
Synthetic Strategies for Peroxide Ring Construction in Artemisinin




Abstract
: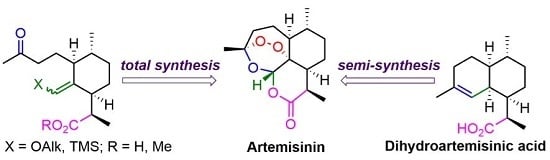
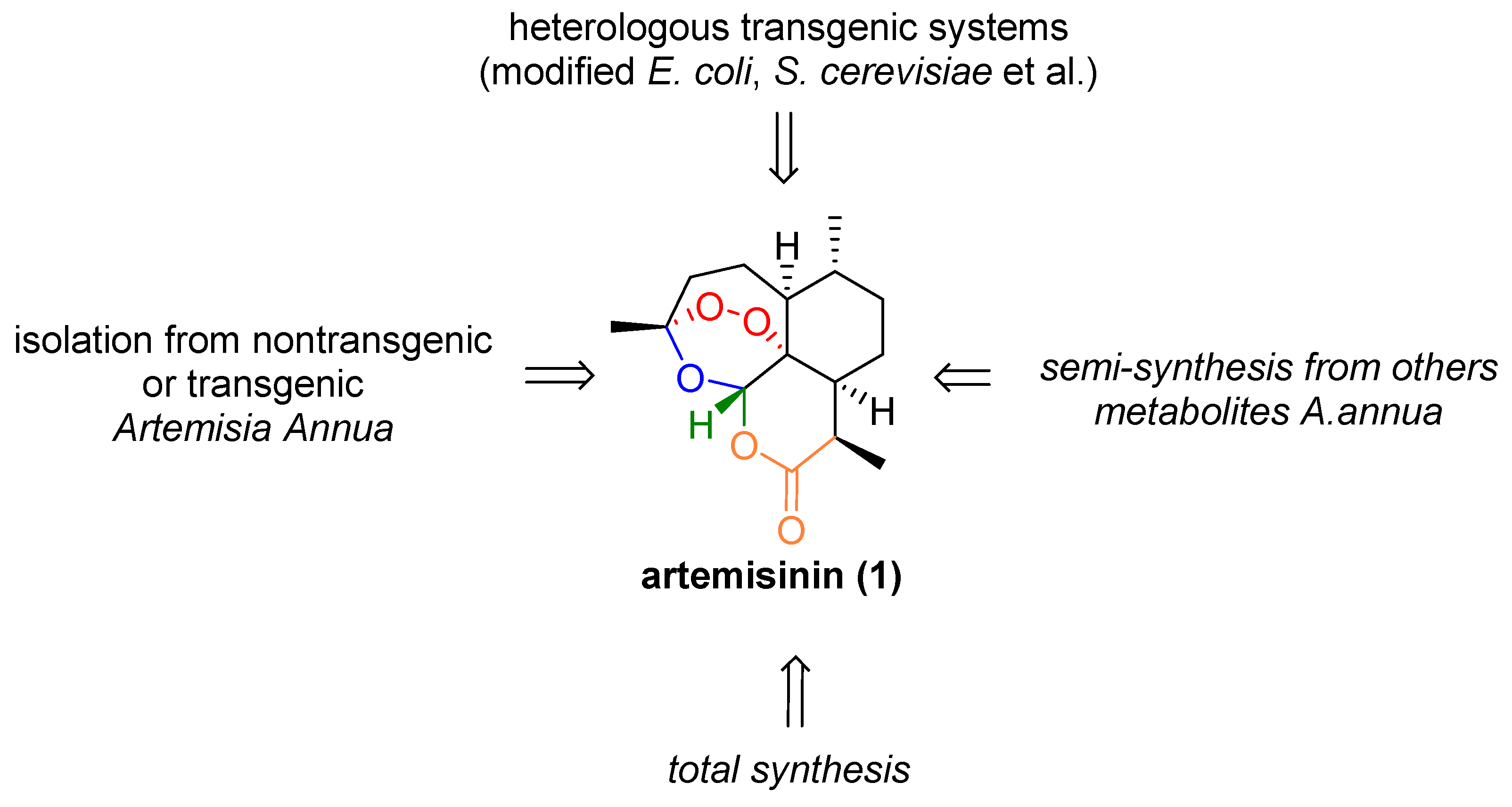
1. Introduction
