Integrative Pathway Analysis of Genes and Metabolites Reveals Metabolism Abnormal Subpathway Regions and Modules in Esophageal Squamous Cell Carcinoma

Abstract
:
1. Introduction
2. Results
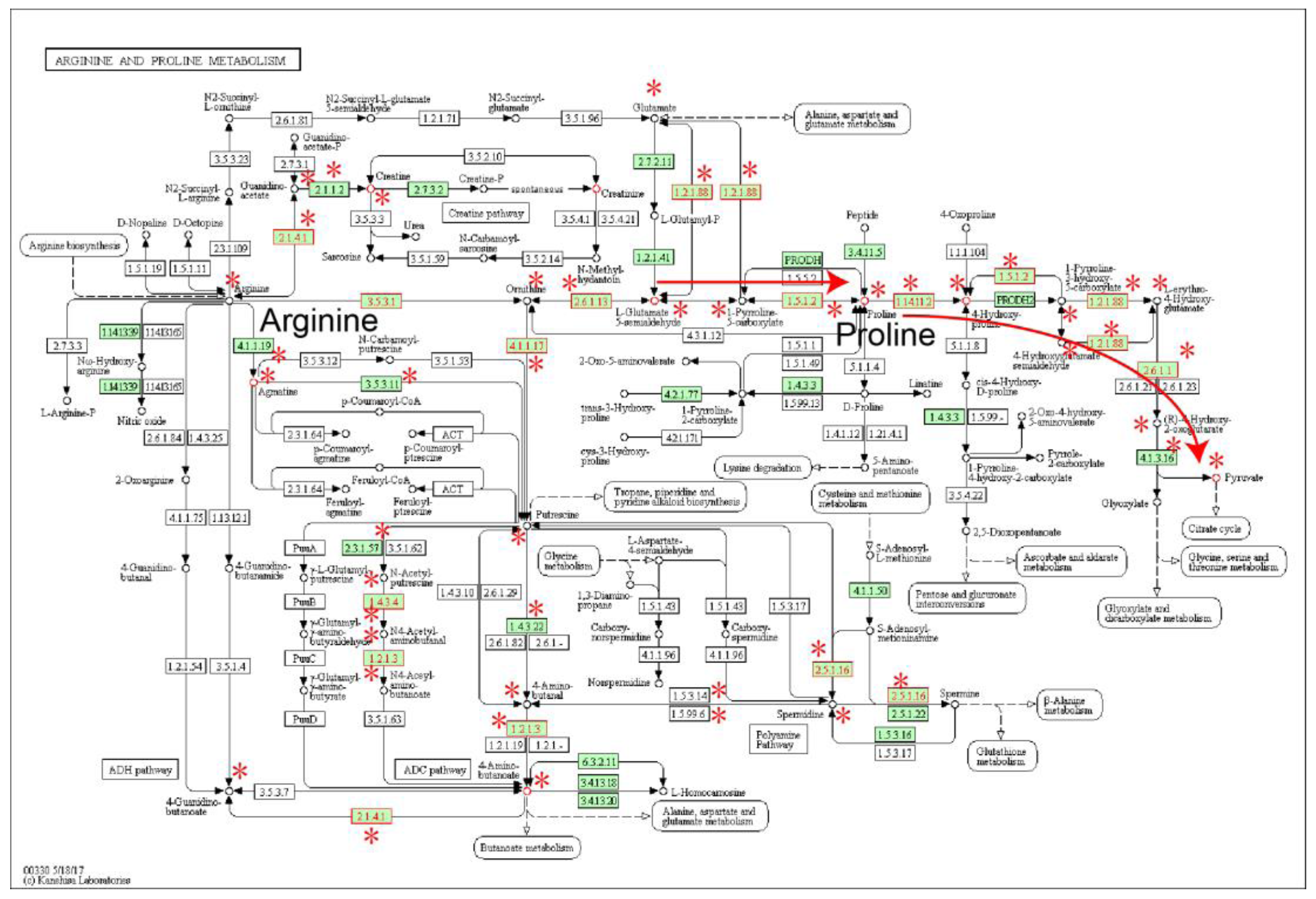
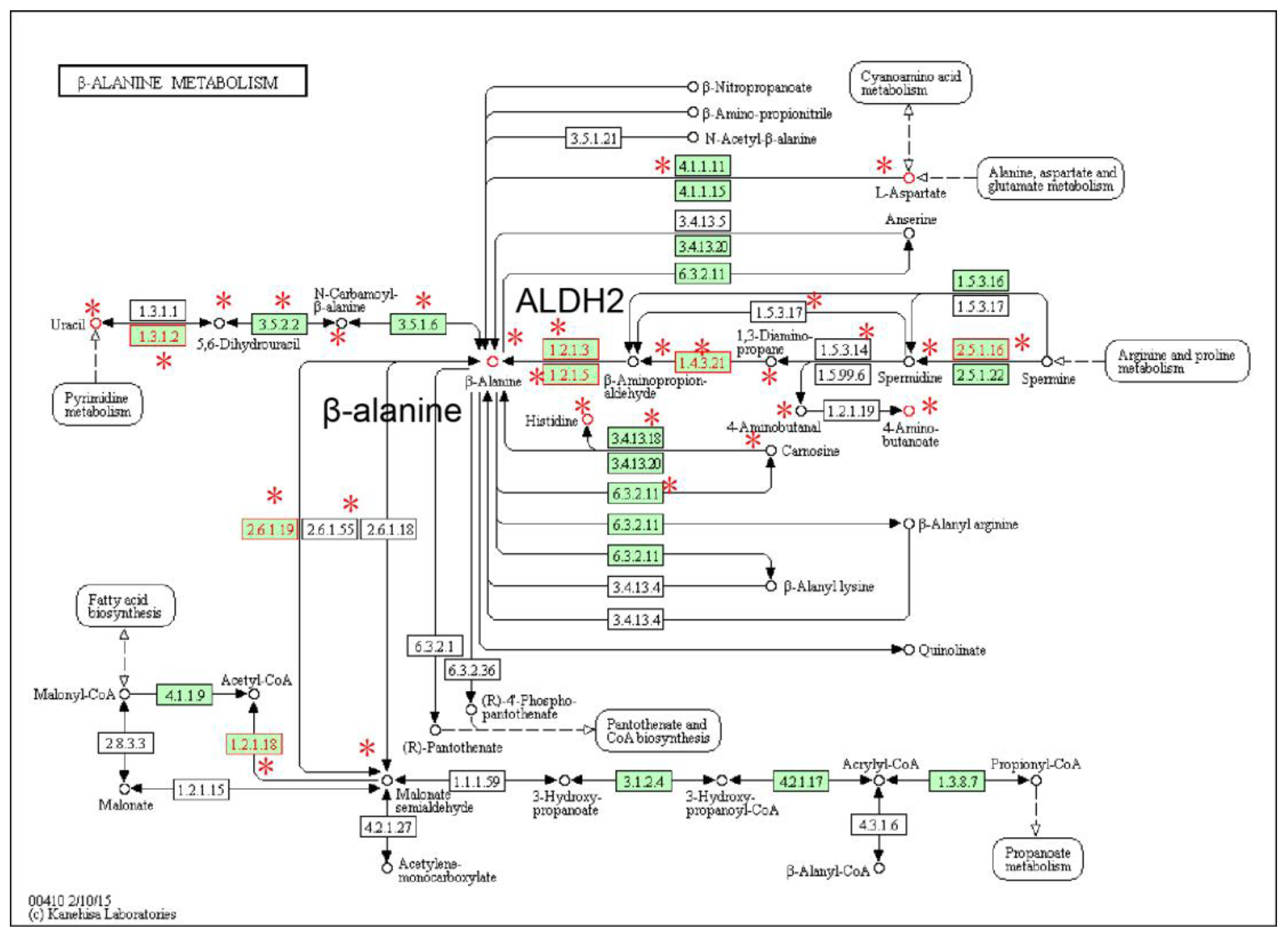
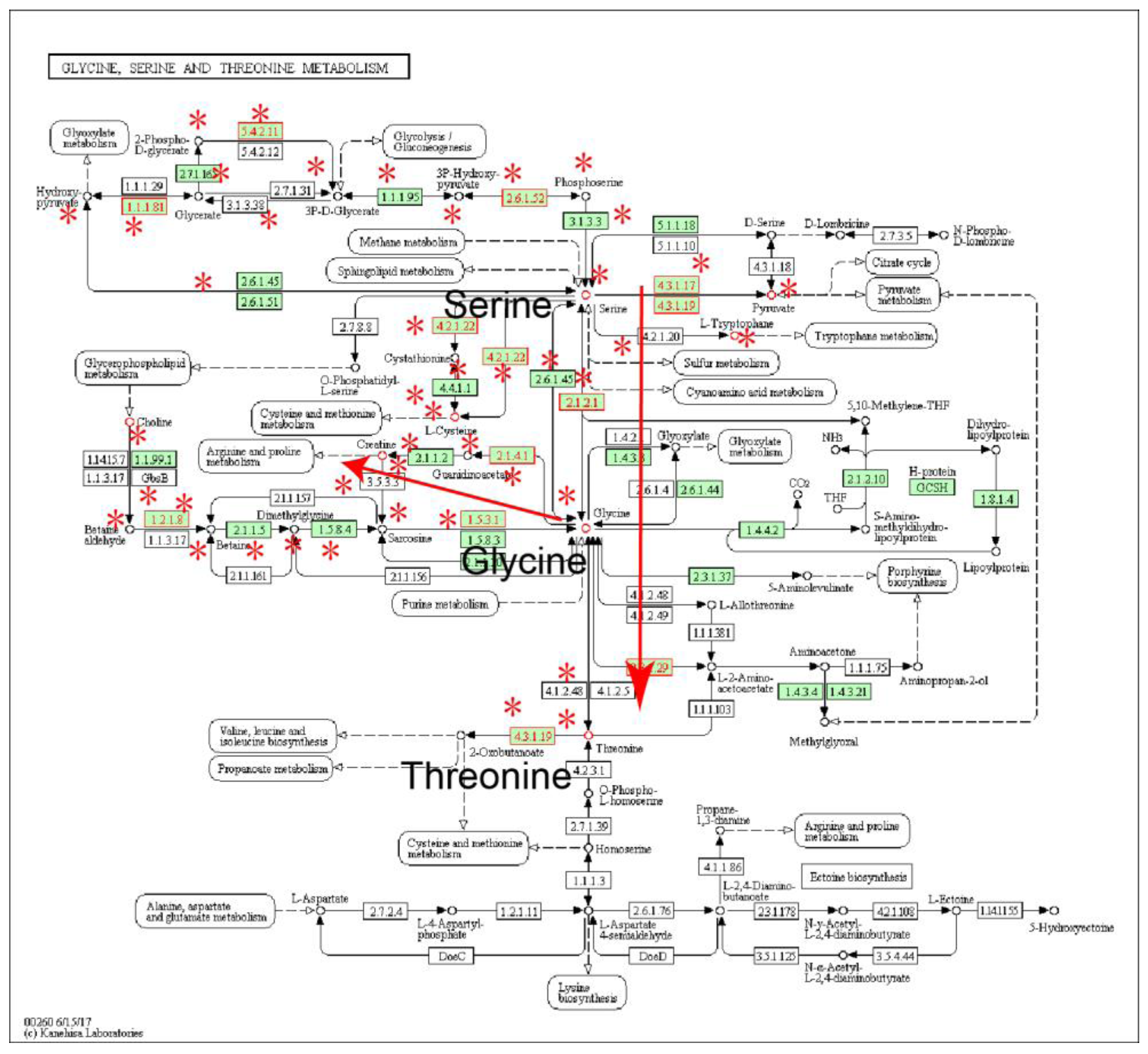
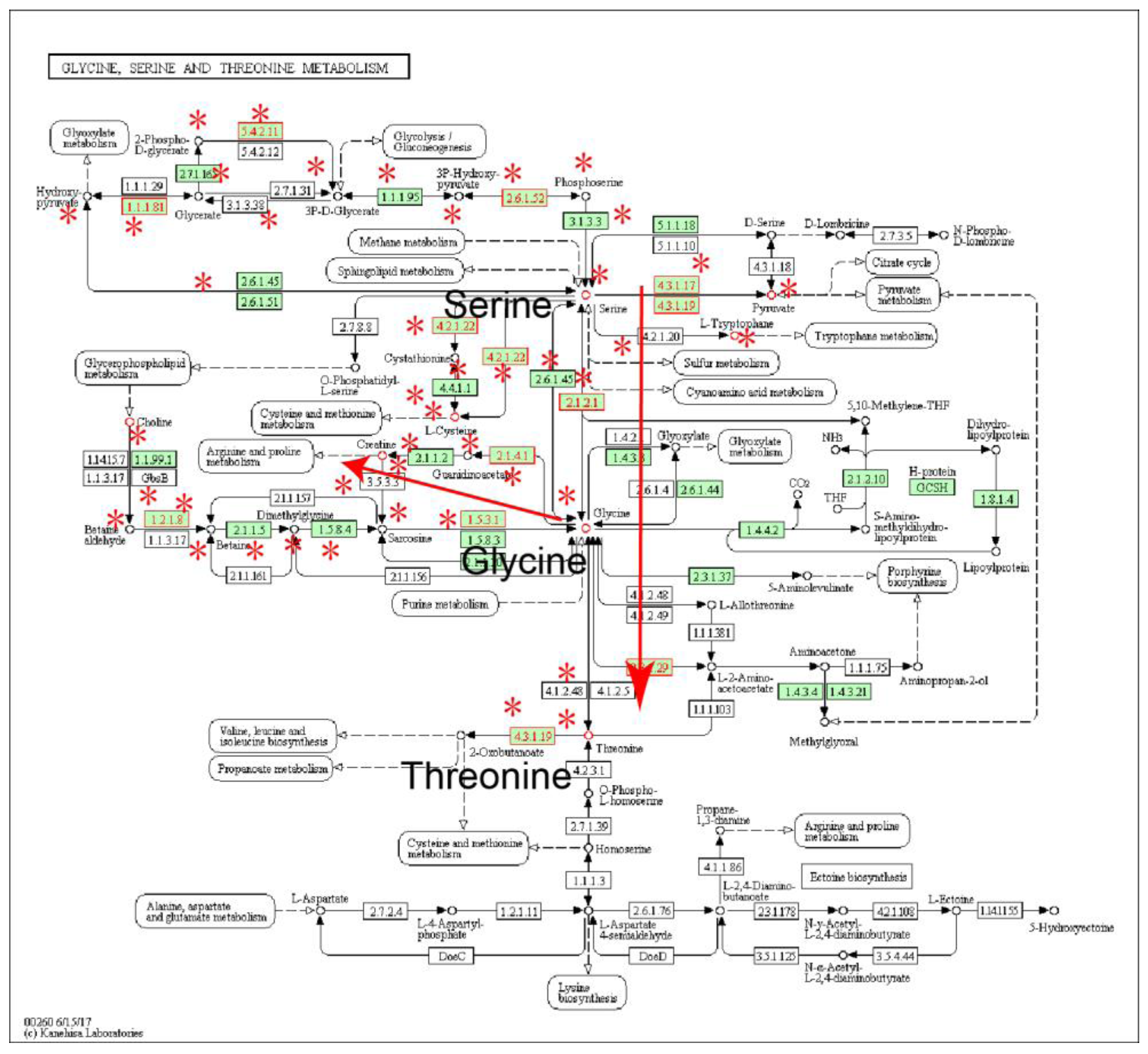
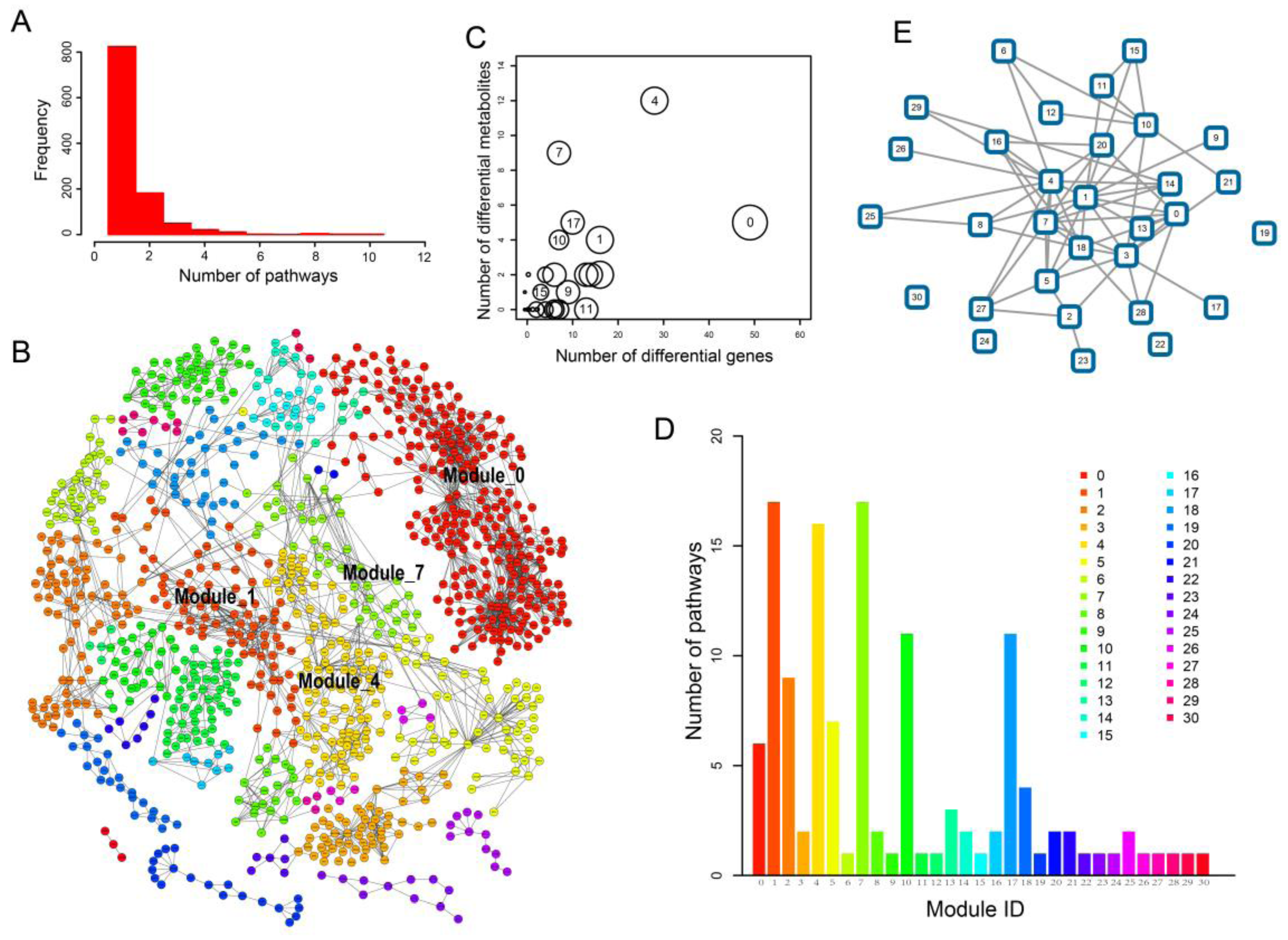
2.1. Identification of Metabolic Subpathways Associated with ESCC
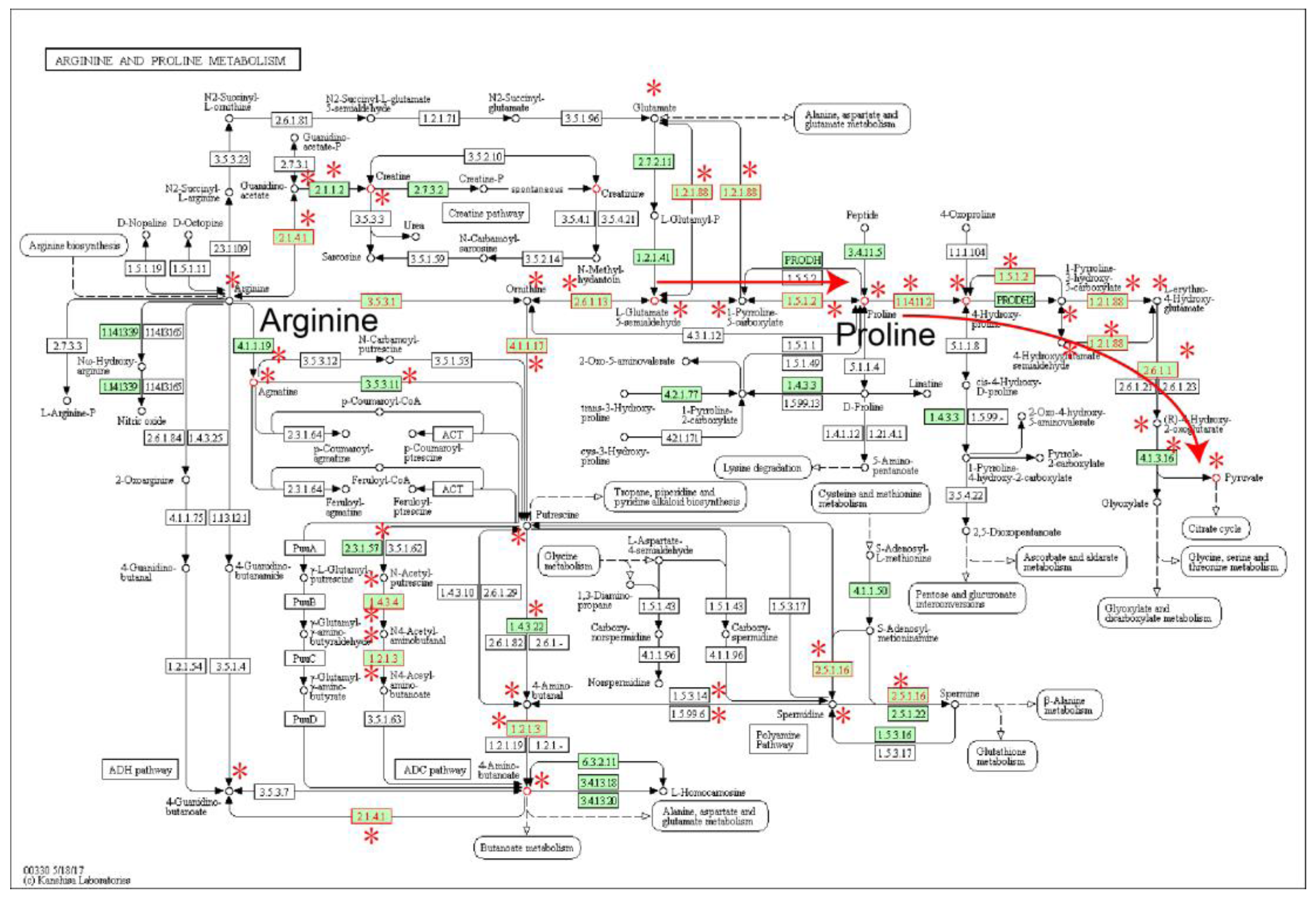
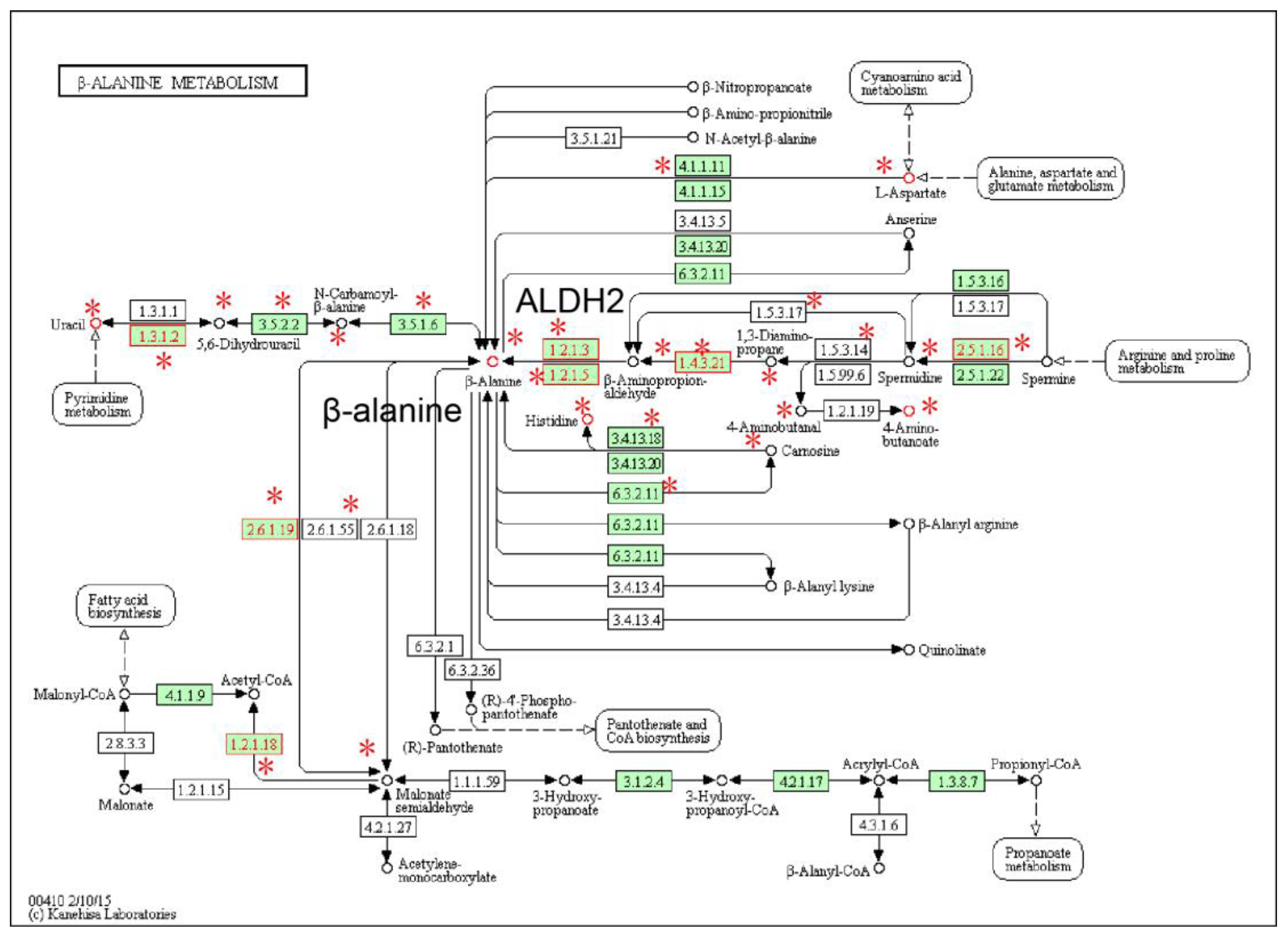
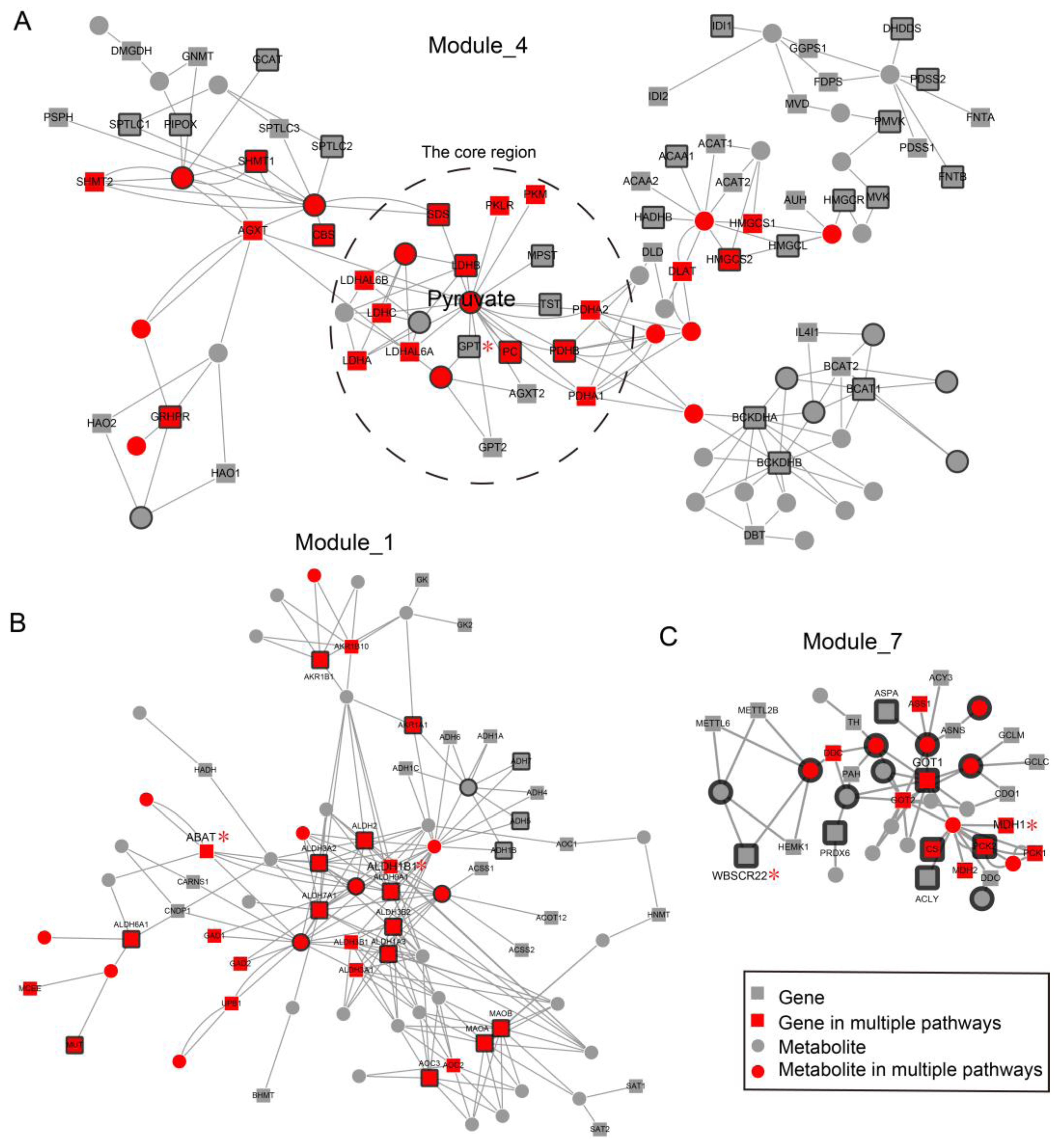
2.2. Local Region Analysis of Subpathways Revealed Important Functional Genes and Metabolites
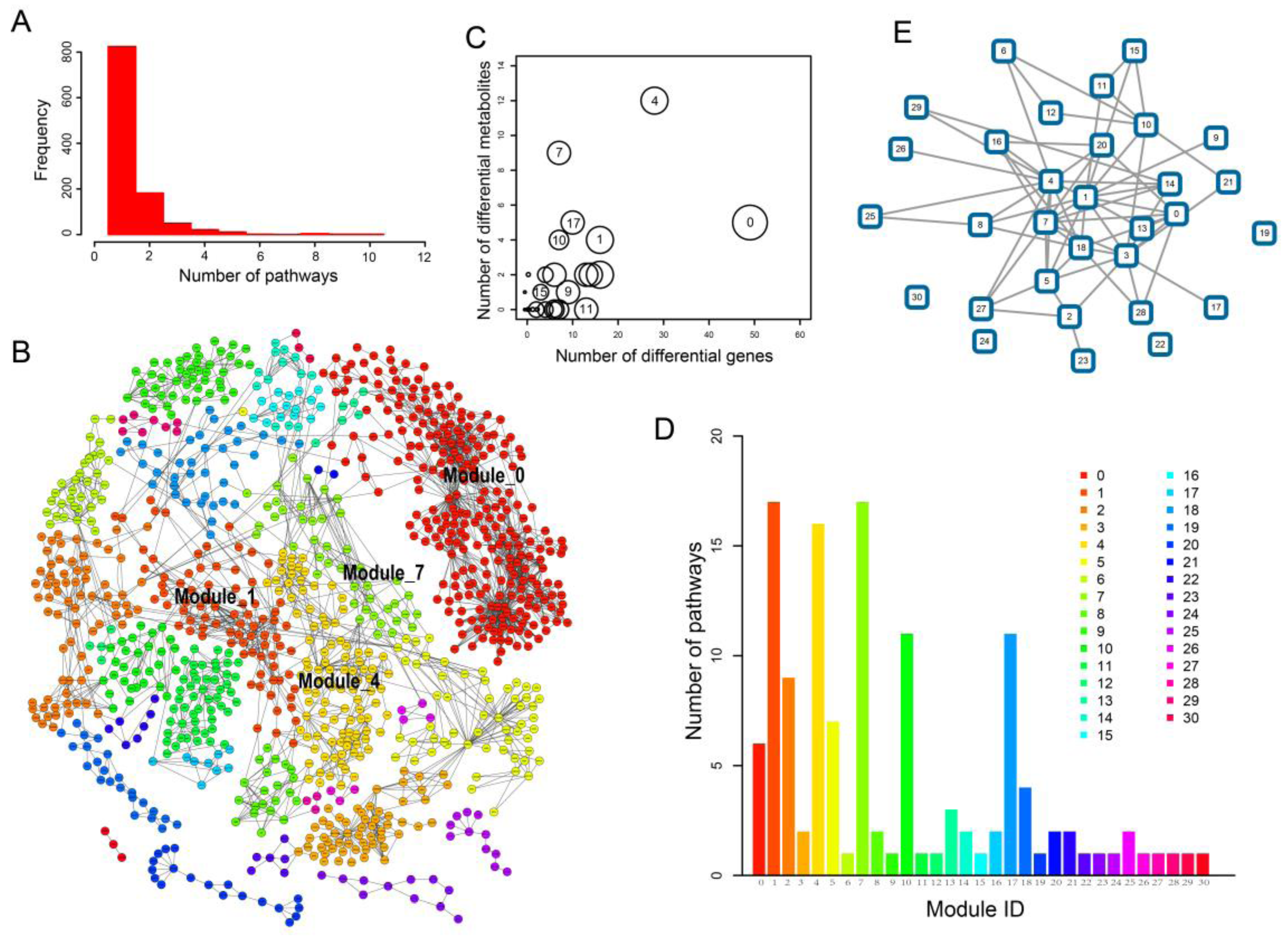
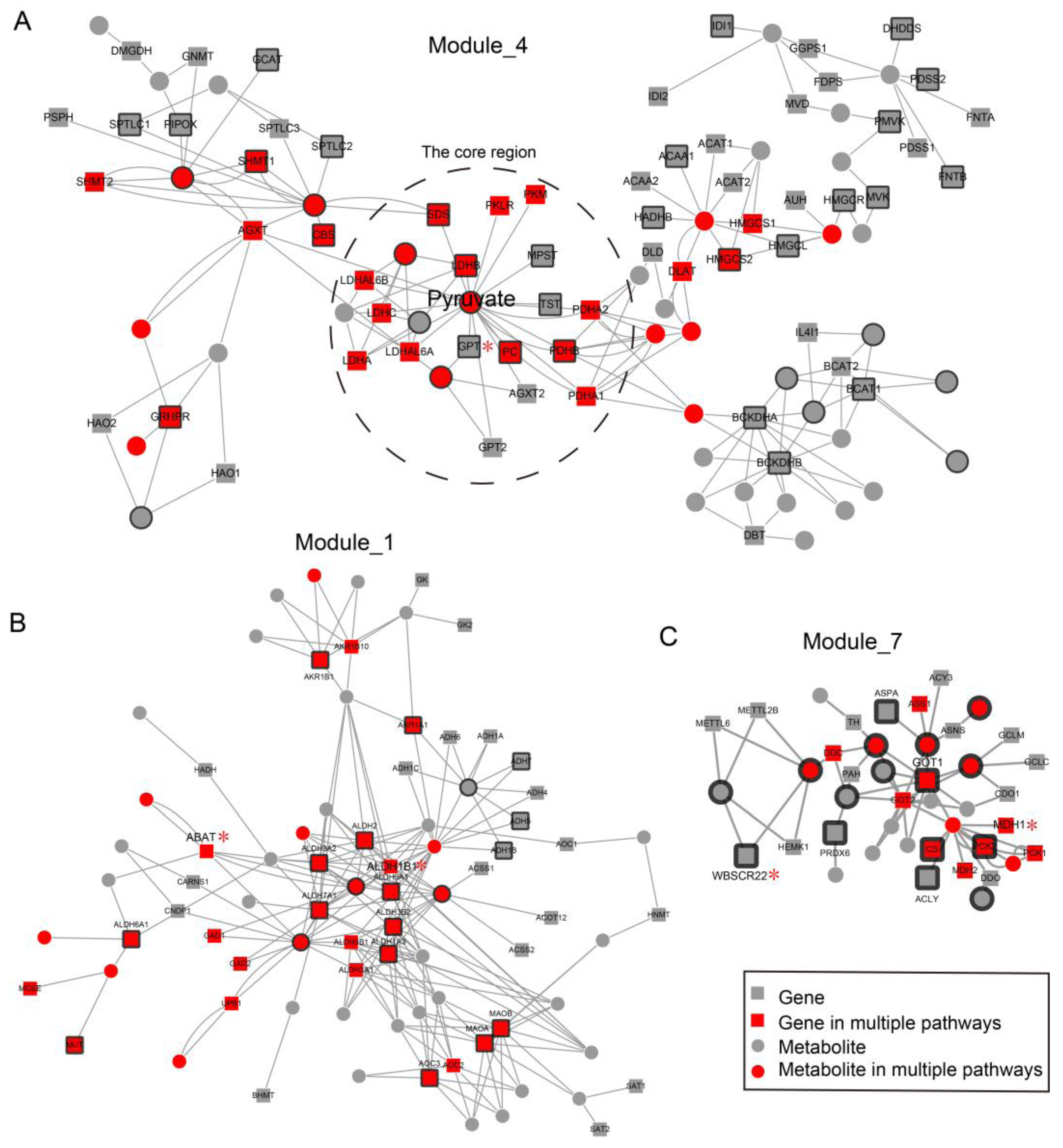
2.3. Network Analysis Revealed Important Functional Modules and Genes in ESCC
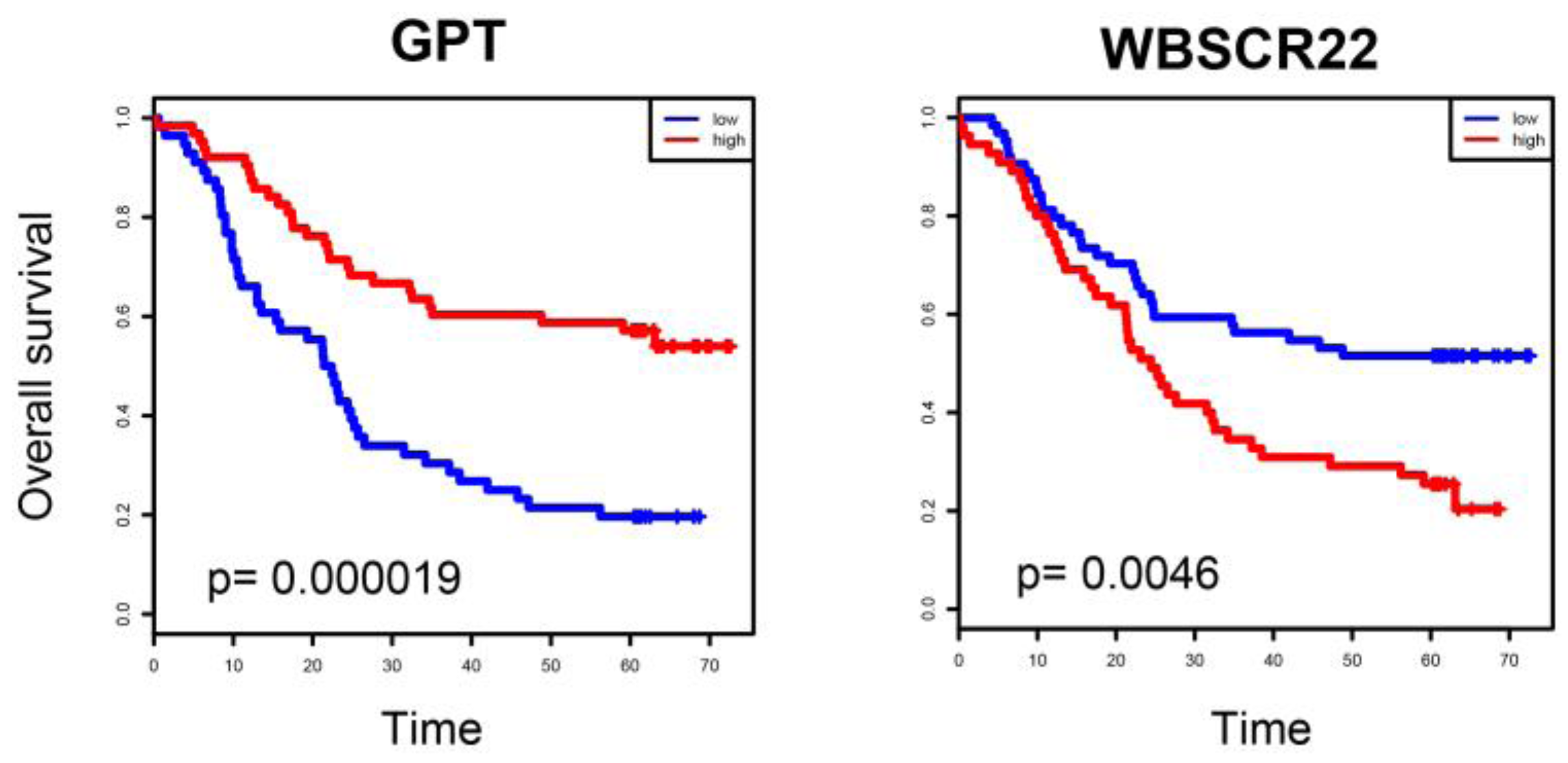
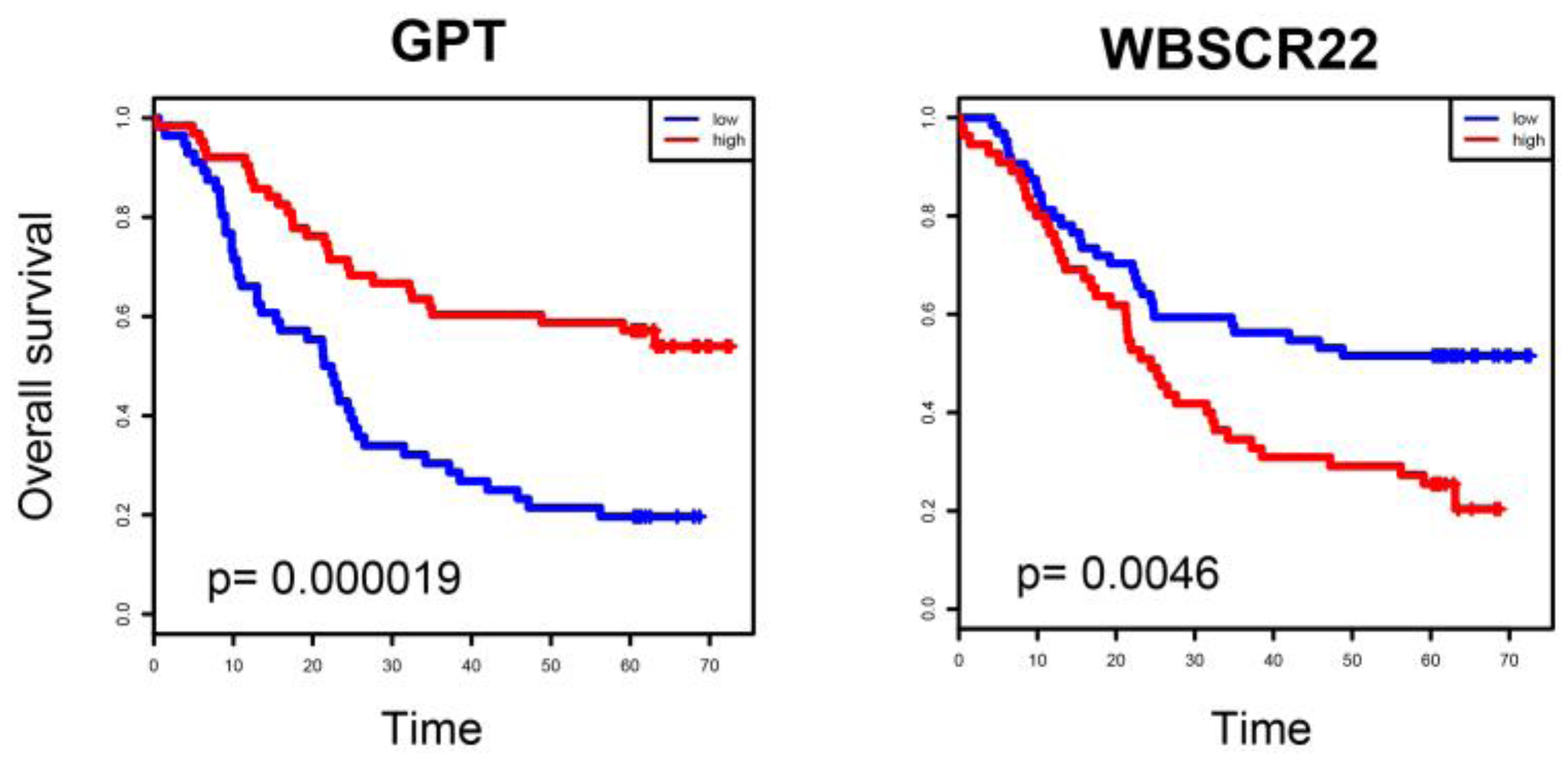
2.4. ESCC-Related Prognostic Genes Mainly Located in Peripheral Regions of Network
2.5. Prognostic Genes that Appeared in the Center Modules of the Network
3. Discussion
4. Materials and Methods
4.1. Metabolites Highly Related with ESCC
4.2. Gene Expression Profiles and Differential Genes Related with ESCC
4.3. Integrative Pathway Analysis Pipeline
4.4. Identification of Network Modules
4.5. Survival Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, J.; Chen, Y.; Zhang, R.; Song, Y.; Cao, J.; Bi, N.; Wang, J.; He, J.; Bai, J.; Dong, L.; et al. Global and targeted metabolomics of esophageal squamous cell carcinoma discovers potential diagnostic and therapeutic biomarkers. Mol. Cell. Proteom. 