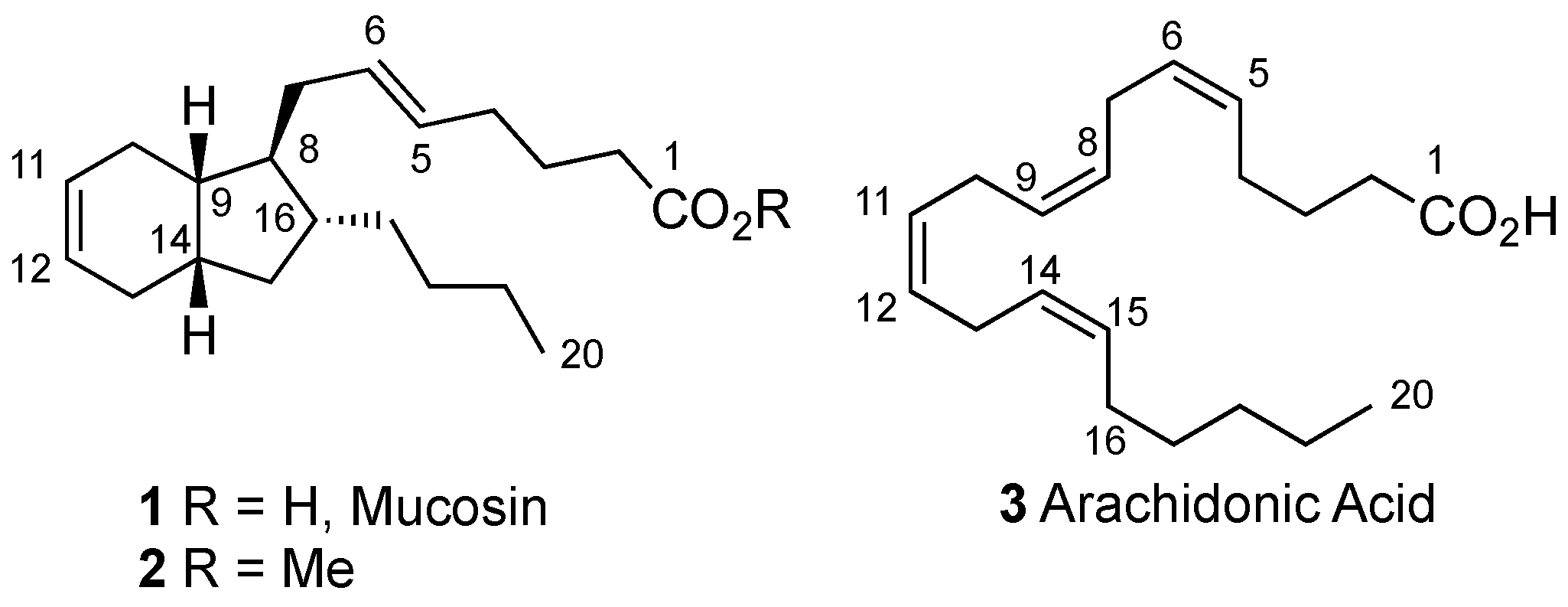
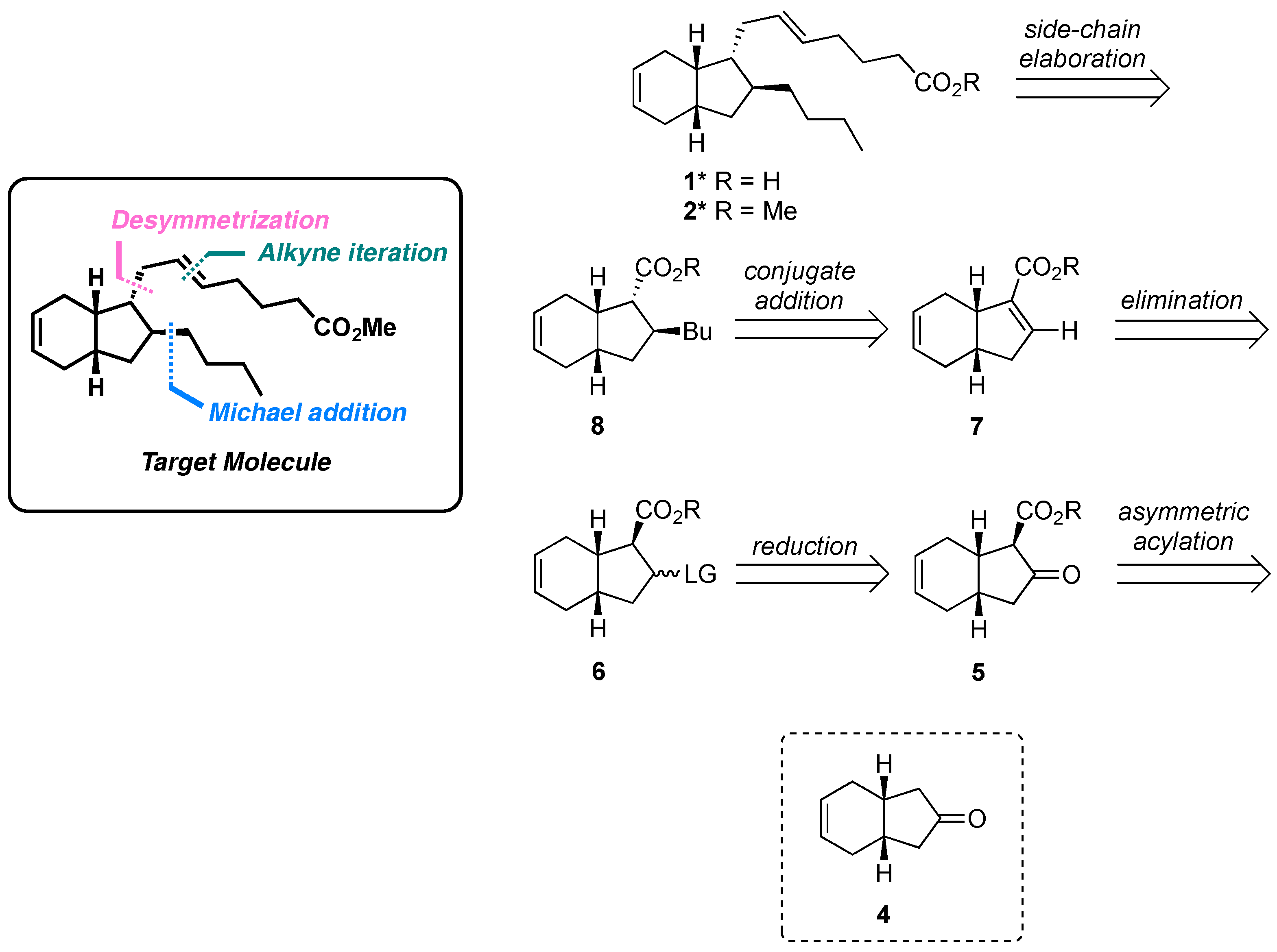
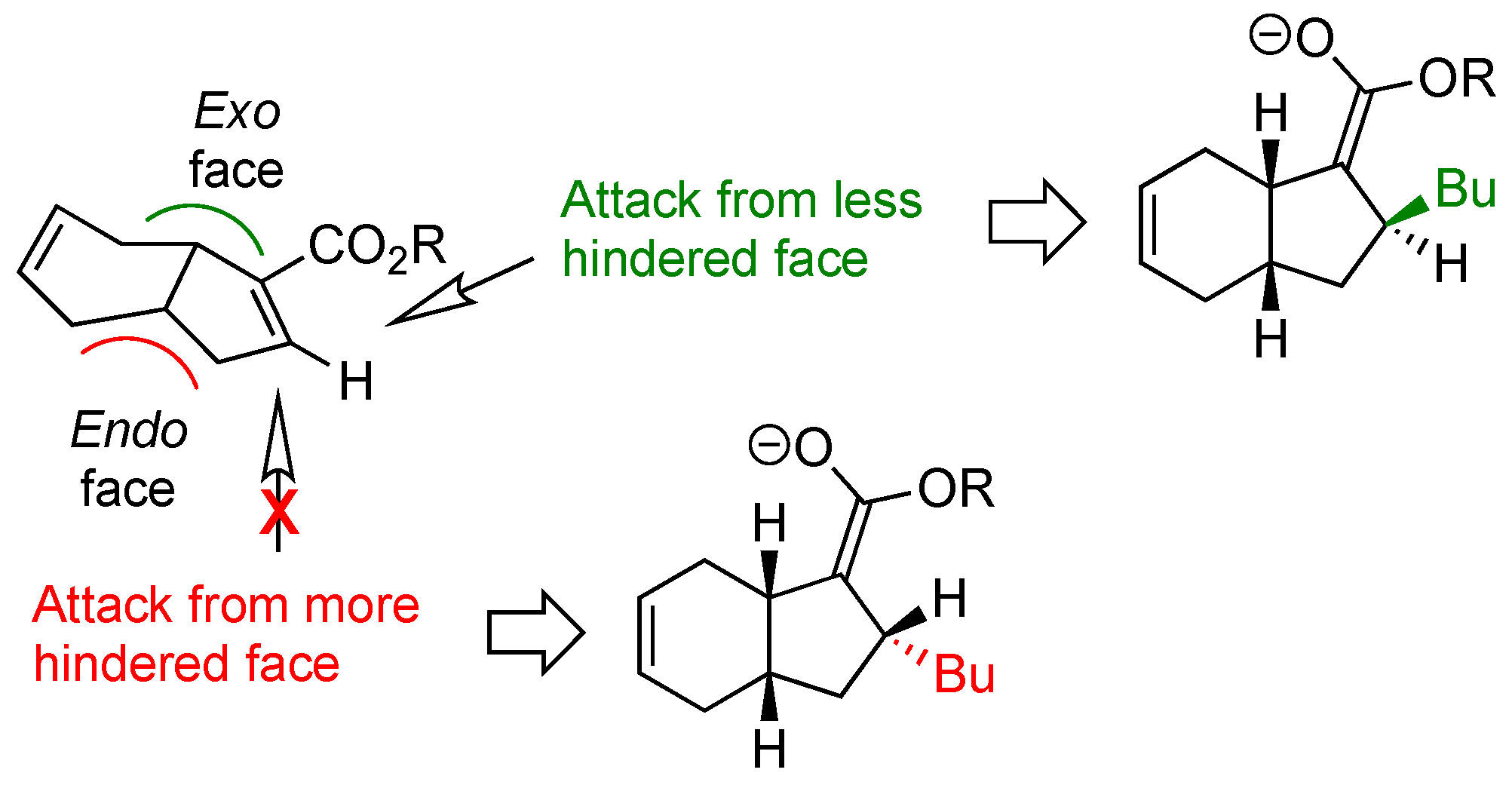
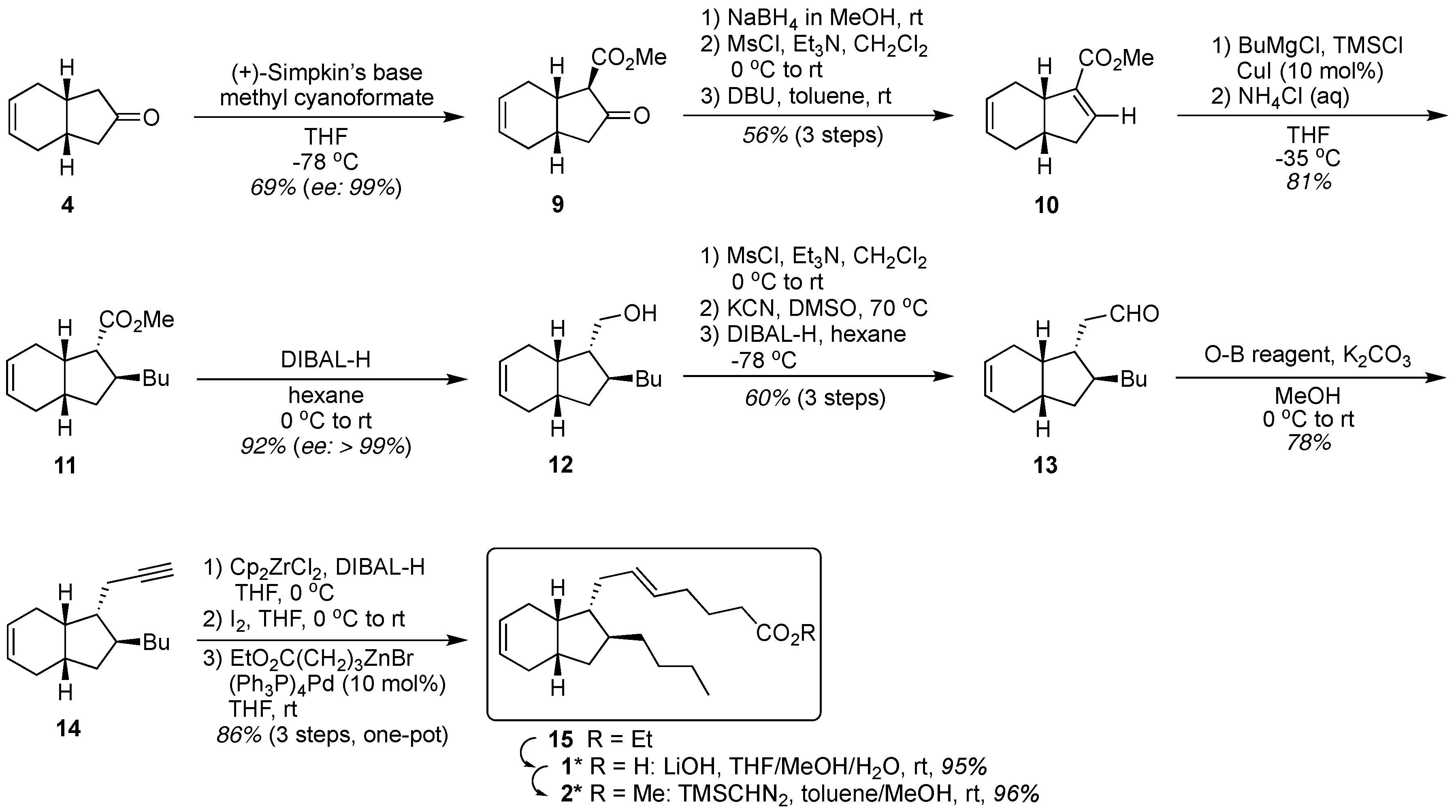
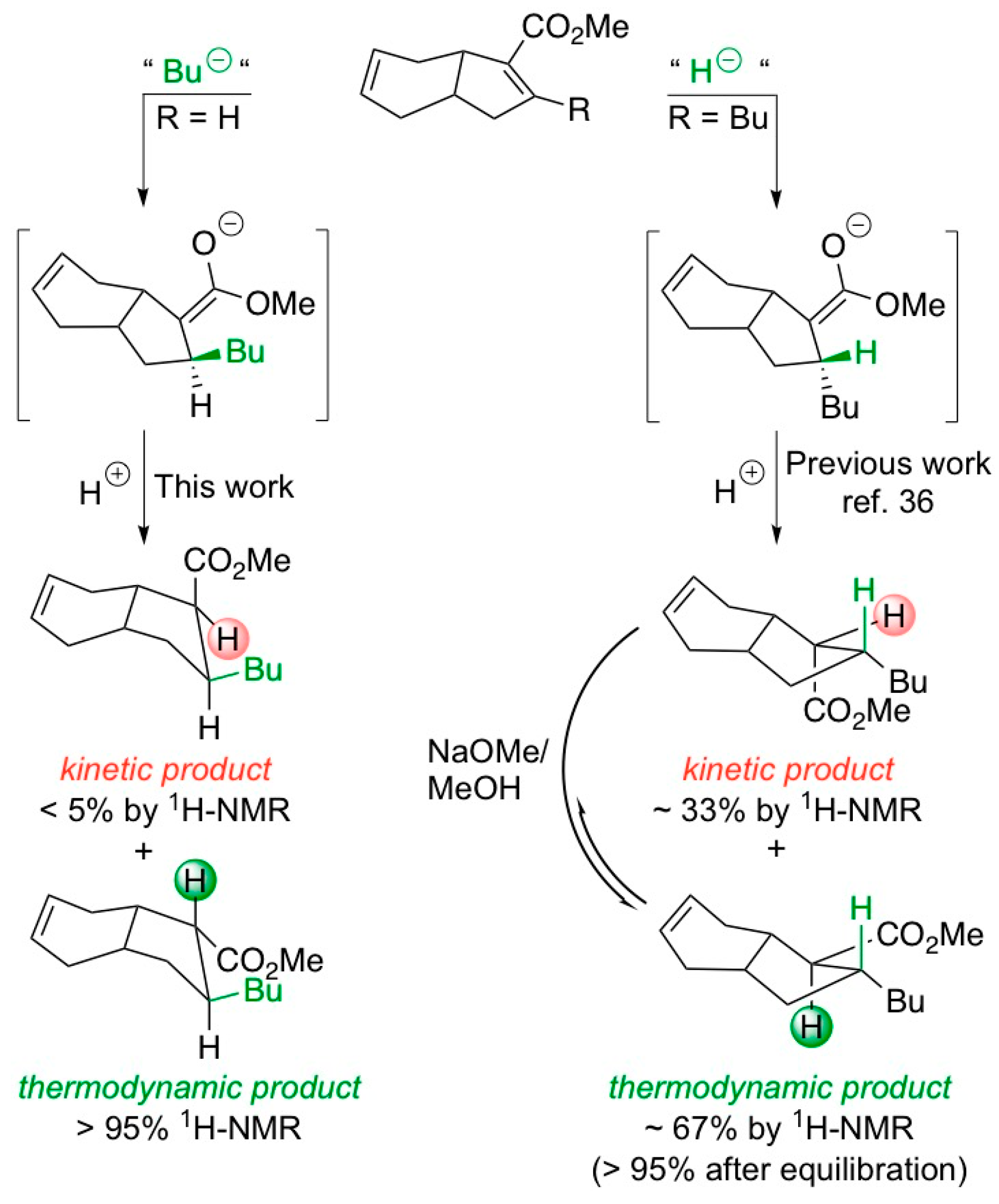
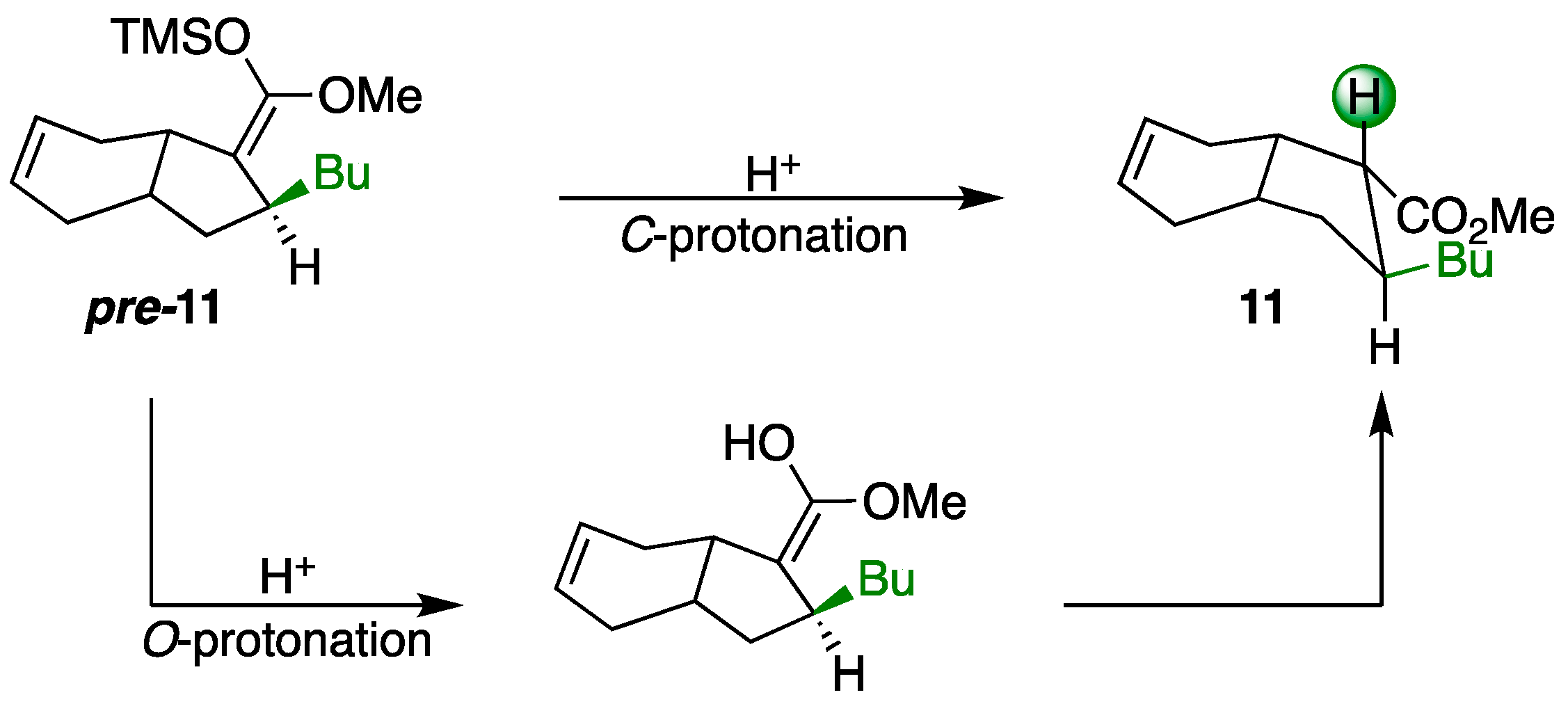
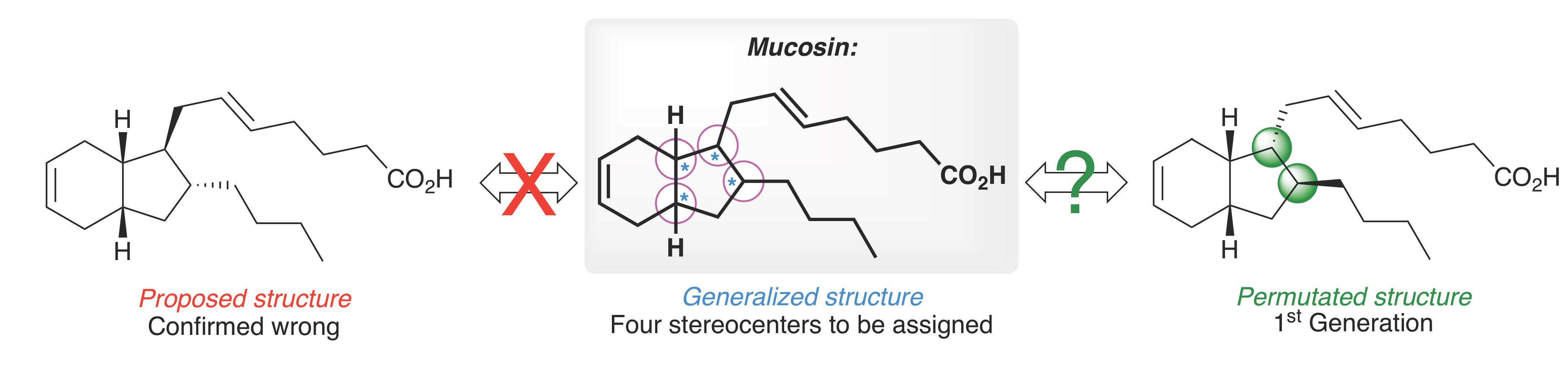
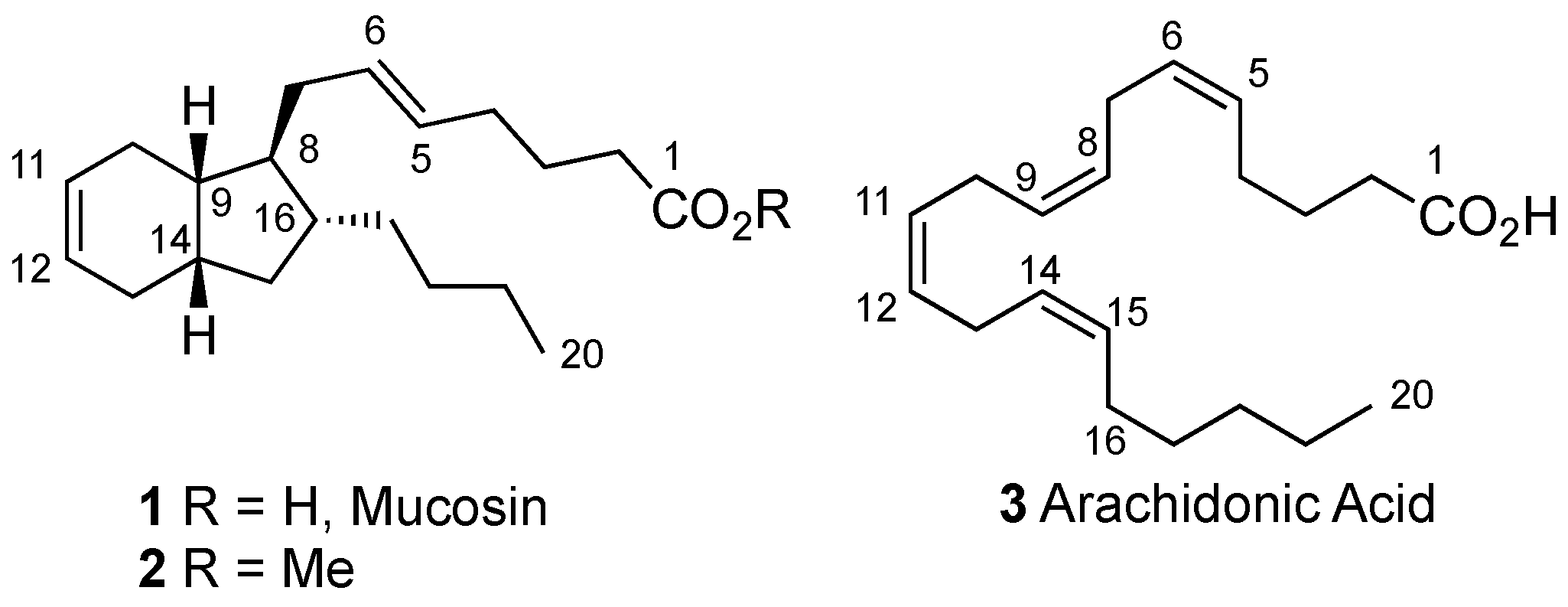
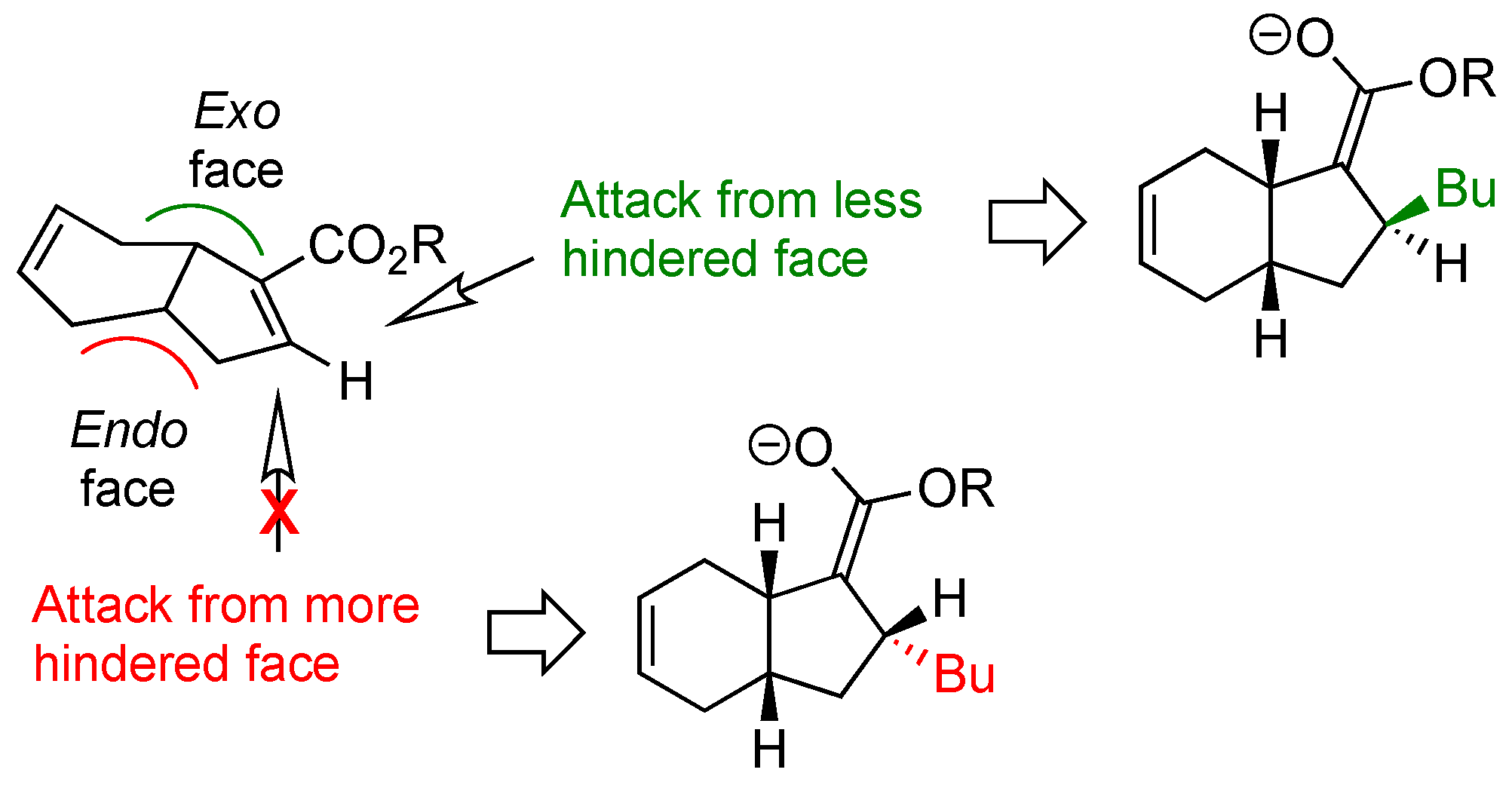
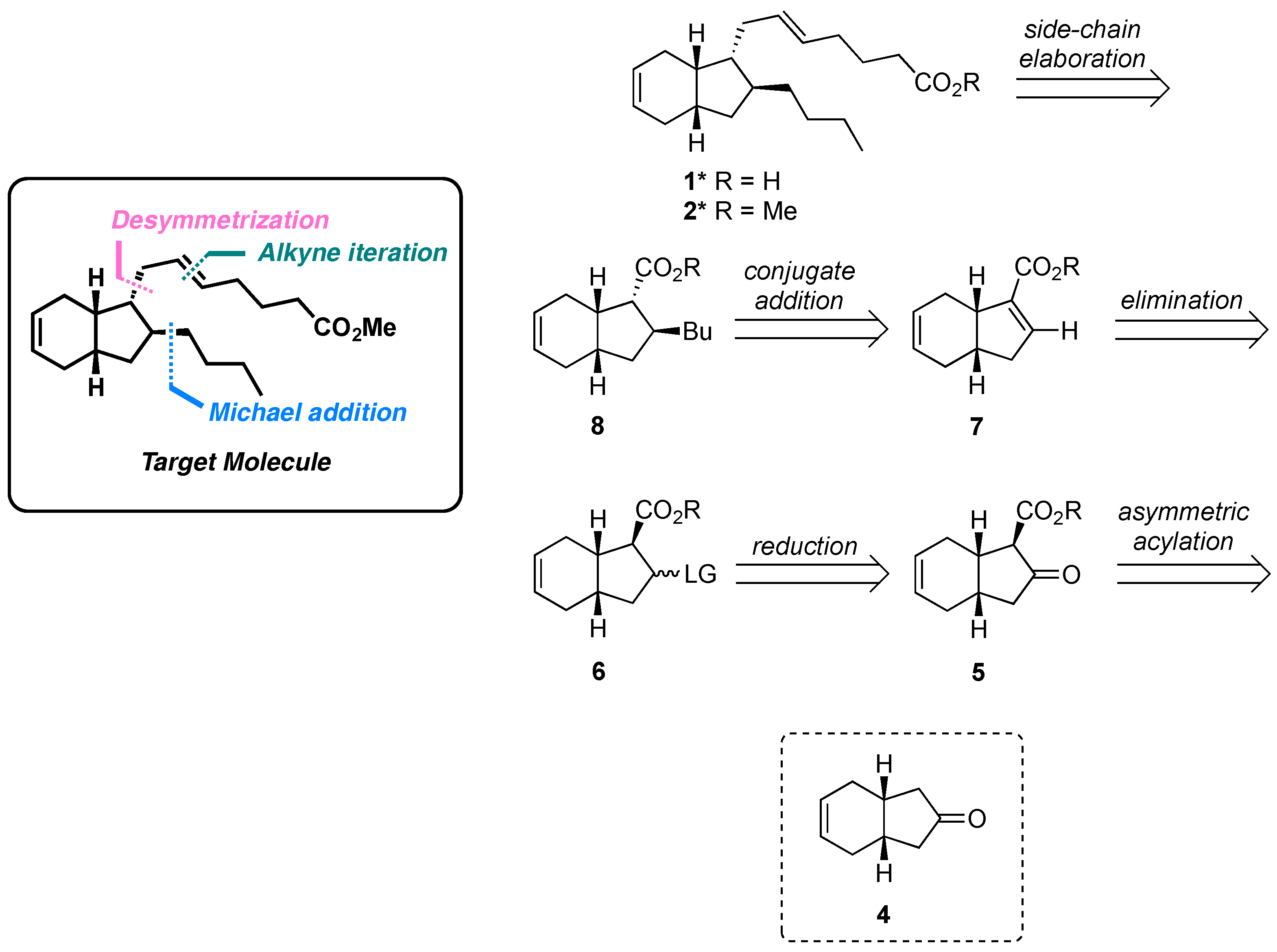
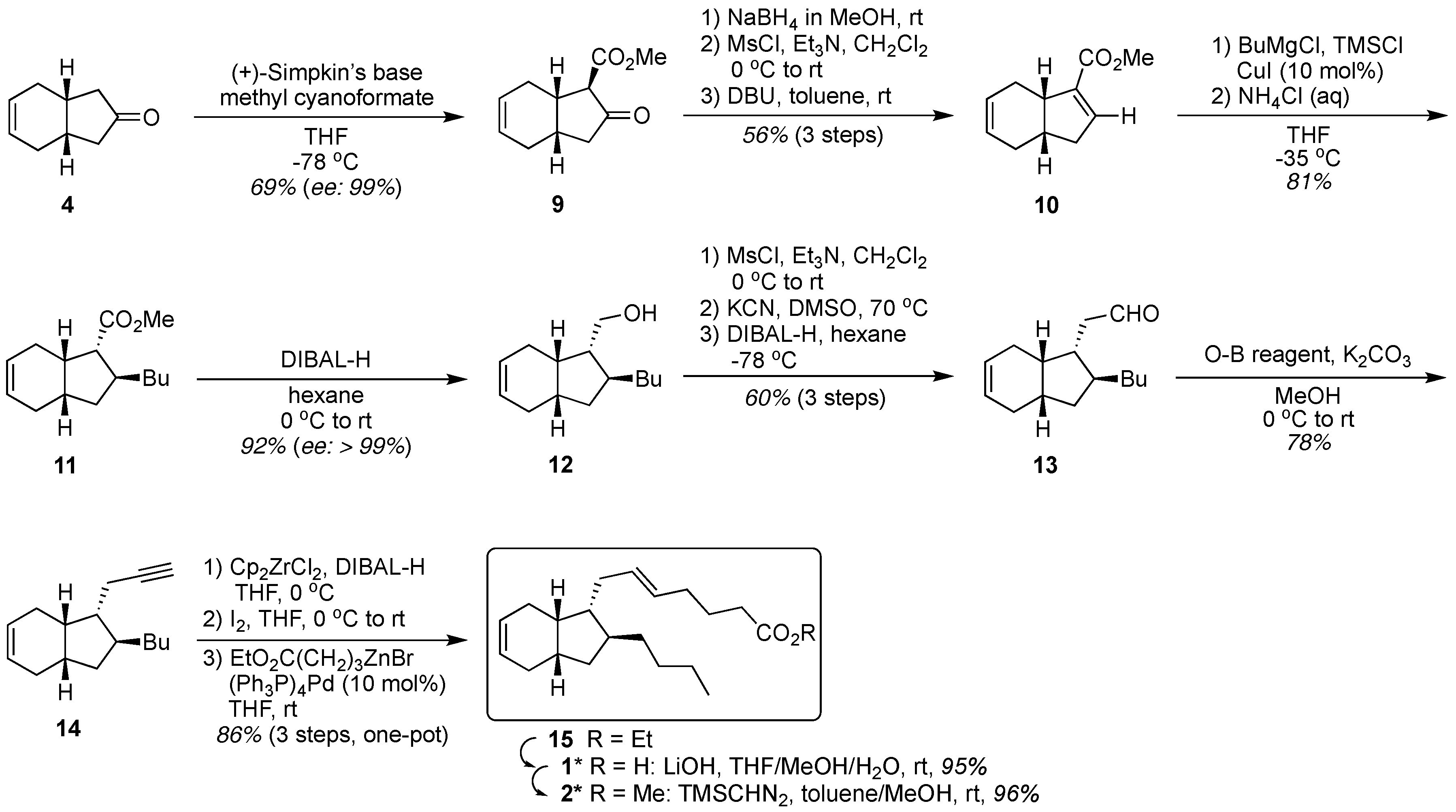
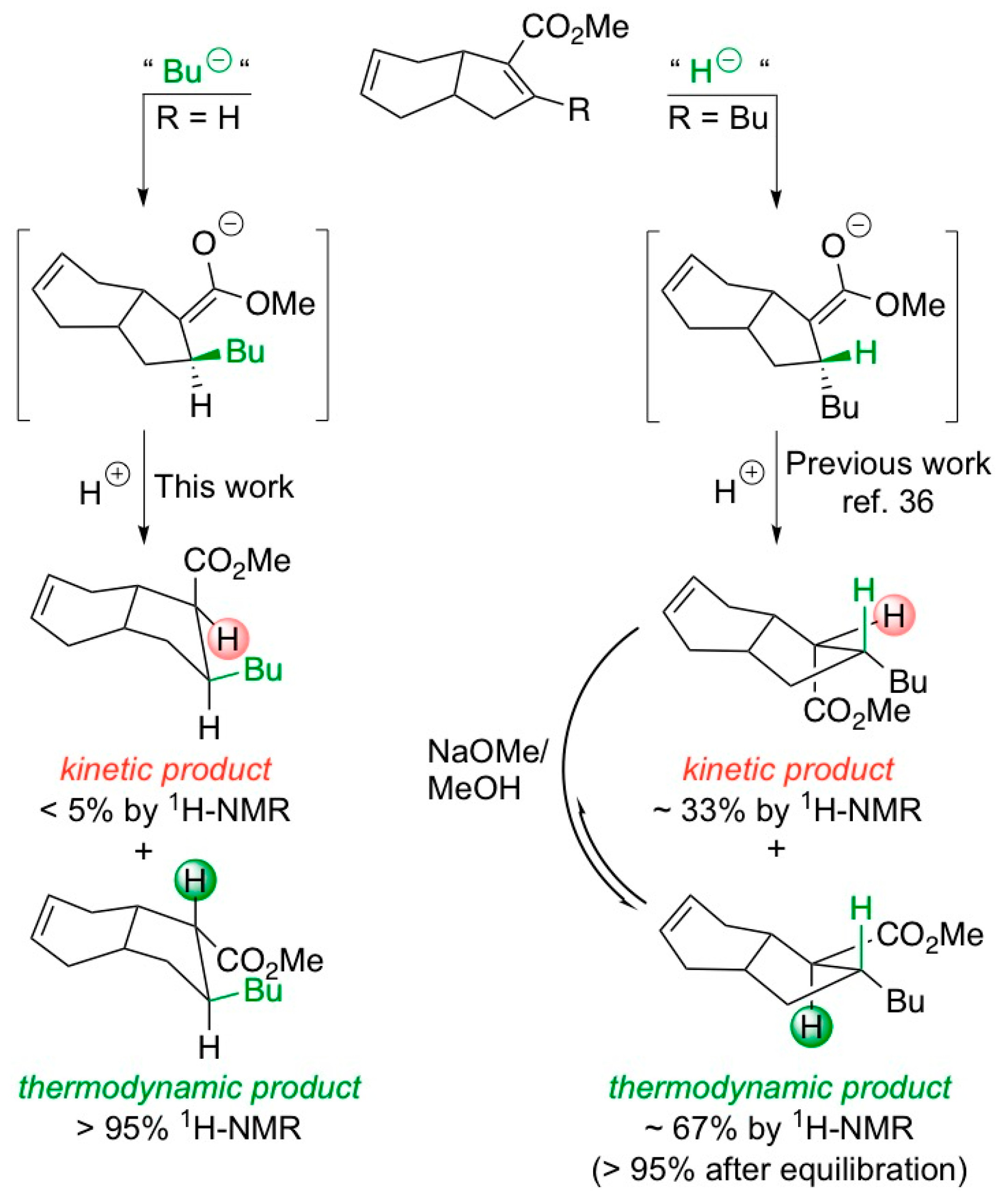
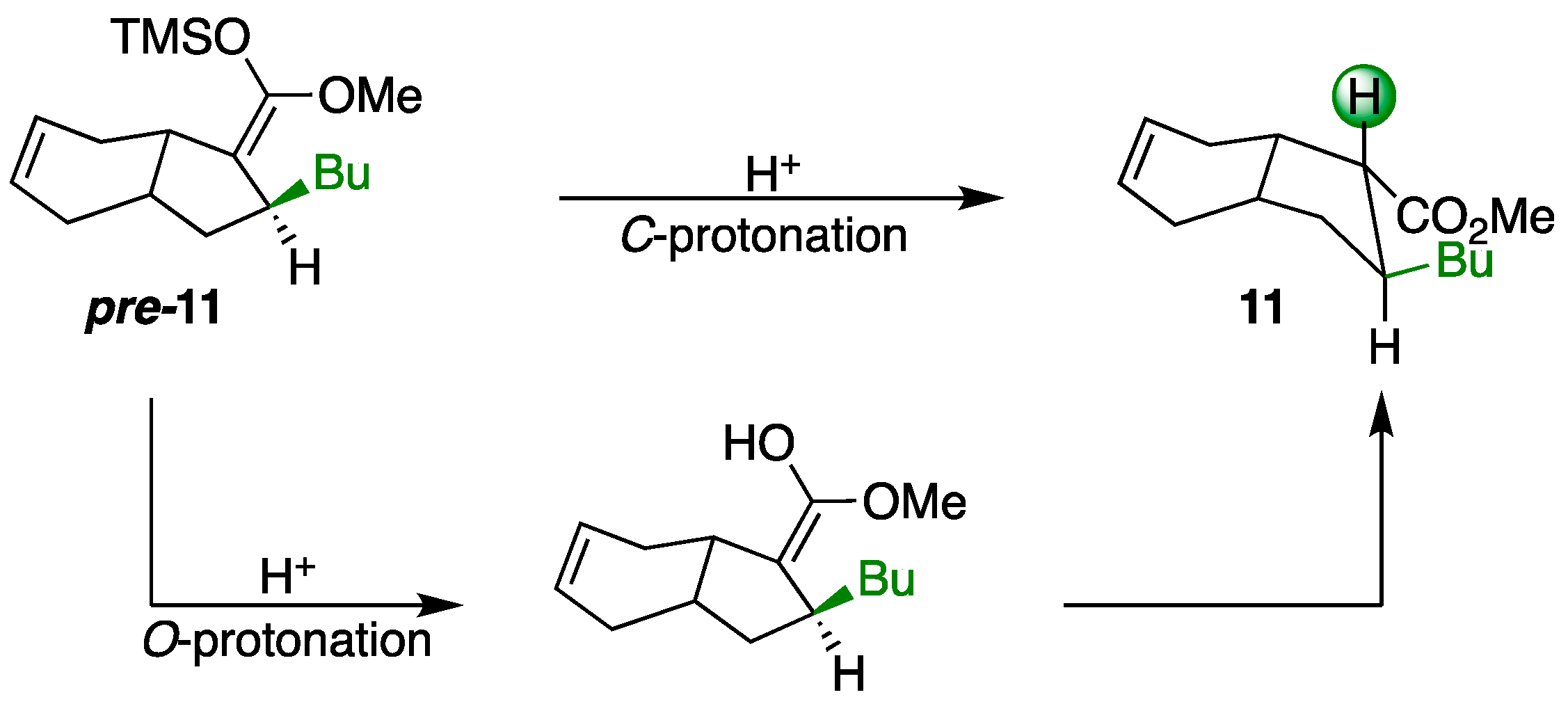
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis of Keto Ester 9
3.3. Synthesis of Michael Acceptor 10
3.3.1. Synthesis of α-Hydroxy Ester pre-10a
3.3.2. Synthesis of Mesylate pre-10b
3.3.3. Synthesis of Michael Acceptor 10
3.4. Synthesis of Michael Adduct 11
3.5. Synthesis of Carbinol 12
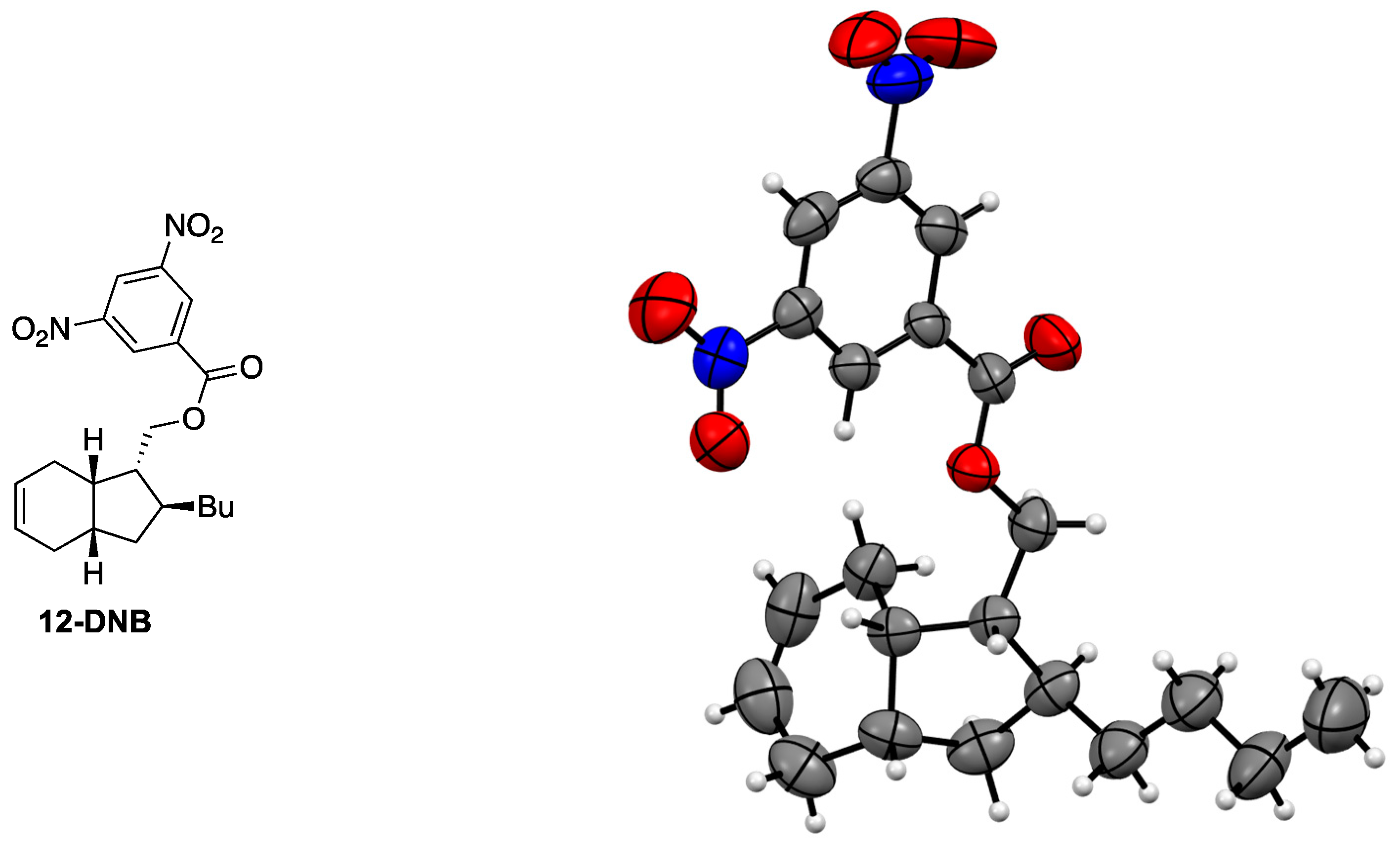
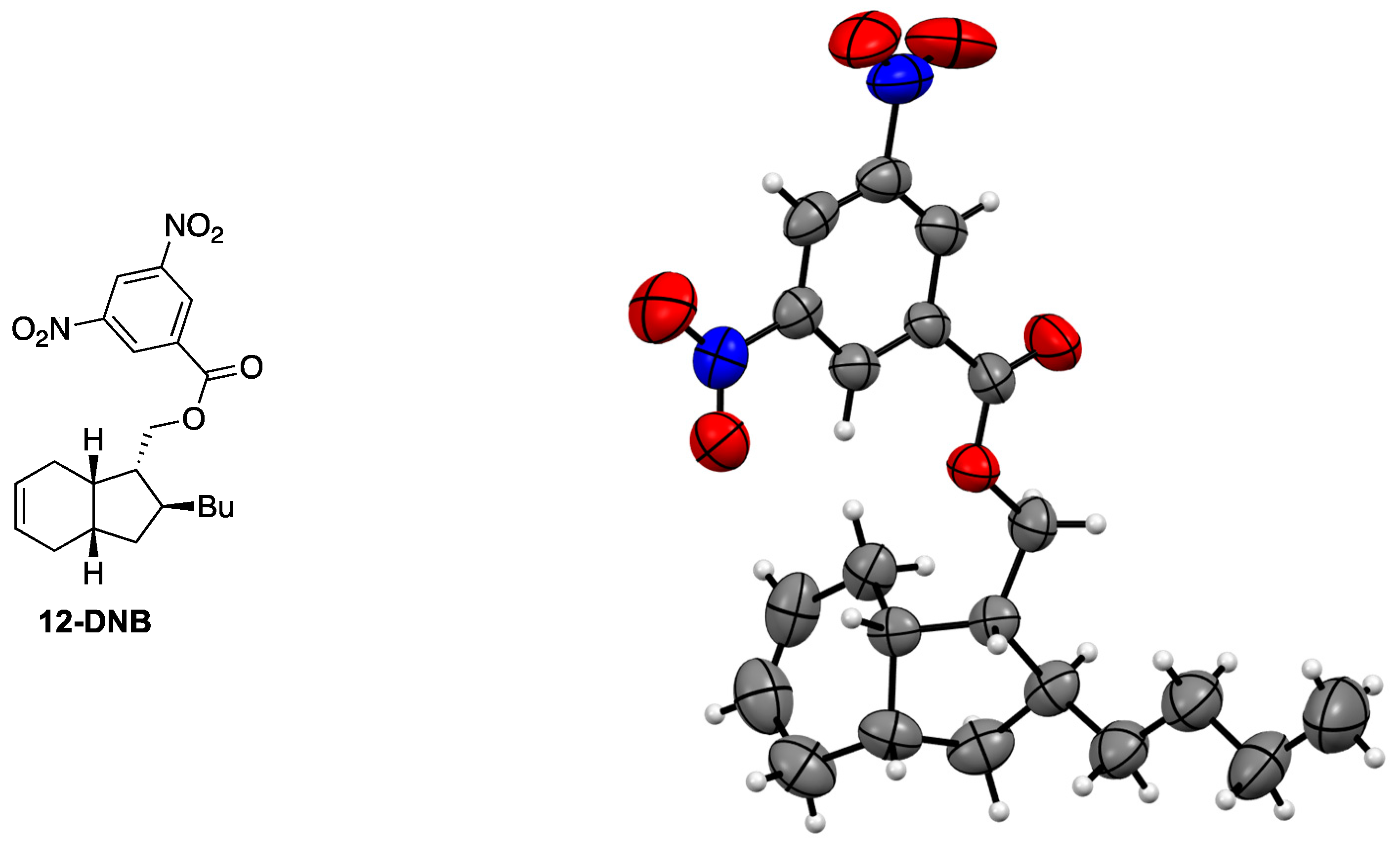
3.6. Synthesis of 3,5-Dinitrobenzoate 12-DNB
3.7. Synthesis of Aldehyde 13
3.7.1. Synthesis of Mesylate pre-13a
3.7.2. Synthesis of Nitrile pre-13b
3.7.3. Synthesis of Aldehyde 13
3.8. Synthesis of Alkyne 14
3.9. Synthesis of exo-Mucosin 1*
3.9.1. Synthesis of Ethyl Ester 15
3.9.2. Synthesis of exo-Mucosin 1*
3.9.3. Synthesis of Methyl Ester 2*
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080. [Google Scholar] [CrossRef] [PubMed]
- Flower, R.J. Prostaglandins, bioassay and inflammation. Br. J. Pharmacol. 2006, 147, S182–S192. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, M.W. A Depressor substance in seminal fluid. Chem. Ind. 1933, 52, 1056–1057. [Google Scholar]
- Von Euler, U.S. Über die spezifische blutdrucksenkende substanz des menschlichen prostata- und samenblasensekretes. Klin. Wochenschr. 1935, 14, 1182–1183. [Google Scholar] [CrossRef]
- Bergström, S.; Sjövall, J. The isolation of prostaglandin. Acta Chem. Scand. 1957, 11, 1086. [Google Scholar] [CrossRef]
- Bergström, S.; Ryhage, R.; Samuelsson, B.; Sjövall, J. Prostaglandins and related factors: 15. The structures of prostaglandin E1, F1β and F1β. J. Biol. Chem. 1963, 238, 3555–3564. [Google Scholar]
- Hamberg, M.; Samuelsson, B. On the mechanism of the biosynthesis of prostaglandins E1 and F1β. J. Biol. Chem. 1967, 242, 5336–5343. [Google Scholar] [PubMed]
- Corey, E.J.; Weinshenker, N.M.; Schaaf, T.K.; Huber, W. Stereo-controlled synthesis of prostaglandins F2β and E2 (dl). J. Am. Chem. Soc. 1969, 91, 5675–5677. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Lands, W.E.M. Stimulation and blockade of prostaglandin biosynthesis. J. Biol. Chem. 1971, 246, 6700–6702. [Google Scholar] [PubMed]