2. Construction of Artemisinin Peroxide Fragment in Total Synthesis
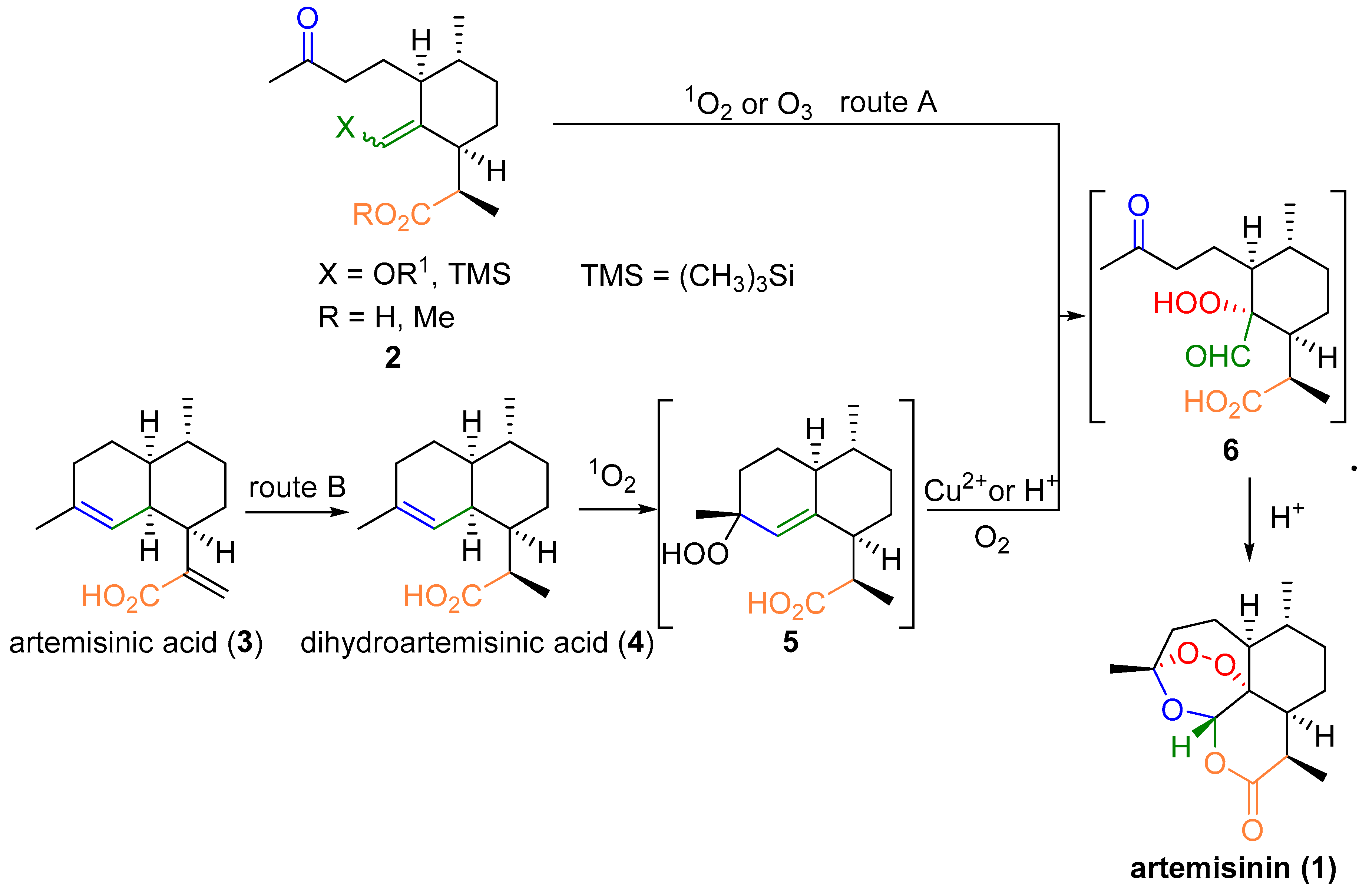
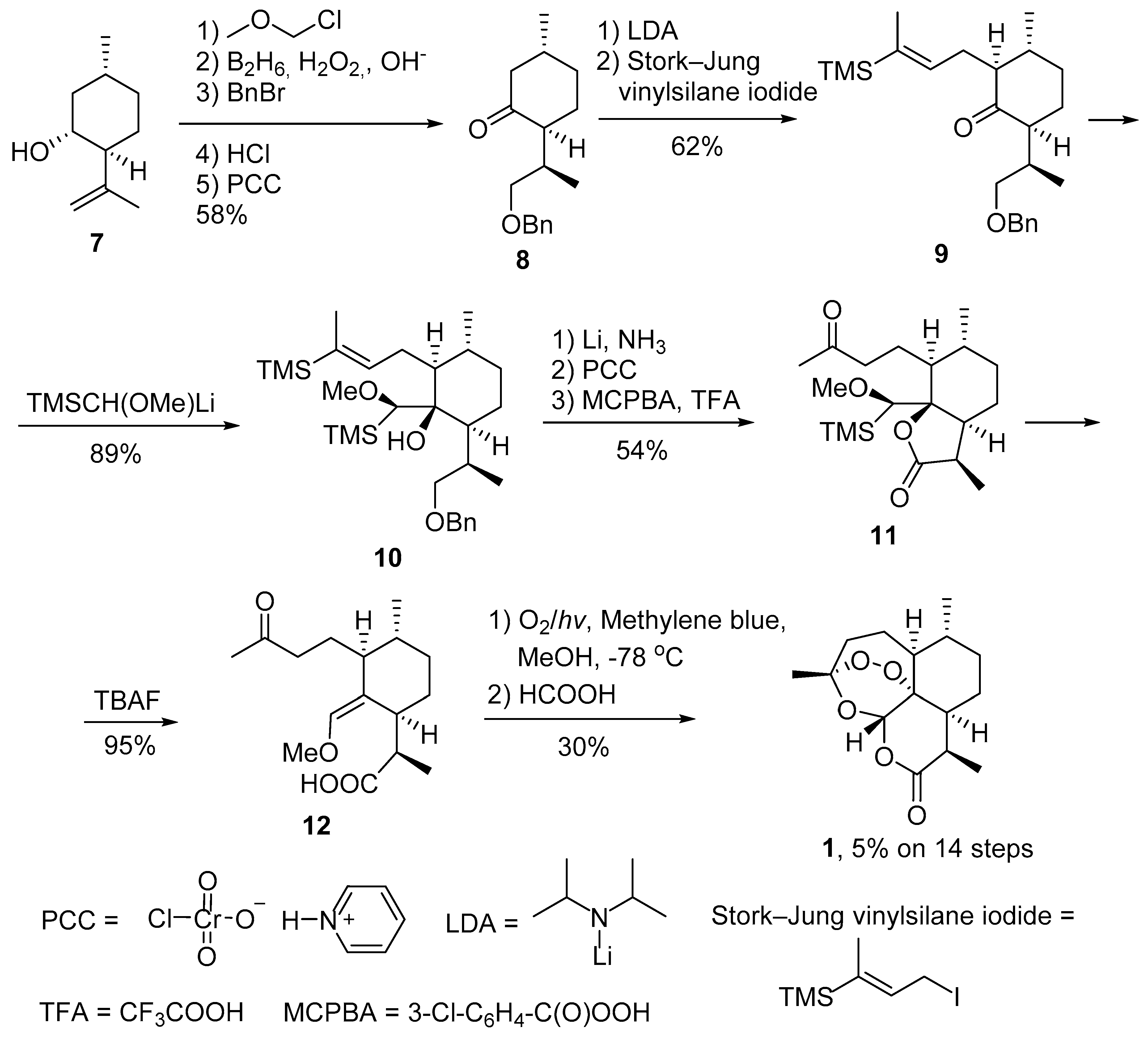
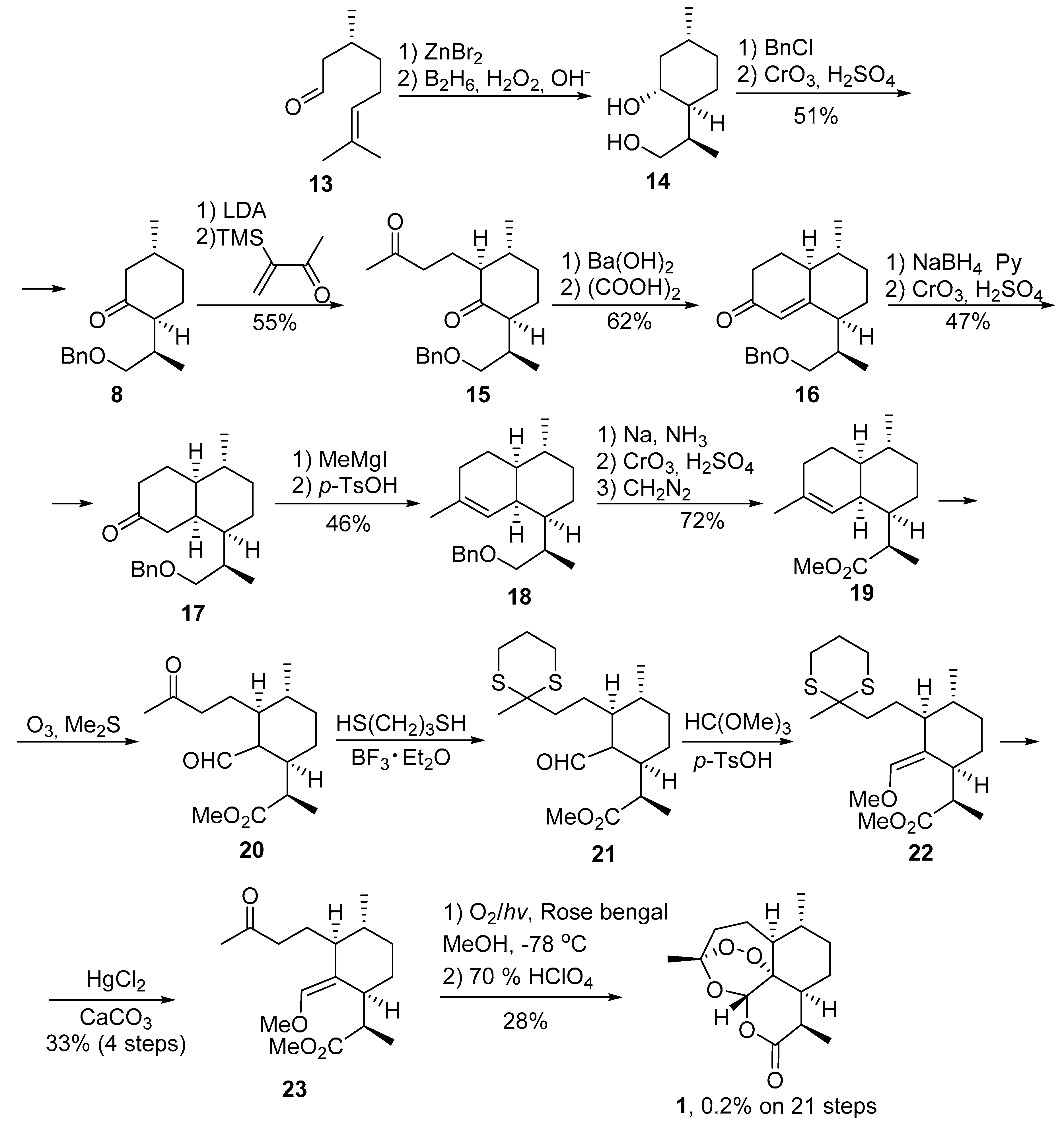
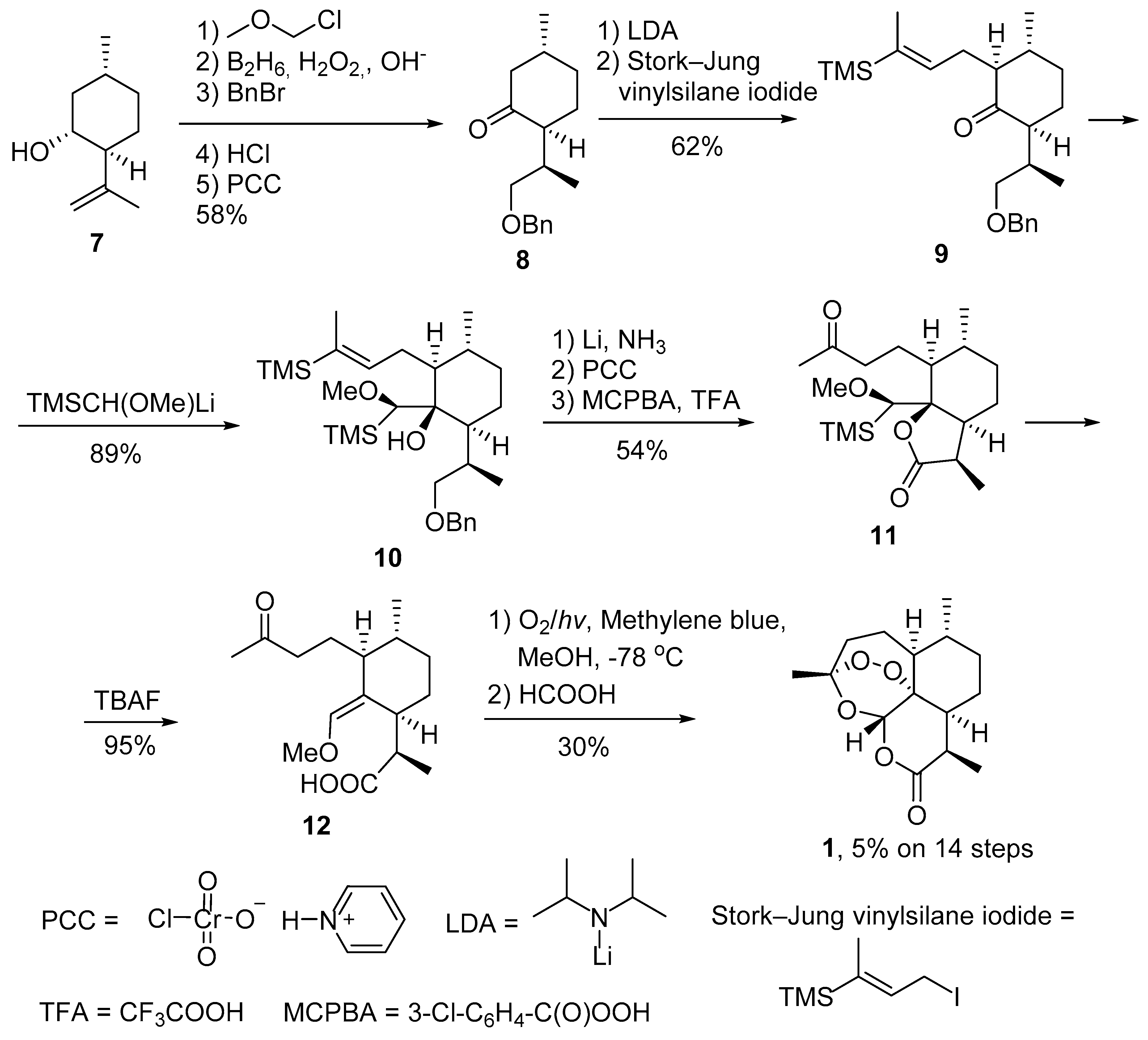