2013, 12, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Peng, Y.; Li, X.; Wang, Y.; Pan, E.; Guo, W.; Pu, Y.; Yin, L. Identification of plasma metabolomic profiling for diagnosis of esophageal squamous-cell carcinoma using an UPLC/TOF/MS platform. Int. J. Mol. Sci. 2013, 14, 8899–8911. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; Huang, G.W.; Wu, Z.Y.; Xu, Y.J.; Li, X.C.; Xue, Y.J.; Zhu, Y.; Zhao, J.M.; Li, M.; Zhang, J.; et al. Integrative analyses of transcriptome sequencing identify novel functional lncRNAs in esophageal squamous cell carcinoma. Oncogenesis 2017, 6, e297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, Y.; Shang, D.; Yang, H.; Liu, W.; Han, J.; Sun, Z.; Yao, Q.; Zhang, C.; Ma, J.; et al. MPINet: Metabolite pathway identification via coupling of global metabolite network structure and metabolomic profile. BioMed Res. Int. 2014, 2014, 325697. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Han, J.; Yao, Q.; Zou, C.; Xu, Y.; Zhang, C.; Shang, D.; Zhou, L.; Sun, Z.; Li, J.; et al. Subpathway-GM: Identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic Acids Res. 2013, 41, e101. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Liao, Y.; Pan, L. Prediction and validation of association between microRNAs and diseases by multipath methods. Biochim. Biophys. Acta 2016, 1860, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Alaimo, S.; Marceca, G.P.; Ferro, A.; Pulvirenti, A. Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach. Non-Coding RNA 2017, 3, 20. [Google Scholar] [CrossRef]