- Corey, E.J.; Ensley, H.E. Preparation of an optically active prostaglandin intermediate via asymmetric induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, D.L.; El-Harith, E.A.; Kraemer, S.A.; Andrews, M.J.; Yao, E.F.; Armstrong, R.L.; Smith, W.L. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. 1990, 265, 5192–5198. [Google Scholar] [PubMed]
- Noverr, M.C.; Erb-Downward, J.R.; Huffnagle, G.B. Production of eicosanoids and other oxylipins by pathogenic eukaryotic microbes. Clin. Microbiol. Rev. 2003, 16, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. The resolution of inflammation: The devil in the flask and in the details. FASEB J. 2011, 25, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Vito, S.; Inceoglu, B.; Hammock, B.D. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 2014, 113–115, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Aursnes, M.; Tungen, J.E.; Vik, A.; Colas, R.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Total synthesis of the lipid mediator PD1n-3 DPA: Configurational assignments and anti-inflammatory and pro-resolving actions. J. Nat. Prod. 2014, 77, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Vik, A.; Ramon, S.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Synthesis and anti-inflammatory and pro-resolving activities of 22-OH-PD1, a monohydroxylated metabolite of protectin D1. J. Nat. Prod. 2014, 77, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Primdahl, K.G.; Aursnes, M.; Walker, M.E.; Colas, R.A.; Serhan, C.N.; Dalli, J.; Hansen, T.V.; Vik, A. Synthesis of 13(R)-hydroxy-7Z,10Z,13R,14E,16Z,19Z docosapentaenoic acid (13R-HDPA) and its biosynthetic conversion to the 13-Series Resolvins. J. Nat. Prod. 2016, 79, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Ramon, S.; Dalli, J.; Sanger, J.M.; Winkler, J.W.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Serhan, C.N. The protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am. J. Pathol. 2016, 186, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- McGovern, P.E.; Michel, R.H. Royal purple dye: Tracing the chemical origins of the industry. Anal. Chem. 1985, 57, 1514A–1522A. [Google Scholar]
- Cooksey, C.J. Tyrian purple: 6,6′-Dibromoindigo and related compounds. Molecules 2001, 6, 736–769. [Google Scholar] [CrossRef]
- Leal, M.C.; Puga, J.; Serôdio, J.; Gomes, N.C.M.; Calado, R. Trends in the discovery of new marine natural products from invertebrates over the last two decades—Where and what are we bioprospecting? PLoS ONE 2012, 7, e30580. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2005, 22, 15–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Casapullo, A.; Scognamiglio, G.; Cimino, G. Mucosin: A new bicyclic eicosanoid from the Mediterranean sponge Reniera mucosa. Tetrahedron Lett. 1997, 38, 3643–3646. [Google Scholar] [CrossRef]