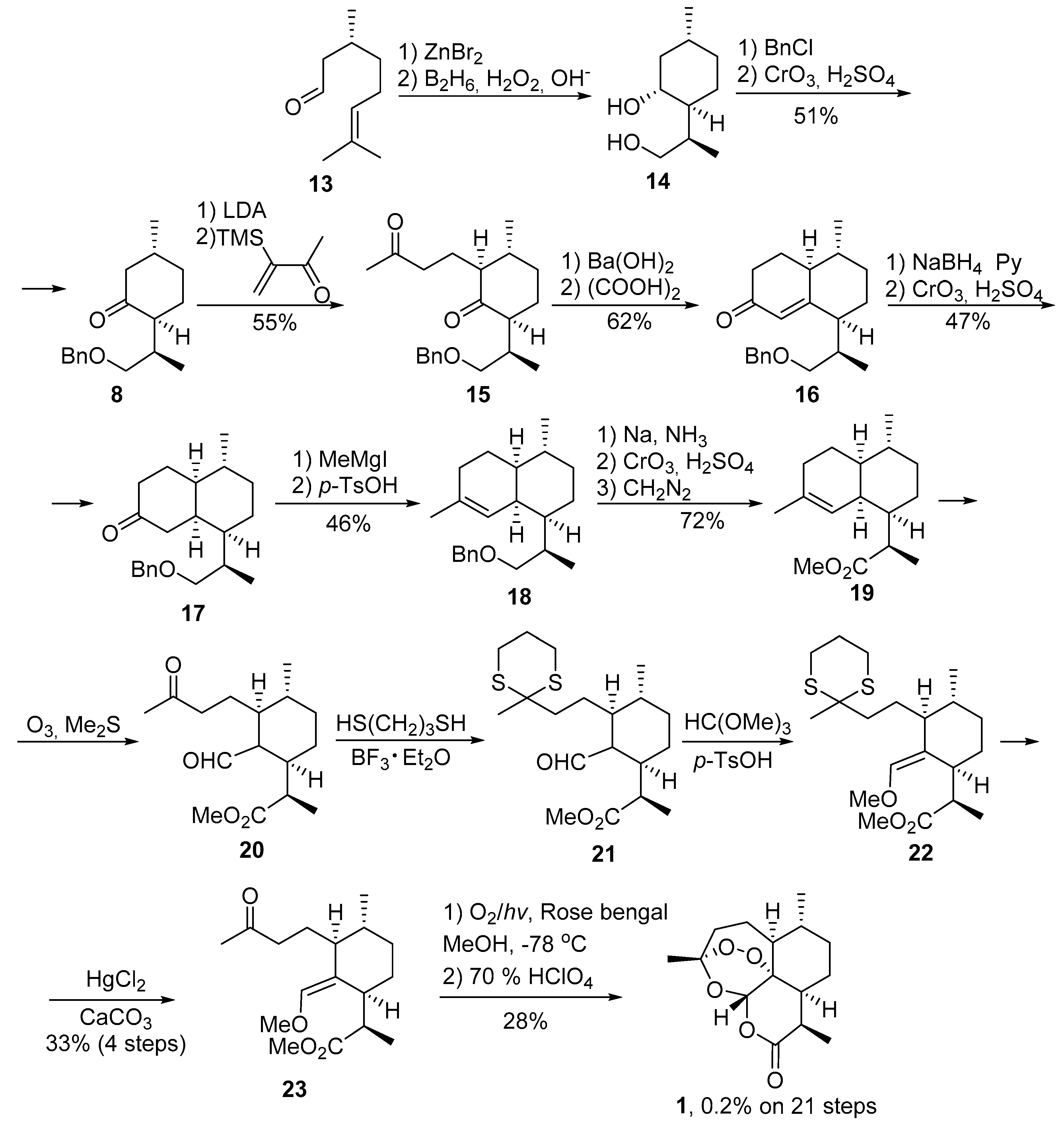
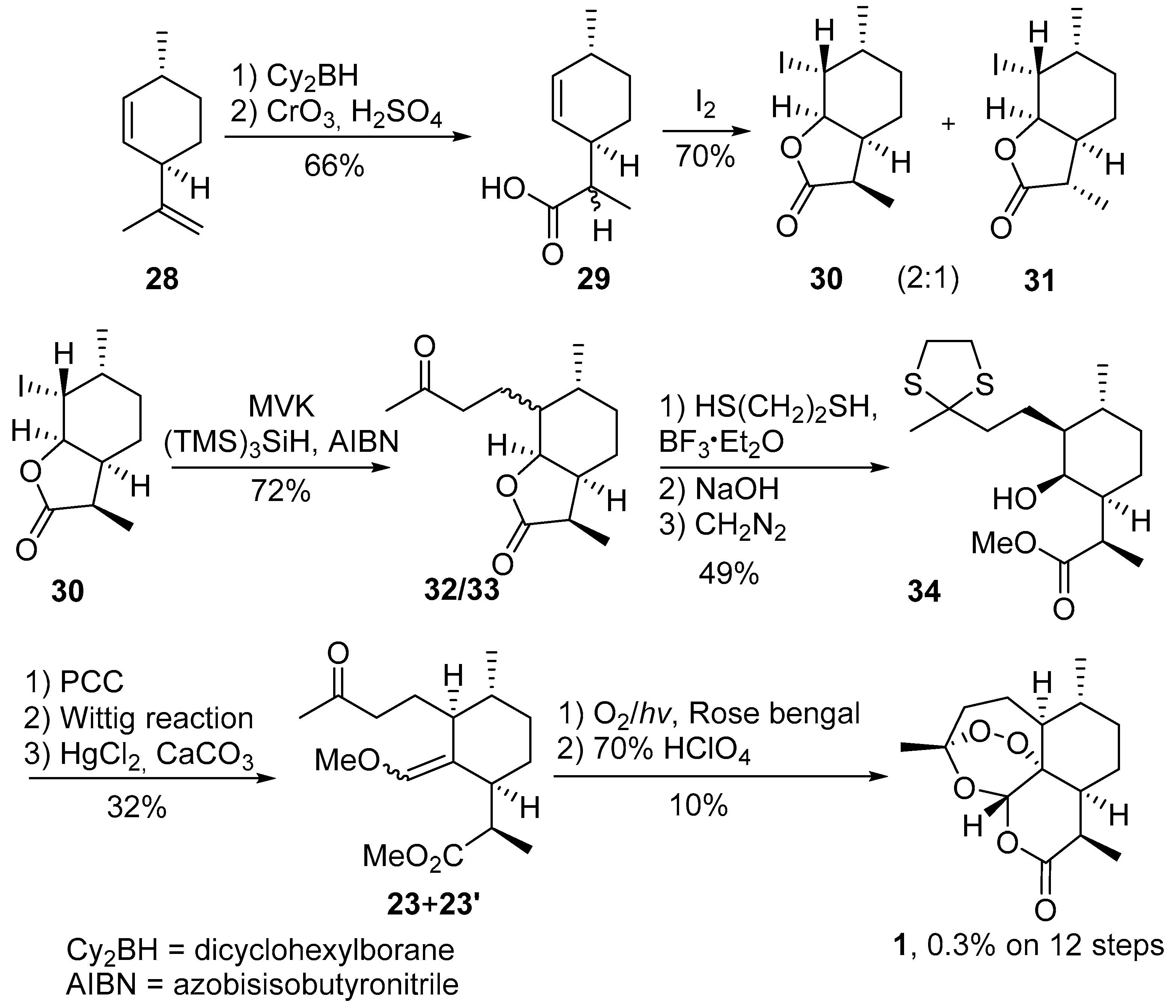
2.1. Using Singlet Oxygen
2.2. Other Methods
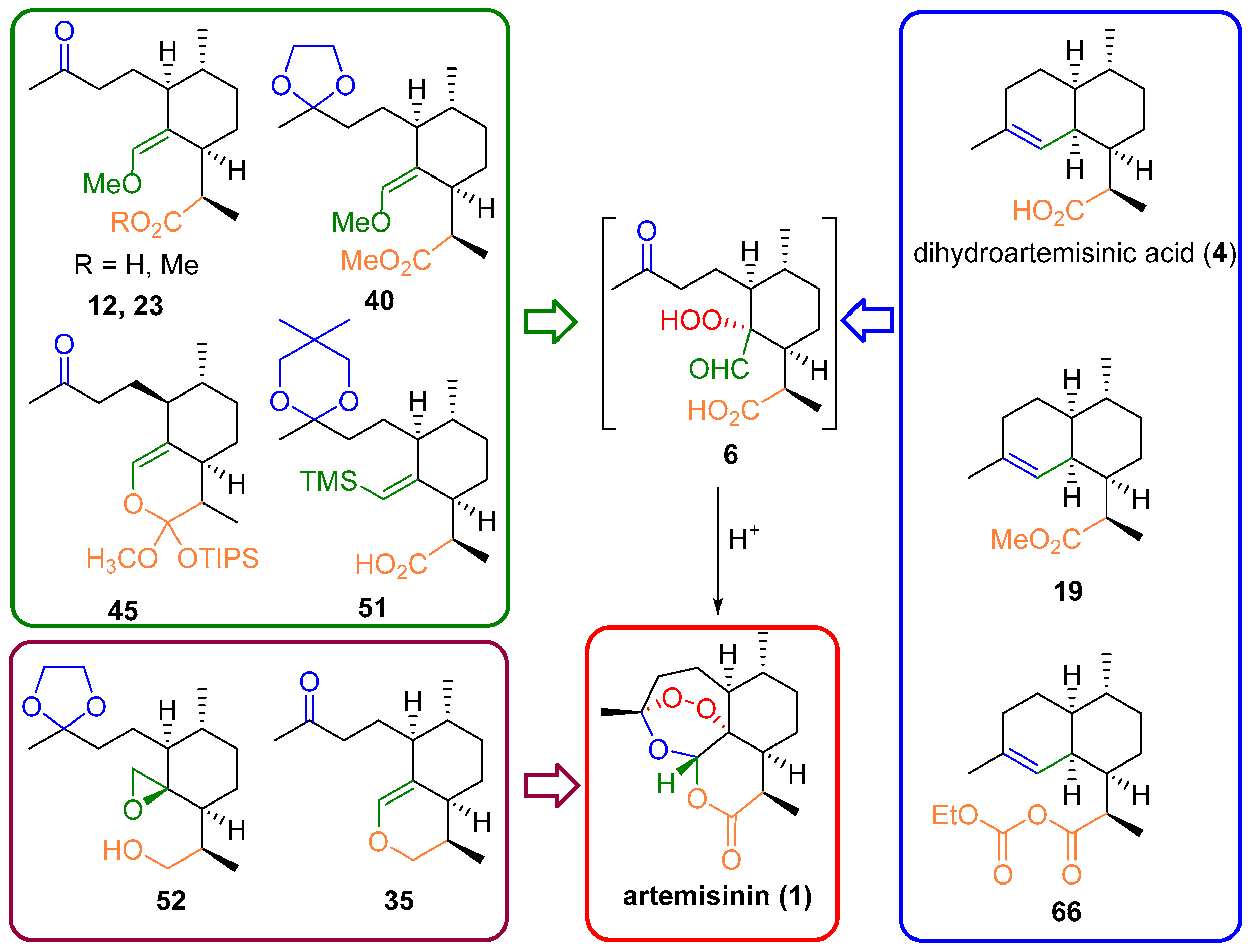
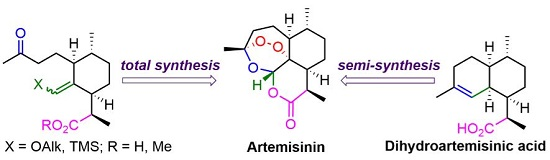