- Togashi, Y.; Arao, T.; Kato, H.; Matsumoto, K.; Terashima, M.; Hayashi, H.; de Velasco, M.A.; Fujita, Y.; Kimura, H.; Yasuda, T.; et al. Frequent amplification of ORAOV1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ROS production. Oncotarget 2014, 5, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Qiao, F.; Chen, L.; Lu, C.; Xu, L.; Gao, X. Serum metabolomic signatures of lymph node metastasis of esophageal squamous cell carcinoma. J. Proteome Res. 2014, 13, 4091–4103. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, M.; Enkhtaivan, G.; Mistry, B.; Patel, R.V.; Moon, S.; Kim, D.H. β-Alanine intercede metabolic recovery for amelioration of human cervical and renal tumors. Amino Acids 2017, 49, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Kamatani, Y.; Takahashi, A.; Usami, M.; Hosono, N.; Kawaguchi, T.; Tsunoda, T.; Kamatani, N.; Kubo, M.; Nakamura, Y.; et al. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology 2009, 137, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; McKay, J.D.; Curado, M.P.; Oliveira, J.C.; Koifman, S.; Koifman, R.; Zaridze, D.; Shangina, O.; Wunsch-Filho, V.; Eluf-Neto, J.; et al. Multiple ADH genes are associated with upper aerodigestive cancers. Nat. Genet. 2008, 40, 707–709. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; Truong, T.; Gaborieau, V.; Chabrier, A.; Chuang, S.C.; Byrnes, G.; Zaridze, D.; Shangina, O.; Szeszenia-Dabrowska, N.; Lissowska, J.; et al. A genome-wide association study of upper aerodigestive tract cancers conducted within the INHANCE consortium. PLoS Genet. 2011, 7, e1001333. [Google Scholar] [CrossRef]