- Gallantree-Smith, H.C.; Antonsen, S.G.; Görbitz, C.H.; Hansen, T.V.; Nolsøe, J.M.J.; Stenstrøm, Y.H. Total synthesis based on the originally claimed structure of mucosin. Org. Biomol. Chem. 2016, 14, 8433–8437. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R.; Stec, J.; Owen, D.R.; Whitby, R.J. The first total synthesis of (+)-mucosin. Chem. Commun. 2012, 48, 3409–3411. [Google Scholar] [CrossRef] [PubMed]
- Bunn, B.J.; Simpkins, N.S.; Spavold, Z.; Crimmin, M.J. The effect of added salts on enantioselective transformations of cyclic ketones by chiral lithium amide bases. J. Chem. Soc. Perkin Trans. 1 1993, 24, 3113–3116. [Google Scholar] [CrossRef]
- Bunn, B.J.; Cox, P.J.; Simpkins, N.S. Enantioselective deprotonation of 8-oxabicyclo[3.2.1]octan-3-one systems using homochiral lithium amide bases. Tetrahedron 1993, 49, 207–218. [Google Scholar] [CrossRef]
- Rodeschini, V.; Simpkins, N.S.; Wilson, C. Kinetic resolution in a bridgehead lithiation mediated by a chiral Bis-lithium amide: Assignment of the absolute configuration of clusianone. J. Org. Chem. 2007, 72, 4265–4267. [Google Scholar] [CrossRef] [PubMed]
- Normant, J.F. Organocopper(I) compounds and organocuprates in synthesis. Synthesis 1972, 2, 63–80. [Google Scholar] [CrossRef]
- Posner, G.H. Conjugate addition reactions of organocopper reagents. Org. Reac. 1972, 19, 1–113. [Google Scholar]
- Lipshutz, B.H. Applications of higher-order mixed organocuprates to organic synthesis. Synthesis 1987, 4, 325–341. [Google Scholar] [CrossRef]
- Dambrin, V.; Villiéras, M.; Lebreton, J.; Toupet, L.; Amri, H.; Villiéras, J. Copper (I) mediated highly diastereoselective conjugate addition of Grignard reagents to functionalised 6-membered ring cycloalkenols. Synthesis of cyclohexanes derivatives substituted on three contiguous atoms. Tetrahedron Lett. 1999, 40, 871–874. [Google Scholar] [CrossRef]
- Dambrin, V.; Villiéras, M.; Janvier, P.; Toupet, L.; Amri, H.; Lebreton, J.; Villiéras, J. Copper(I) mediated highly diastereoselective conjugate addition of Grignard reagents to functionalised cycloalkenols: A general and efficient route for the stereoselective synthesis of 5- and 6-membered ring trisubstituted cycloalkanols. Tetrahedron 2001, 57, 2155–2170. [Google Scholar] [CrossRef]
- Harutyunyan, S.R.; López, F.; Browne, W.R.; Correa, A.; Peña, D.; Badorrey, R.; Meetsma, A.; Minnaard, A.J.; Feringa, B.L. On the mechanism of the copper-catalyzed enantioselective 1,4-addition of Grignard reagents to α,β-unsaturated carbonyl compounds. J. Am. Chem. Soc. 2006, 128, 9103–9118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohira, S. Methanolysis of dimethyl (1-diazo-2-oxopropyl) phosphonate: Generation of dimethyl (diazomethyl) phosphonate and reaction with carbonyl compounds. Synth. Commun. 1989, 19, 561–564. [Google Scholar] [CrossRef]