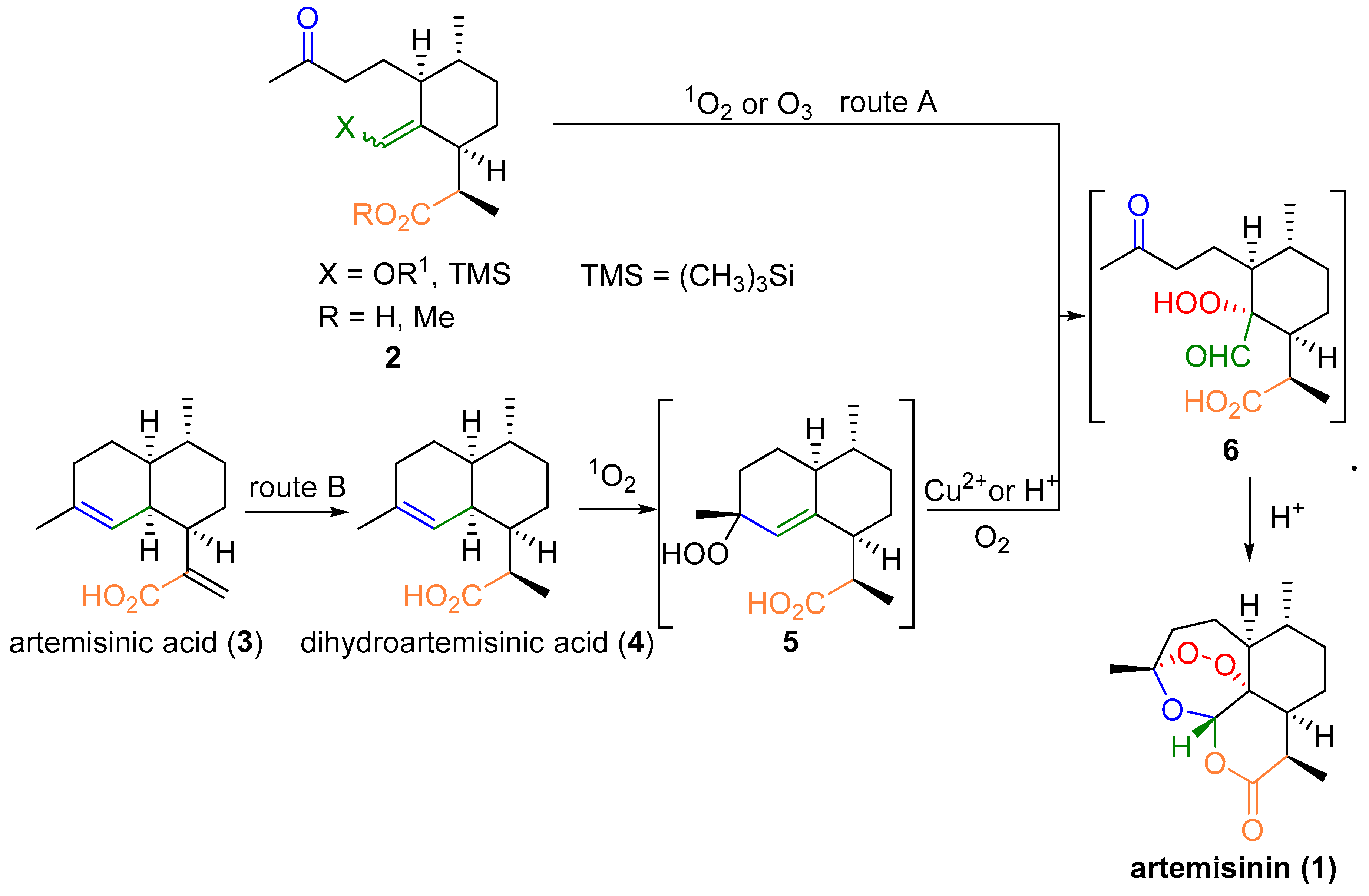
3. Artemisinin Synthesis Based on Dihydroartemisinic Acid
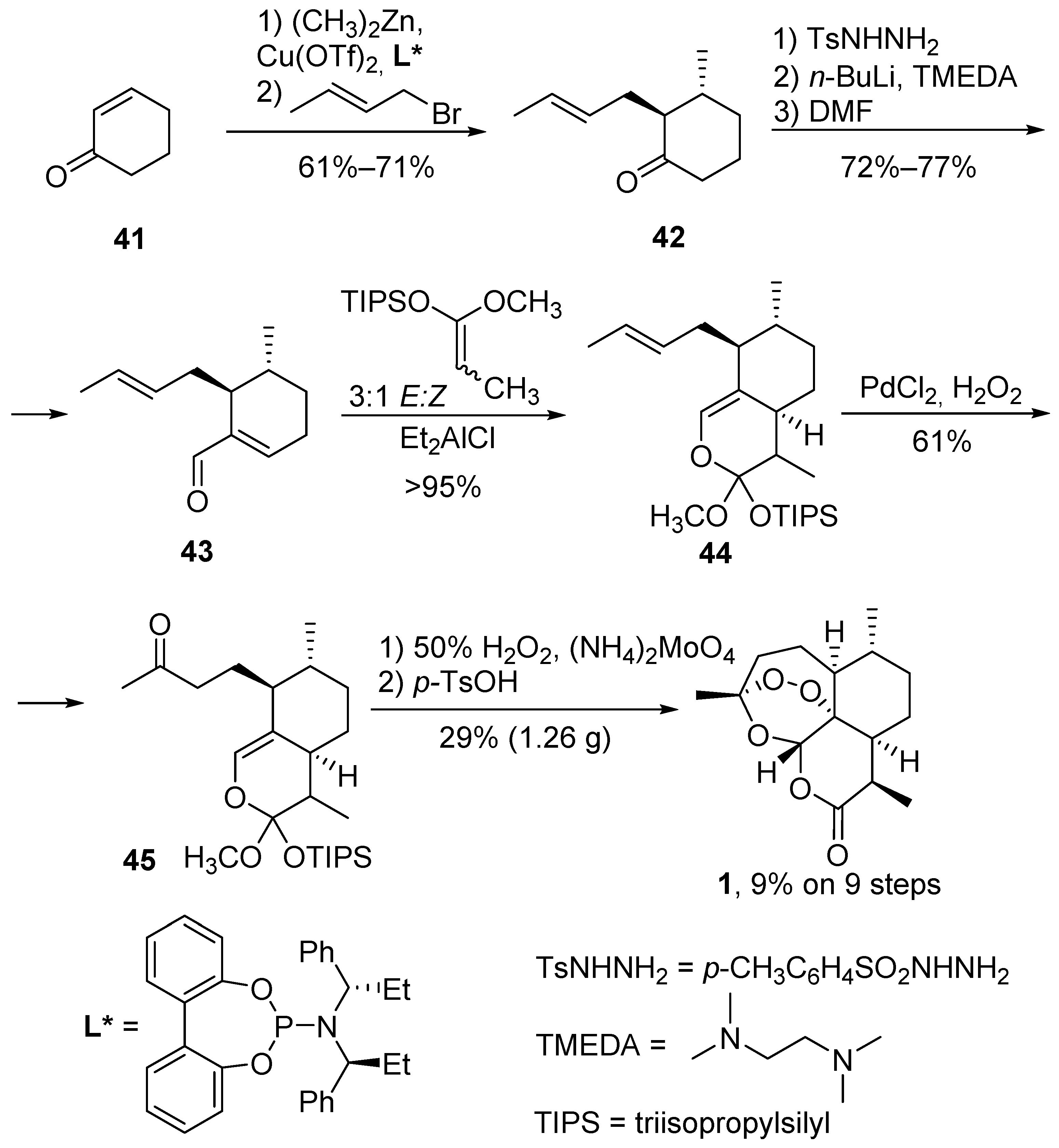
3.1. Preparation of Dihydroartemisinic Acid