- Islami, F.; Fedirko, V.; Tramacere, I.; Bagnardi, V.; Jenab, M.; Scotti, L.; Rota, M.; Corrao, G.; Garavello, W.; Schuz, J.; et al. Alcohol drinking and esophageal squamous cell carcinoma with focus on light-drinkers and never-smokers: A systematic review and meta-analysis. Int. J. Cancer 2011, 129, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Guo, W.; Zhang, X.F.; Li, Y.; Wang, N.; Ge, H.; Wei, L.Z.; Wen, D.G.; Zhang, J.H. Correlations between serine hydroxymethyltransferase1 C1420T polymorphisms and susceptibilities to esophageal squamous cell carcinoma and gastric cardiac adenocarcinoma. Chin. J. Cancer 2006, 25, 281–286. [Google Scholar]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tong, L.; Wei, J.; Pan, W.; Li, L.; Ge, Y.; Zhou, L.; Yuan, Q.; Zhou, C.; Yang, M. The ALDH7A1 genetic polymorphisms contribute to development of esophageal squamous cell carcinoma. Tumour Biol. 2014, 35, 12665–12670. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kraft, P.; Zhai, K.; Chang, J.; Wang, Z.; Li, Y.; Hu, Z.; He, Z.; Jia, W.; Abnet, C.C.; et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat. Genet. 2012, 44, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, Y.; Ohashi, S.; Amanuma, Y.; Nakai, Y.; Tsurumaki, M.; Kikuchi, O.; Miyamoto, S.; Oyama, T.; Kawamoto, T.; Chiba, T.; et al. Impairment of aldehyde dehydrogenase 2 increases accumulation of acetaldehyde-derived DNA damage in the esophagus after ethanol ingestion. Am. J. Cancer Res. 2014, 4, 279–284. [Google Scholar] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed]
- Scuoppo, C.; Miething, C.; Lindqvist, L.; Reyes, J.; Ruse, C.; Appelmann, I.; Yoon, S.; Krasnitz, A.; Teruya-Feldstein, J.; Pappin, D.; et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature 2012, 487, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Ramus, S.J.; Tyrer, J.; Lee, A.; Shen, H.C.; Beesley, J.; Lawrenson, K.; McGuffog, L.; Healey, S.; Lee, J.M.; et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015, 47, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.F.; Razvi, M.; Chen, H.; Washington, K.; Roessner, A.; Schneider-Stock, R.; El-Rifai, W. DNA hypermethylation regulates the expression of members of the Mu-class glutathione S-transferases and glutathione peroxidases in Barrett's adenocarcinoma. Gut 2009, 58, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Stiebler, A.C.; Freitag, J.; Schink, K.O.; Stehlik, T.; Tillmann, B.A.; Ast, J.; Bolker, M. Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in Fungi and animals. PLoS Genet. 2014, 10, e1004685. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Zhang, S.; Ding, N.; Zeng, X.; Zhang, X. Complex Network Clustering by a Multi-objective Evolutionary Algorithm Based on Decomposition and Membrane Structure. Sci. Rep. 2016, 6, 33870. [Google Scholar] [CrossRef] [PubMed]
- Szalay-Beko, M.; Palotai, R.; Szappanos, B.; Kovacs, I.A.; Papp, B.; Csermely, P. ModuLand plug-in for Cytoscape: Determination of hierarchical layers of overlapping network modules and community centrality. Bioinformatics 2012, 28, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xue, R.; Lu, C.; Deng, C.; Liu, T.; Zeng, H.; Wang, Q.; Shen, X. Metabolomic study for diagnostic model of oesophageal cancer using gas chromatography/mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 3111–3117. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Hasim, A.; Mamtimin, B.; Kong, B.; Zhang, H.P.; Sheyhidin, I. Plasma free amino acid profiling of esophageal cancer using high-performance liquid chromatography spectroscopy. World J. Gastroenterol. 2014, 20, 8653–8659. [Google Scholar] [CrossRef] [PubMed]