- Müller, S.; Liepold, B.; Roth, G.J.; Bestmann, H.J. An improved one-pot procedure for the synthesis of alkynes from aldehydes. Synlett 1996, 6, 521–522. [Google Scholar] [CrossRef]
- Habrant, D.; Rauhala, V.; Koskinen, A.M.P. Conversion of carbonyl compounds to alkynes: General overview and recent developments. Chem. Soc. Rev. 2010, 39, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Taber, D.F.; Bai, S.; Guo, P.-F. A convenient reagent for aldehyde to alkyne homologation. Tetrahedron Lett. 2008, 49, 6904–6906. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.; Müller, H. Dialkylaluminiumhydride als sterisch spezifische reduktionsmittel für acetylene. Chem. Ber. 1956, 89, 444–447. [Google Scholar] [CrossRef]
- Hart, D.W.; Blackburn, T.F.; Schwartz, J. Hydrozirconation. III. Stereospecific and regioselective functionalization of alkylacetylenes via vinylzirconium(IV) intermediates. J. Am. Chem. Soc. 1975, 97, 679–680. [Google Scholar] [CrossRef]
- Brown, H.C.; Gupta, S.K. Hydroboration. XXXIX. 1,3,2-Benzodioxaborole (catecholborane) as a new hydroboration reagent for alkenes and alkynes. General synthesis of alkane- and alkeneboronic acids and esters via hydroboration. Directive effects in the hydroboration of alkenes and alkynes with catecholborane. J. Am. Chem. Soc. 1975, 97, 5249–5255. [Google Scholar]
- Zhao, Y.; Snieckus, V. A Practical in situ generation of the Schwartz reagent. Reduction of Tertiary amides to aldehydes and hydrozirconation. Org. Lett. 2014, 16, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, M.; Ma, X.; Nijesh, K.; De, S.; Parameswaran, P.; Roesky, H.W. An aluminum dihydride working as a catalyst in hydroboration and dehydrocoupling. J. Am. Chem. Soc. 2016, 138, 2548–2551. [Google Scholar] [CrossRef] [PubMed]
- Palei, B.A.; Gavrilenko, V.V.; Zakharkin, L.I. Comparative reactivity of alkyl, vinyl, and acetylene derivatives of aluminum. Russ. Chem. Bull. 1969, 18, 2590–2595. [Google Scholar] [CrossRef]
- Diederich, F.; Stang, P. (Eds.) Metal-Catalyzed Cross Coupling Reactions; J. Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Huo, S. Highly efficient, general procedure for the preparation of alkylzinc reagents from unactivated alkyl bromides and chlorides. Org. Lett. 2003, 5, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Pohnert, G.; Boland, W. The oxylipin chemistry of attraction and defense in brown algae and diatoms. Nat. Prod. Rep. 2002, 19, 108–122. [Google Scholar] [PubMed]