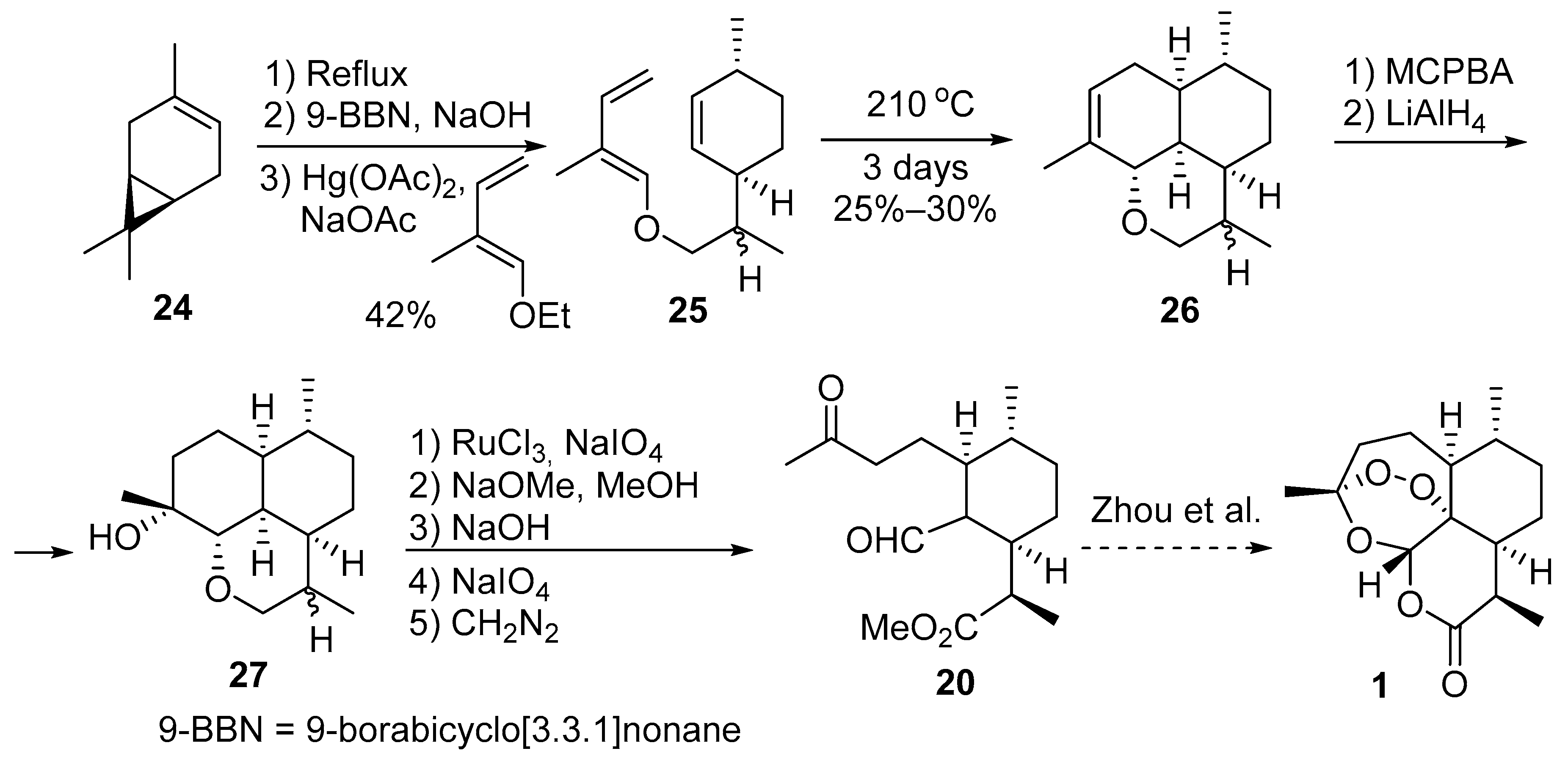
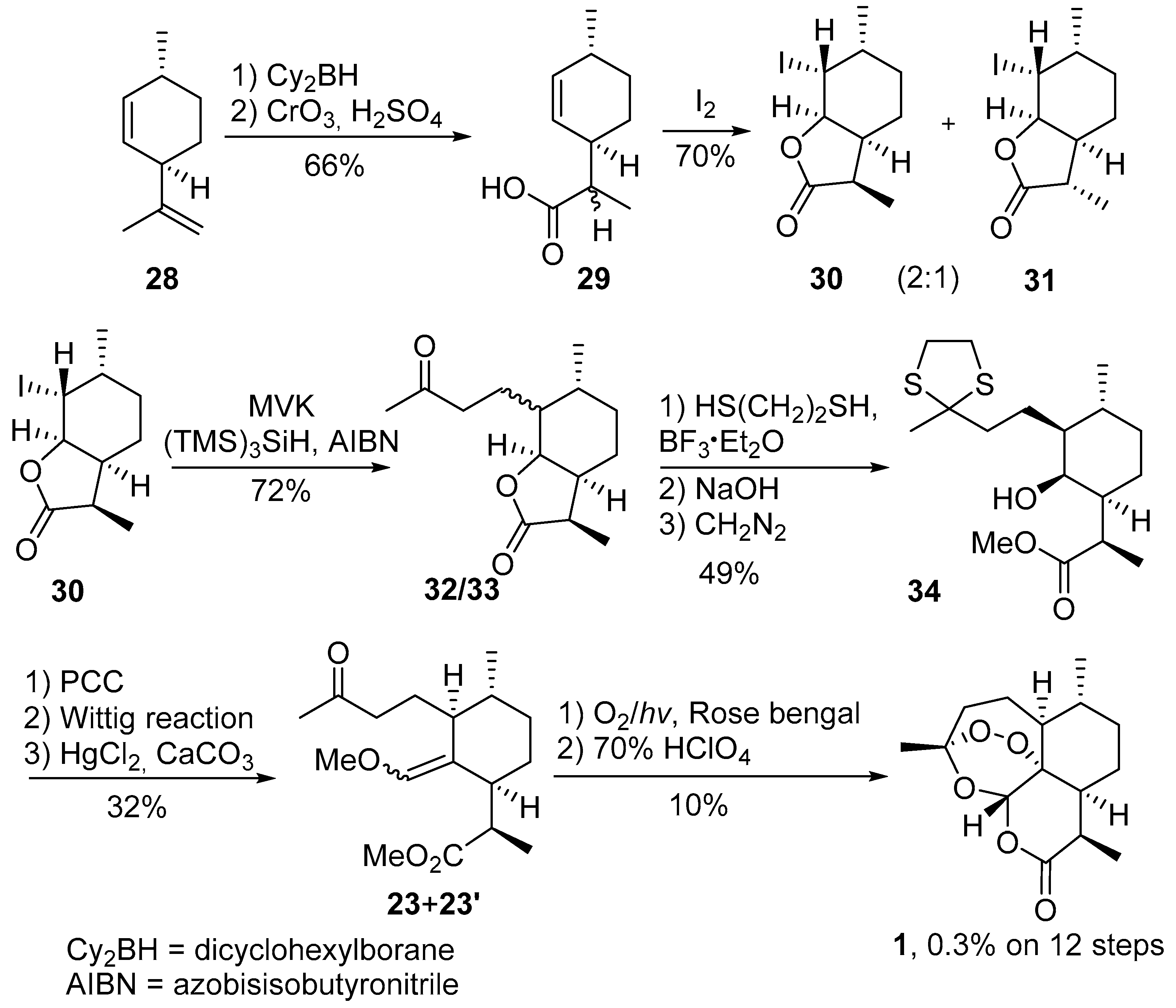
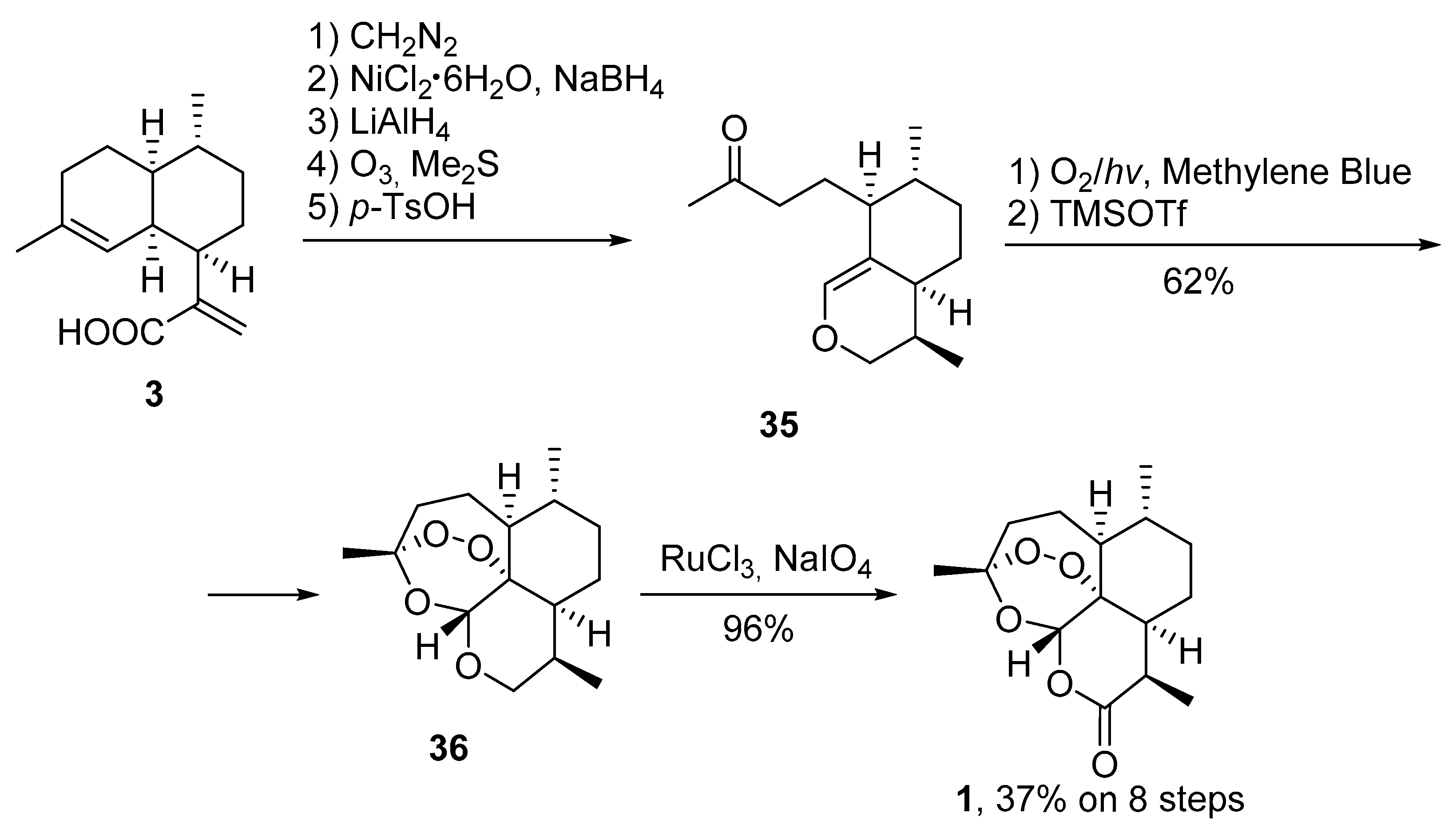
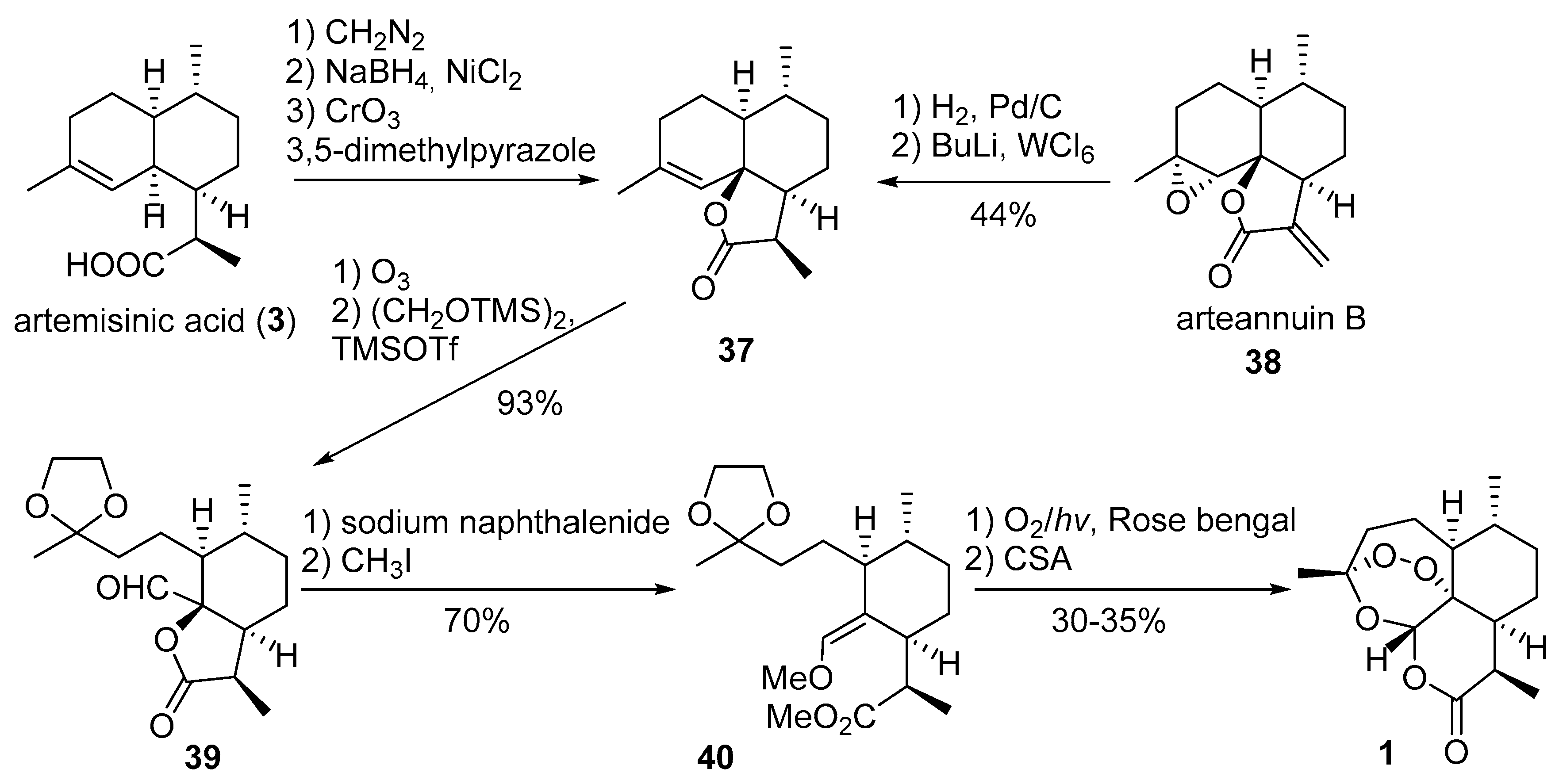
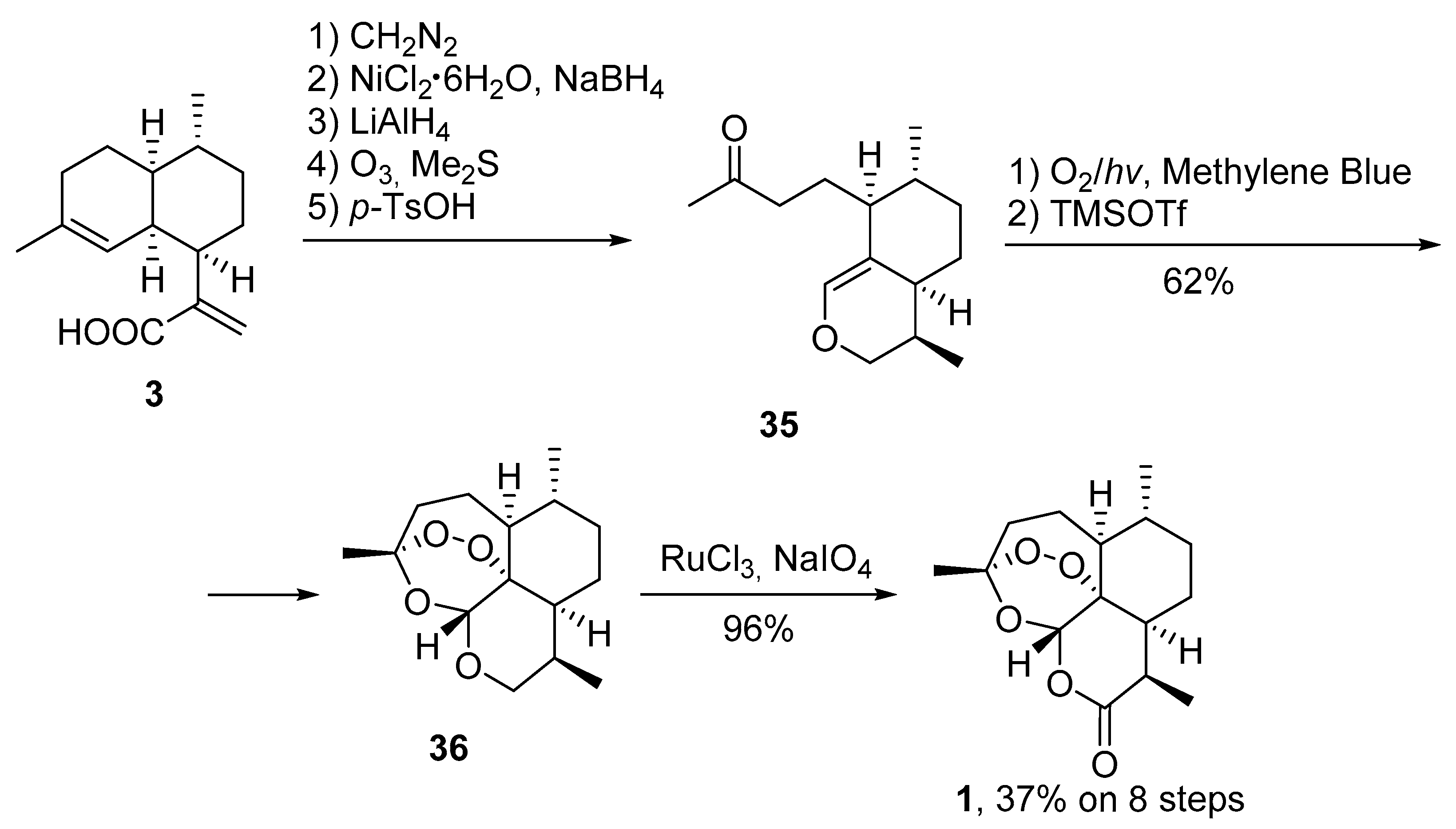