- Hasim, A.; Ma, H.; Mamtimin, B.; Abudula, A.; Niyaz, M.; Zhang, L.W.; Anwer, J.; Sheyhidin, I. Revealing the metabonomic variation of EC using 1H-NMR spectroscopy and its association with the clinicopathological characteristics. Mol. Biol. Rep. 2012, 39, 8955–8964. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, J.; Chen, L.; Deng, P.; Bu, Q.; Xiang, P.; Li, M.; Lu, W.; Xu, Y.; Lin, H.; et al. 1H-NMR based metabonomic profiling of human esophageal cancer tissue. Mol. Cancer 2013, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Miao, Y.; Wang, Q.; Jiang, W.; Xu, C.; Li, J.; Han, J.; Zhang, F.; Gong, B.; et al. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 2009, 37, e131. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Z.; Tian, L.; Zhou, C.; He, M.Y.; Gao, Y.; Wang, S.; Zhou, F.; Shi, S.; Feng, X.; et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 2014, 63, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Berty, H.P.; Shi, H.; Lyons-Weiler, J. Determining the statistical significance of survivorship prediction models. J. Eval. Clin. Pract. 2010, 16, 155–165. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subpathway Id | Pathway Name | p Value | FDR |
|---|---|---|---|
| path:00330_1 | Arginine and proline metabolism | 4.93 × 10−12 | 4.19 × 10−10 |
| path:00280_1 | Valine, leucine and isoleucine degradation | 3.12 × 10−7 | 1.32 × 10−5 |
| path:00410_2 | beta-Alanine metabolism | 1.19 × 10−6 | 3.38 × 10−5 |
| path:00260_1 | Glycine, serine and threonine metabolism | 2.16 × 10−6 | 4.60 × 10−5 |