- Andreou, A.; Brodhun, F.; Feussner, I. Biosynthesis of oxylipins in non-mammals. Prog. Lipid Res. 2009, 48, 148–170. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.; Valentao, P.; Andrade, P.A. Biologically active oxylipins from enzymatic and nonenzymatic routes in macroalgae. Mar. Drugs 2016, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.; Woodward, R.B. The conservation of orbital symmetry. Acc. Chem. Res. 1968, 1, 17–22. [Google Scholar] [CrossRef]
- Klas, K.; Tsukamoto, S.; Sherman, D.H.; Williams, R.M. Natural Diels–Alderases: Elusive and irresistible. J. Org. Chem. 2015, 80, 11672–11685. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.D.; Ketchum, S.O.; Gerwick, W.H. 5-Lipoxygenase-derived oxylipins from the red alga Rhodymenia pertusa. Phytochemistry 2000, 53, 129–133. [Google Scholar] [CrossRef]
- Bernart, M.; Gerwick, W.H. Isolation of 12-(S)-hepe from the red marine alga Murrayella periclados and revision of structure of an acyclic icosanoid from Laurencia hybrida. Implications to the biosynthesis of the marine prostanoid hybridalactone. Tetrahedron Lett. 1988, 29, 2015–2018. [Google Scholar] [CrossRef]
- Jiang, Z.D.; Gerwick, W.H. Novel oxylipins from the temperate red alga Polyneura latissima: Evidence for an arachidonate 9(S)-lipoxygenase. Lipids 1997, 32, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Gerwick, W.H. Two new icosapentaenoic acids from the temperate red seaweed Ptilota filicina J. Agardh. Lipids 1987, 22, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Solem, M.L.; Jiang, Z.D.; Gerwick, W.H. Three new and bioactive icosanoids from the temperate red marine alga Farlowia mollis. Lipids 1989, 24, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, M.; Gerwick, W.H. Biosynthesis of vicinal dihydroxy fatty acids in the red alga Gracilariopsis lemaneiformis: Identification of a sodium-dependent 12-lipoxygenase and a hydroperoxide isomerase. Arch. Biochem. Biophys. 1993, 305, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Åsen, P.A.; Hamberg, M. Biosynthesis of 13R-hydroxyarachidonic acid, an unusual oxylipin from the red alga lithothamnion corallioides. Phytochemistry 1993, 34, 1029–1033. [Google Scholar] [CrossRef]
- Burgess, J.R.; De la Rosa, R.I.; Jacobs, R.S.; Butler, A. A new eicosapentaenoic acid formed from arachidonic acid in the coralline red algae Bossiella orbigniana. Lipids 1991, 26, 162–165. [Google Scholar] [CrossRef]
- Murakami, N.; Shirahashi, H.; Nagatsu, A.; Sakakibara, J. Two unsaturated 9R-hydroxy fatty acids from the cyanobacterium Anabaena flos-aquae f. flos-aquae. Lipids 1992, 27, 776–778. [Google Scholar] [CrossRef]
- Moghaddam, M.F.; Gerwick, W.H.; Ballantine, D.L. Discovery of 12-(S)-hydroxy-5,8,10,14-icosatetraenoic acid [12-(S)-HETE] in the tropical red alga Platysiphonia miniata. Prostaglandines 1989, 37, 303–308. [Google Scholar] [CrossRef]