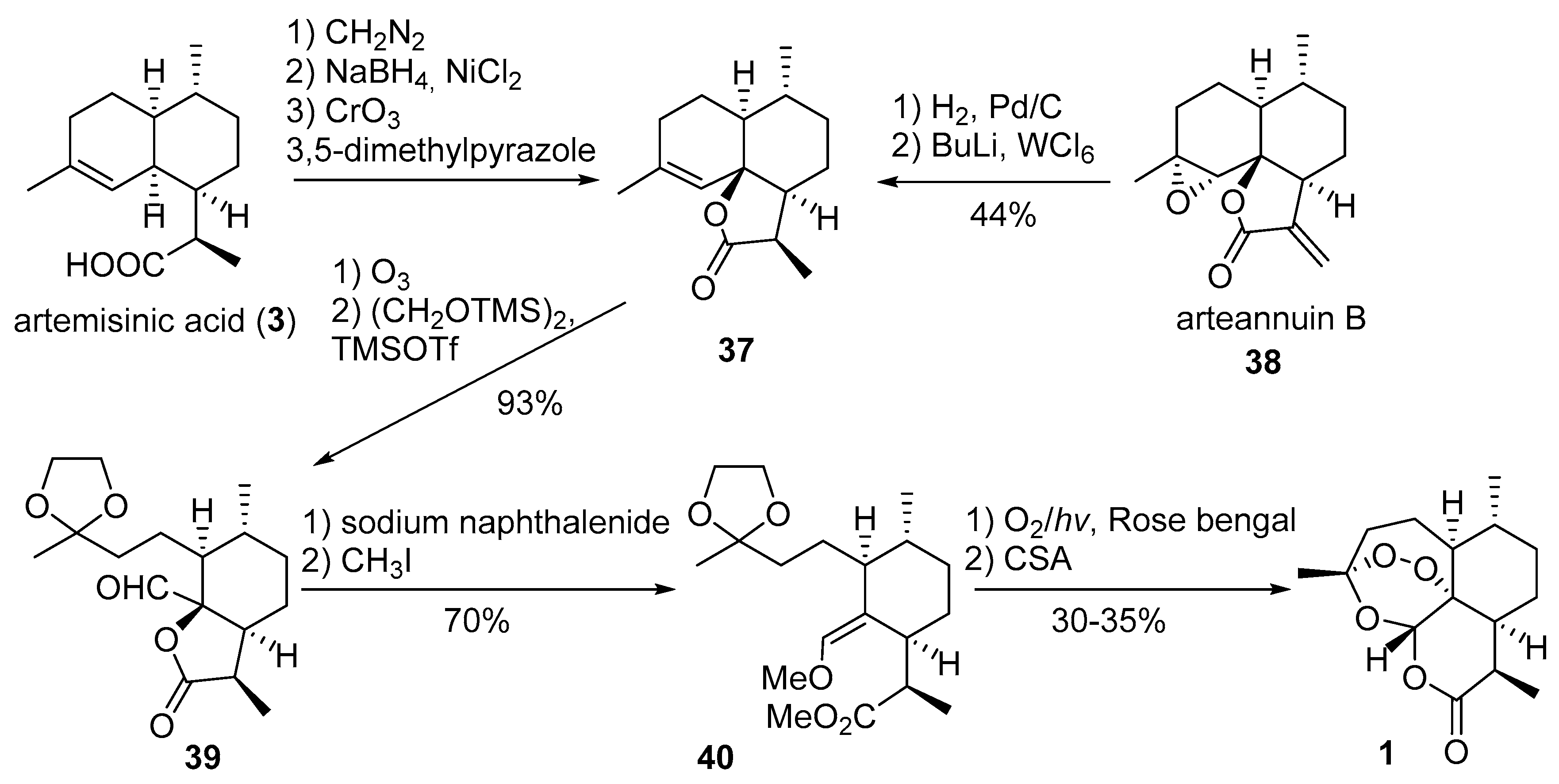
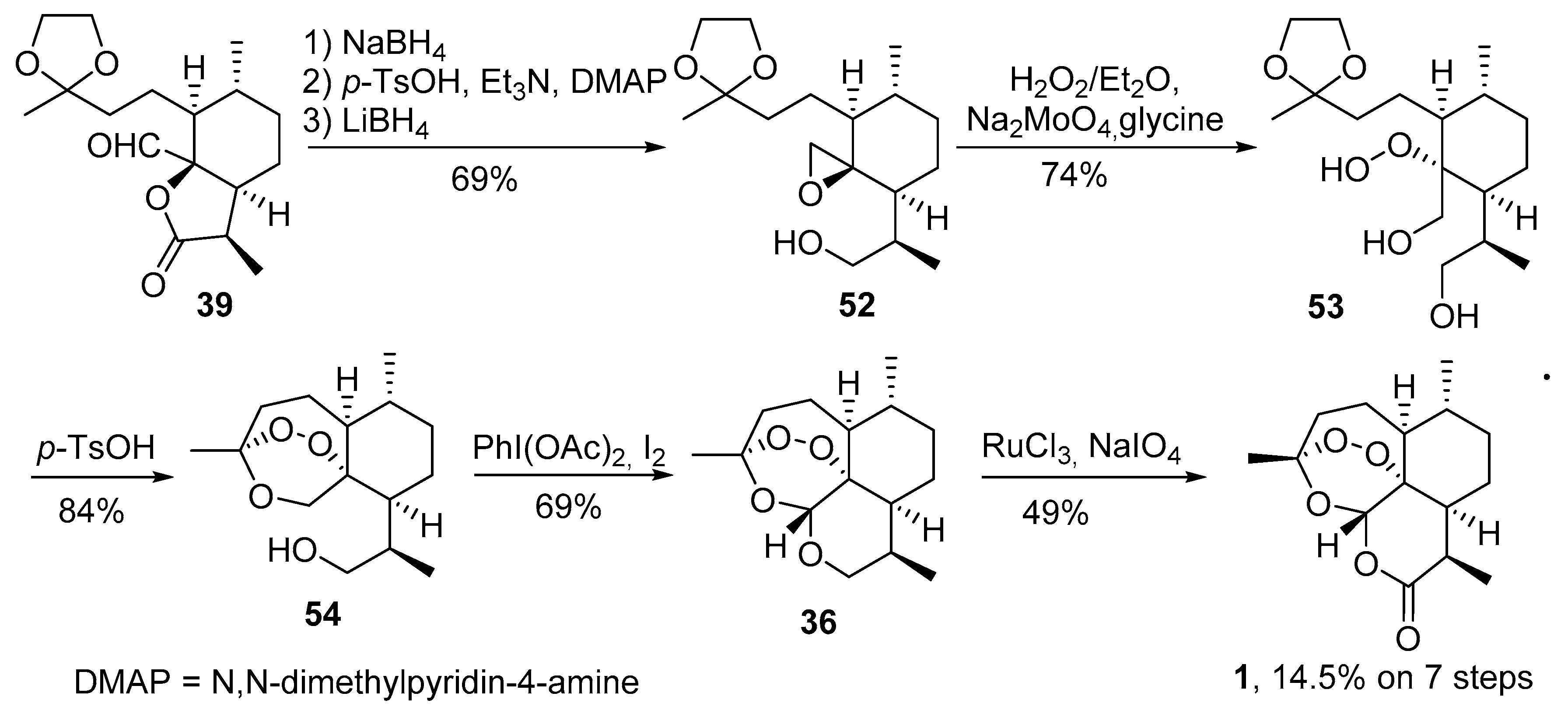
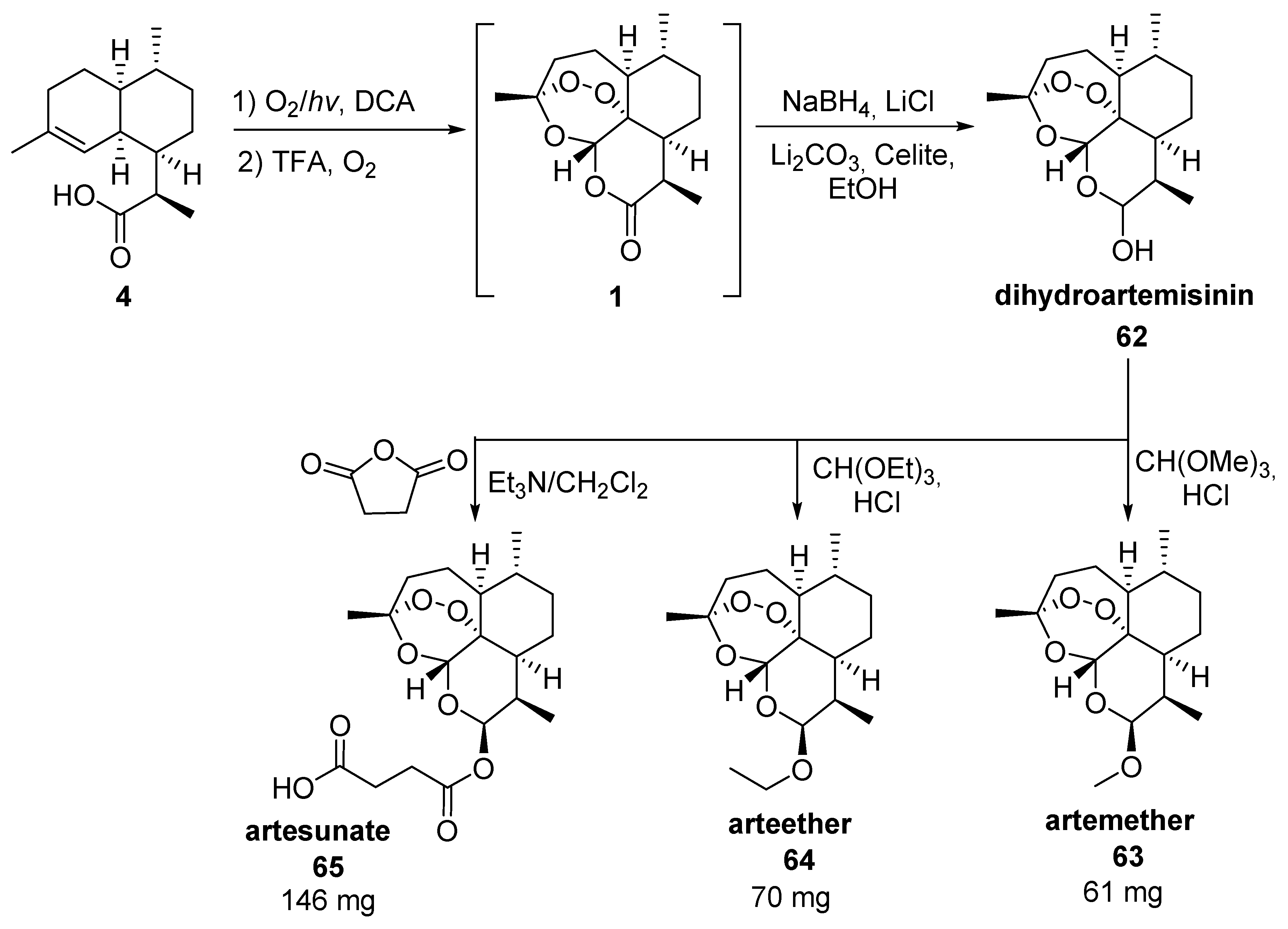
3.2. Transformation of Dihydroartemisinic Acid into Artemisinin via Hydroperoxidation and Rearrangements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2015. Available online: http://www.who.int/malaria/publications/world-malaria-report-2015/en/ (accessed on 8 December 2016).