| path:00010_1 | Glycolysis/Gluconeogenesis | 1.45 × 10−5 | 0.00021 |
| path:00270_1 | Cysteine and methionine metabolism | 1.59 × 10−5 | 0.00021 |
| path:00250_2 | Alanine, aspartate and glutamate metabolism | 1.99 × 10−5 | 0.00021 |
| path:00250_1 | Alanine, aspartate and glutamate metabolism | 2.05 × 10−5 | 0.00021 |
| path:00604_1 | Glycosphingolipid biosynthesis—ganglio series | 2.80 × 10−5 | 0.00026 |
| path:00531_2 | Glycosaminoglycan degradation | 3.50 × 10−5 | 0.00029 |
| path:00240_1 | Pyrimidine metabolism | 5.39 × 10−5 | 0.00041 |
| path:00340_1 | Histidine metabolism | 7.77 × 10−5 | 0.00053 |
| path:00052_2 | Galactose metabolism | 8.18 × 10−5 | 0.00053 |
| path:00520_1 | Amino sugar and nucleotide sugar metabolism | 0.00011 | 0.00067 |
| path:00562_1 | Inositol phosphate metabolism | 0.00011 | 0.00067 |
| path:00620_1 | Pyruvate metabolism | 0.00015 | 0.00081 |
| path:00640_1 | Propanoate metabolism | 0.00028 | 0.0014 |
| path:00230_1 | Purine metabolism | 0.00031 | 0.0014 |
| path:00630_1 | Glyoxylate and dicarboxylate metabolism | 0.00040 | 0.0017 |
| path:00360_1 | Phenylalanine metabolism | 0.00041 | 0.0017 |
| path:00600_1 | Sphingolipid metabolism | 0.00046 | 0.0018 |
| path:00532_1 | Glycosaminoglycan biosynthesis—chondroitin sulfate | 0.00048 | 0.0018 |
| path:00510_1 | N-Glycan biosynthesis | 0.00063 | 0.0023 |
| path:00480_1 | Glutathione metabolism | 0.00073 | 0.0025 |
| path:00030_1 | Pentose phosphate pathway | 0.0010 | 0.0035 |
| path:00100_4 | Steroid biosynthesis | 0.0011 | 0.0038 |
| path:00020_1 | Citrate cycle (TCA cycle) | 0.0013 | 0.0042 |
| path:00062_2 | Fatty acid elongation | 0.0014 | 0.0043 |
| path:00350_2 | Tyrosine metabolism | 0.0014 | 0.0043 |