- Weinberger, F.; Lion, U.; Delage, L.; Kloareg, B.; Potin, P.; Beltran, J.; Flores, V.; Faugeron, S.; Correa, J.; Pohnert, G. Up-regulation of lipoxygenase, phospholipase, and oxylipin-production in the induced chemical defense of the red alga Gracilaria chilensis against epiphytes. J. Chem. Ecol. 2011, 37, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.-M.; Hwang, A.; Yeh, D. Purification, substrate specificity, and products of a Ca2+-stimulating lipoxygenase from sea algae (Ulva lactuca). J. Agric. Food Chem. 1997, 45, 2055–2060. [Google Scholar] [CrossRef]
- Mohamed, Y.M.A.; Hansen, T.V. Synthesis of methyl (5Z,8Z,10E,12E,14Z)-eicosapentaenoate. Tetrahedron Lett. 2011, 52, 1057–1059. [Google Scholar] [CrossRef]
- Wise, M.L.; Hamberg, M.; Gerwick, W. Biosynthesis of conjugated triene-containing fatty acids by a novel isomerase from the red marine alga Ptilota filicina. Biochemistry 1994, 33, 15223–15232. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Auclair, K.; Kendrew, S.G.; Park, C.; Vederas, J.C.; Hutchinson, C.R. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 1999, 284, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Auclair, K.; Sutherland, A.; Kennedy, J.; Witter, D.J.; Van den Heever, J.P.; Hutchinson, C.R.; Vederas, J.C. Lovastatin nonaketide synthase catalyzes an intramolecular Diels−Alder reaction of a substrate analogue. J. Am. Chem. Soc. 2000, 122, 11519–11520. [Google Scholar] [CrossRef]
- Alt, S.; Wilkinson, B. Biosynthesis of the novel macrolide antibiotic anthracimycin. Chem. Biol. 2015, 10, 2468–2479. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, K.; Katayama, K.; Suzuki, Y.; Ichihara, A. Enzymatic activity catalysing exo-selective Diels–Alder reaction in solanapyrone biosynthesis. J. Chem. Soc. Chem. Commun. 1995, 13, 1321–1322. [Google Scholar] [CrossRef]
- Kasahara, K.; Miyamaoto, T.; Fujimoto, T.; Oguri, H.; Tokiwano, T.; Oikawa, H.; Ebizuka, Y.; Fujii, I. Solanapyrone synthase, a possible Diels-Alderase and iterative Type I polyketide synthase encoded in a biosynthetic gene cluster from Alternaria solani. ChemBioChem 2010, 11, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ruszczycky, M.W.; Choi, S.-H.; Liu, Y.-N.; Liu, H.-W. Enzyme-catalysed [4+2] cycloaddition is a key step in the biosynthesis of spinosyn A. Nature 2011, 473, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, H.; Yagi, K.; Watanabe, K.; Honma, M.; Ichihara, A. Biosynthesis of macrophomic acid: Plausible involvement of intermolecular Diels–Alder reaction. Chem. Commun. 1997, 1, 97–98. [Google Scholar] [CrossRef]
- Ose, T.; Watanabe, K.; Mie, T.; Honma, M.; Watanabe, H.; Yao, M.; Oikawa, H.; Tanaka, I. Insight into a natural Diels–Alder reaction from the structure of macrophomate synthase. Nature 2003, 422, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H. Carbocyclic oxylipins of marine origin. Chem. Rev. 1993, 93, 1807–1823. [Google Scholar] [CrossRef]
- Gerwick, W.H. Epoxy allylic carbocations as conceptual intermediates in the biogenesis of diverse marine oxylipins. Lipids 1996, 31, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Singh, I.P. Structural diversity of marine oxylipins. In Lipid Biotechnology; Kuo, T.M., Gardner, H.W., Eds.; Marcel Dekker: New York, NY, USA, 2002; pp. 249–275. [Google Scholar]
- Li, L.; Yu, P.; Tang, M.-C.; Zou, Y.; Gao, S.-S.; Hung, Y.-S.; Zhao, M.; Watanabe, K.; Houk, K.N.; Tang, Y. The catalytic mechanism of a natural Diels–Alderase revealed in molecular detail. J. Am. Chem. Soc. 2016, 138, 15837–15840. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, N.; Itoh, S.; Fujita, K.; Sakanishi, K.; Sugiyama, R.; Terajima, Y.; Iwabuchi, Y.; Nishimura, S.; Kakeya, H. Asymmetric total synthesis of heronamides A–C: Stereochemical Confirmation and impact of long-range stereochemical communication on the biological activity. Chem. Eur. J. 2016, 22, 8586–8595. [Google Scholar] [CrossRef] [PubMed]
- Booth, T.J.; Alt, S.; Capon, R.J.; Wilkinson, B. Synchronous intramolecular cycloadditions of the polyene macrolactam polyketide heronamide C. Chem. Commun. 2016, 52, 6383–6386. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Patel, A.; Houk, K.N. Transannular [6+4] and ambimodal cycloaddition in the biosynthesis of heronamide A. J. Am. Chem. Soc. 2015, 137, 13518–13523. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-I.; McCarty, R.M.; Liu, H.-W. The enzymology of organic transformations: A survey of name reactions in biological systems. Angew. Chem. Int. Ed. 2017, 56, 3446–3489. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.-s.; Ruszczycky, M.W.; Russell, W.K.; Lin, G.-M.; Kim, N.; Choi, S.-h.; Wang, S.-A.; Liu, Y.-N.; Patrick, J.W.; Russell, D.H.; et al. Investigation of the mechanism of the SpnF-catalyzed [4+2]-cycloaddition reaction in the biosynthesis of spinosyn A. Proc. Natl. Acad. Natl. USA 2017, 114, 10408–10413. [Google Scholar] [CrossRef] [PubMed]
- Cavelier, F.; Gomez, S.; Jacquier, R.; Verducci, J. Deracemization of silyl enol ethers. Tetrahedron Asymmetry 1993, 4, 2501–2505. [Google Scholar] [CrossRef]
- Cavelier, F.; Gomez, S.; Jacquier, R.; Verducci, J. A first approach to asymmetric protonation via a polymer supported chiral proton donor. Tetrahedron Lett. 1994, 35, 2891–2894. [Google Scholar] [CrossRef]
- Diba, A.K.; Noll, C.; Richter, M.; Gieseler, M.T.; Kalesse, M. Intramolecular stereoselective protonation of aldehyde-derived enolates. Angew. Chem. 2010, 122, 8545–8547. [Google Scholar] [CrossRef]