| path:00561_1 | Glycerolipid metabolism | 0.0016 | 0.0046 |
| path:00460_1 | Cyanoamino acid metabolism | 0.0020 | 0.0056 |
| path:00053_3 | Ascorbate and aldarate metabolism | 0.0028 | 0.0074 |
| path:00770_2 | Pantothenate and CoA biosynthesis | 0.0031 | 0.0077 |
| path:00900_2 | Terpenoid backbone biosynthesis | 0.0031 | 0.0077 |
| path:00603_1 | Glycosphingolipid biosynthesis—globo series | 0.0032 | 0.0077 |
| path:00650_2 | Butanoate metabolism | 0.0032 | 0.0077 |
| path:00601_1 | Glycosphingolipid biosynthesis—lacto and neolacto series | 0.0034 | 0.0078 |
| path:00760_1 | Nicotinate and nicotinamide metabolism | 0.0043 | 0.0094 |
| path:00590_1 | Arachidonic acid metabolism | 0.0043 | 0.0094 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Wang, Q.; Ma, J.; Shi, S.; Chen, X.; Yang, H.; Han, J. Integrative Pathway Analysis of Genes and Metabolites Reveals Metabolism Abnormal Subpathway Regions and Modules in Esophageal Squamous Cell Carcinoma. Molecules 2017, 22, 1599. https://doi.org/10.3390/molecules22101599
Li C, Wang Q, Ma J, Shi S, Chen X, Yang H, Han J. Integrative Pathway Analysis of Genes and Metabolites Reveals Metabolism Abnormal Subpathway Regions and Modules in Esophageal Squamous Cell Carcinoma. Molecules. 2017; 22(10):1599. https://doi.org/10.3390/molecules22101599
Chicago/Turabian StyleLi, Chunquan, Qiuyu Wang, Jiquan Ma, Shengshu Shi, Xin Chen, Haixiu Yang, and Junwei Han. 2017. "Integrative Pathway Analysis of Genes and Metabolites Reveals Metabolism Abnormal Subpathway Regions and Modules in Esophageal Squamous Cell Carcinoma" Molecules 22, no. 10: 1599. https://doi.org/10.3390/molecules22101599