- Mohr, J.T.; Hong, A.Y.; Stoltz, B.M. Enantioselective protonation. Nat. Chem. 2009, 1, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Bruker. Bruker APEX3, SAINT+ and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Nagao, Y.; Hagiwara, Y.; Tohjo, T.; Hasegawa, Y.; Ochiai, M.; Shiro, M. Highly enantioselective Claisen-type acylation and Dieckmann annulation. J. Org. Chem. 1988, 53, 5983–5986. [Google Scholar] [CrossRef]
- Preparation of the racemic reference compound rac-12 was performed according to the general procedures outlined in Scheme 2, replacing (+)-Simpkin’s base with LDA to obtain rac-9 for the synthetic sequence.
- The publCIF document accompanying compound 12-DNB is uploaded as separate file and the structure is deposited at Cambridge Crystallographic Data Centre and identified as CCDC 1535632.
Sample Availability: Samples of exo-mucosin (1*) and methyl ester 2* are available from the authors. |
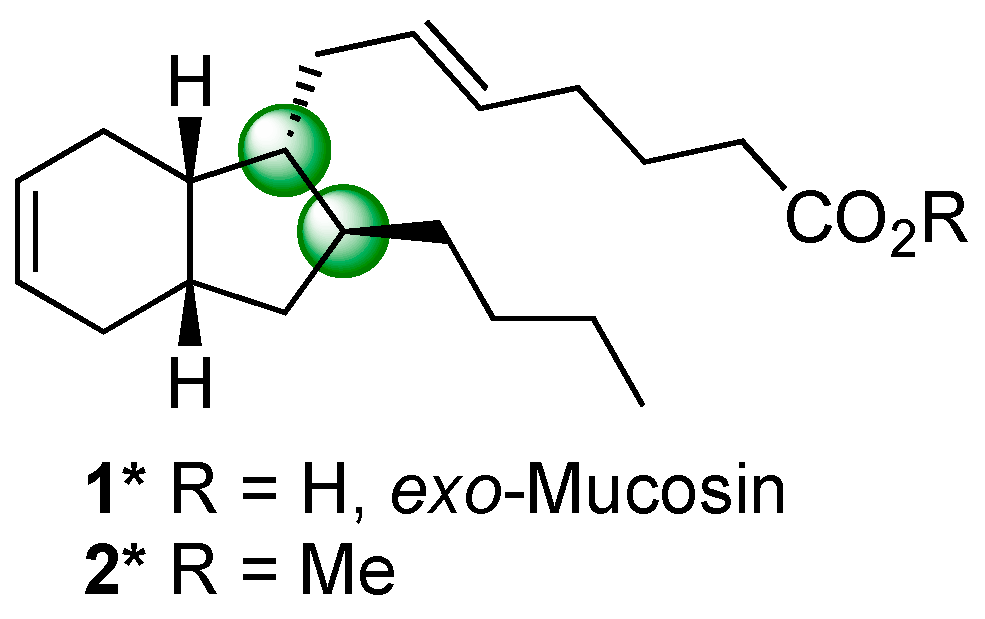

{kind=link}
{kind=link}
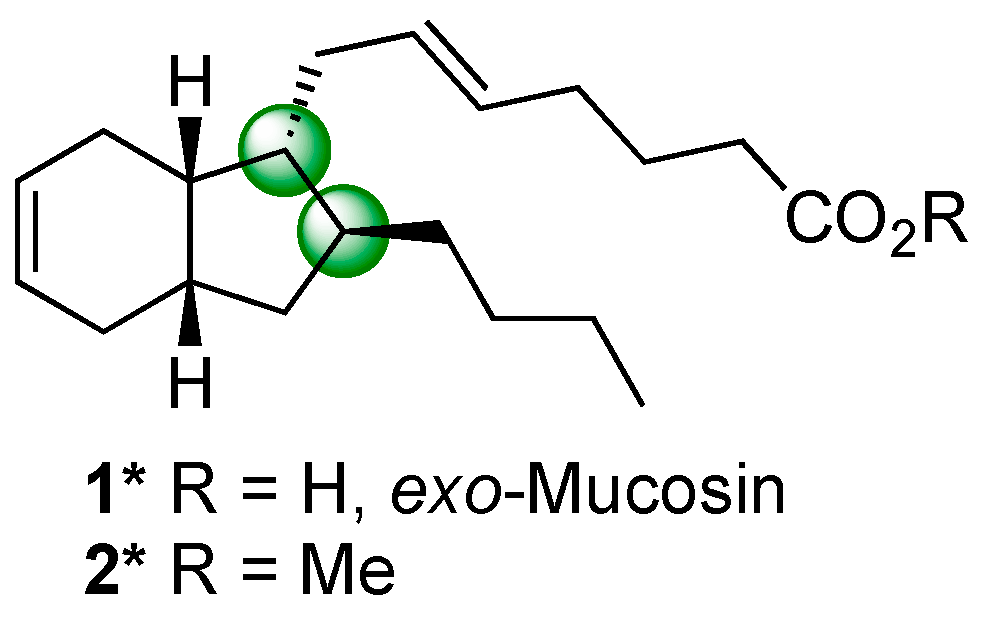{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Casapullo et al. [35] | Whitby et al. [37] | Previous Work [36] | This Work |
|---|---|---|---|---|
| 1 | 174.2 | 174.2 | 174.2 | 174.2 |
| 2 | 130.0 | 130.3 | 130.4 | 131.2 |
| 3 | 129.8 | 129.8 | 129.9 | 129.0 |
| 4 | 127.0 | 127.3 | 126.3 | 125.3 |
| 5 | 127.0 | 127.1 | 126.1 | 125.1 |
| 6 | 52.1 | 52.2 | 51.4 | 51.6 |
| 7 | 51.4 | 51.4 | 51.0 | 51.4 |
| 8 | 47.1 | 47.2 | 44.0 | 41.3 |
| 9 | 42.1 | 42.3 | 40.3 | 37.2 |
| 10 | 39.9 | 40.1 | 38.1 | 36.2 |
| 11 | 36.7 | 37.0 | 37.7 | 35.5 |
| 12 | 36.5 | 36.74 | 37.1 | 35.4 |
| 13 | 36.4 | 36.68 | 34.9 | 33.4 |
| 14 | 33.2 | 33.4 | 33.4 | 33.0 |
| 15 | 32.0 | 32.4 | 31.9 | 31.9 |
| 16 | 31.7 | 31.9 | 31.0 | 31.0 |
| 17 | 31.5 | 31.6 | 27.8 | 26.9 |
| 18 | 30.7 § | 30.7 | 27.7 | 24.7 |
| 19 | 24.5 | 24.7 | 24.8 | 23.0 |
| 20 | 22.6 | 22.9 | 22.9 | 21.7 |
| 21 | 13.8 | 14.1 | 14.1 | 14.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonsen, S.G.; Gallantree-Smith, H.; Görbitz, C.H.; Hansen, T.V.; Stenstrøm, Y.H.; Nolsøe, J.M.J. Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin. Molecules 2017, 22, 1720. https://doi.org/10.3390/molecules22101720
Antonsen SG, Gallantree-Smith H, Görbitz CH, Hansen TV, Stenstrøm YH, Nolsøe JMJ. Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin. Molecules. 2017; 22(10):1720. https://doi.org/10.3390/molecules22101720
Chicago/Turabian StyleAntonsen, Simen G., Harrison Gallantree-Smith, Carl Henrik Görbitz, Trond Vidar Hansen, Yngve H. Stenstrøm, and Jens M. J. Nolsøe. 2017. "Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin" Molecules 22, no. 10: 1720. https://doi.org/10.3390/molecules22101720