- Roberts, D.J.; Chitnis, C.E. Molecular pathogenesis of malaria. In Molecular Hematology; Wiley-Blackwell: Oxford, UK, 2010; pp. 196–207. [Google Scholar]
- Lin Chua, C.L.; Ataíde, R.; Umbers, A.J.; Boeuf, P. Pathogenesis of malarial parasites in humans. In Human Emerging and Re-Emerging Infections; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 393–422. [Google Scholar]
- Meshnick, S.R.; Dobson, M.J. The history of antimalarial drugs. In Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery; Rosenthal, P.J., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 15–25. [Google Scholar]
- Petersen, I.; Eastman, R.; Lanzer, M. Drug-resistant malaria: Molecular mechanisms and implications for public health. FEBS Lett. 2011, 585, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Pitt, S.J. Parasite treatment and control. In Parasitology; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp. 339–373. [Google Scholar]
- Mutabingwa, T.K. Artemisinin-based combination therapies (acts): Best hope for malaria treatment but inaccessible to the needy! Acta Trop. 2005, 95, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Thwing, J.; Eisele, T.P.; Steketee, R.W. Protective efficacy of malaria case management and intermittent preventive treatment for preventing malaria mortality in children: A systematic review for the lives saved tool. BMC Public Health 2011, 11, S14. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S.F. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Qinghaosu (artemisinin): Chemistry and pharmacology. Acta Pharmacol. Sin. 2012, 33, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Zhang, Y.; Zhang, A. Discovery of antimalarial drug artemisinin and beyond. In Case Studies in Modern Drug Discovery and Development; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 227–256. [Google Scholar]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- White, N.J.; Hien, T.T.; Nosten, F.H. A brief history of qinghaosu. Trends Parasitol. 2015, 31, 607–610. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Physiology or Medicine 2015. Nobelprize.Org. Nobel Media ab 2014. Web. 17 October 2016. Available online: http://www.Nobelprize.Org/nobel_prizes/medicine/laureates/2015/ (accessed on 17 November 2016).
- Tu, Y.Y.; Ni, M.Y.; Zhong, Y.R.; Li, L.N.; Cui, S.L.; Zhang, M.Q.; Wang, X.Z.; Liang, X.T. Studies on the constituents of Artemisia annua L. (author’s transl.). Acta Pharm. Sin. 1981, 16, 366–370. [Google Scholar]
- Miller, L.H.; Su, X. Artemisinin: Discovery from the chinese herbal garden. Cell 2011, 146, 855–858. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Qinghaosu (artemisinin): The price of success. Science 2008, 320, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Source of new hope against malaria is in short supply. Science 2005, 307, 33. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef] [PubMed]
- Wallaart, T.E.; van Uden, W.; Lubberink, H.G.M.; Woerdenbag, H.J.; Pras, N.; Quax, W.J. Isolation and identification of dihydroartemisinic acid from Artemisia annua and its possible role in the biosynthesis of artemisinin. J. Nat. Prod. 1999, 62, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, J.C. Agricultural production of artemisinin—A review. Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 21–22. [Google Scholar] [CrossRef]
- Hale, V.; Keasling, J.D.; Renninger, N.; Diagana, T.T. Microbially derived artemisinin: A biotechnology solution to the global problem of access to affordable antimalarial drugs. Am. J. Trop. Med. Hyg. 2007, 77, 198–202. [Google Scholar] [PubMed]
- Jain, D.C.; Mathur, A.K.; Gupta, M.M.; Singh, A.K.; Verma, R.K.; Gupta, A.P.; Kumar, S. Isolation of high artemisinin-yielding clones of Artemisia annua. Phytochemistry 1996, 43, 993–1001. [Google Scholar] [CrossRef]
- Weathers, P.J.; Arsenault, P.R.; Covello, P.S.; McMickle, A.; Teoh, K.H.; Reed, D.W. Artemisinin production in Artemisia annua: Studies in planta and results of a novel delivery method for treating malaria and other neglected diseases. Phytochem. Rev.: Proc. Phytochem. Soc. Eur. 2011, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.S.R.; Acton, N.; Klayman, D.L.; Kendrick, K.; Basile, D.V.; Mante, S. Production of artemisinin in tissue cultures of Artemisia annua. J. Nat. Prod. 1986, 49, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. The biosynthesis of artemisinin (qinghaosu) and the phytochemistry of Artemisia annua L. (qinghao). Molecules 2010, 15, 7603–7698. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, P.R.; Wobbe, K.K.; Weathers, P.J. Recent advances in artemisinin production through heterologous expression. Curr. Med. Chem. 2008, 15, 2886–2896. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Qiu, F.; Yuan, L. Production of artemisinin by genetically-modified microbes. Biotechnol. Lett. 2008, 30, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.J.; Acton, N. A simple conversion of artemisinic acid into artemisinin. J. Nat. Prod. 1989, 52, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Schmid, G.; Hofheinz, W. Total synthesis of qinghaosu. J. Am. Chem. Soc. 1983, 105, 624–625. [Google Scholar] [CrossRef]
- Zhou, W.-S.; Xu, X.-X. Total synthesis of the antimalarial sesquiterpene peroxide qinghaosu and yingzhaosu A. Acc. Chem. Res. 1994, 27, 211–216. [Google Scholar] [CrossRef]
- Kim, B.J.; Sasaki, T. Recent progress in the synthesis of artemisinin and its derivatives. Org. Prep. Proced. Int. 2006, 38, 1–80. [Google Scholar] [CrossRef]
- Cook, S.P. Artemisinin: A case study in the evolution of synthetic strategy. Synlett 2014, 25, 751–759. [Google Scholar] [CrossRef]
- Kumar, V.; Mahajan, A.; Chibale, K. Synthetic medicinal chemistry of selected antimalarial natural products. Bioorgan. Med. Chem. 2009, 17, 2236–2275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, L.; Yang, X.; Zhang, X. Advances in the chemical synthesis of artemisinin. Synth. Commun. 2014, 44, 1987–2003. [Google Scholar] [CrossRef]
- Abdin, M.Z.; Israr, M.; Rehman, R.U.; Jain, S.K. Artemisinin, a novel antimalarial drug: Biochemical and molecular approaches for enhanced production. Planta Med. 2003, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Corsello, M.A.; Garg, N.K. Synthetic chemistry fuels interdisciplinary approaches to the production of artemisinin. Nat. Prod. Rep. 2015, 32, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Vonwiller, S.C. From qinghao, marvelous herb of antiquity, to the antimalarial trioxane qinghaosuand some remarkable new chemistry. Acc. Chem. Res. 1997, 30, 73–79. [Google Scholar] [CrossRef]
- Kong, J.; Yang, Y.; Wang, W.; Cheng, K.; Zhu, P. Artemisinic acid: A promising molecule potentially suitable for the semi-synthesis of artemisinin. RSC Adv. 2013, 3, 7622–7641. [Google Scholar] [CrossRef]
- Dhaintaut, J.; Dlubala, A.; Guevel, R.; Medard, A.; Oddon, G.; Raymond, N.; Turconi, J. Photochemical Process for Producing Artemisinin. U.S. Patent US20110065933 A1, 17 March 2011. [Google Scholar]
- Pantjushenko, E.; Lemonde-San, F. Sanofi and Path Announce the Launch of Large-Scale Production of Semisynthetic Artemisinin against Malaria. Available online: http://en.sanofi.com/Images/32474_20130411_ARTEMISININE_en.pdf (accessed on 20 November 2016).
- Xu, X.-X.; Zhu, J.; Huang, D.-Z.; Zhou, W.-S. Total synthesis of arteannuin and deoxyarteannuin. Tetrahedron 1986, 42, 819–828. [Google Scholar]
- Ravindranathan, T.; Anil Kumar, M.; Menon, R.B.; Hiremath, S.V. Stereoselective synthesis of artemisinin. Tetrahedron Lett. 1990, 31, 755–758. [Google Scholar] [CrossRef]
- Yadav, J.S.; Satheesh Babu, R.; Sabitha, G. Stereoselective total synthesis of (+)-artemisinin. Tetrahedron Lett. 2003, 44, 387–389. [Google Scholar] [CrossRef]
- Ye, B.; Wu, Y.-L. An efficient synthesis of qinghaosu and deoxoginghaosu from arteannuic acid. Chem. Commun. 1990, 726–727. [Google Scholar] [CrossRef]
- Lansbury, P.T.; Nowak, D.M. An efficient partial synthesis of (+)-artemisinin and (+)-deoxoartemisinin. Tetrahedron Lett. 1992, 33, 1029–1032. [Google Scholar] [CrossRef]
- Nowak, D.M.; Lansbury, P.T. Synthesis of (+)-artemisinin and (+)-deoxoartemisinin from arteannuin b and arteannuic acid. Tetrahedron 1998, 54, 319–336. [Google Scholar] [CrossRef]
- Zhu, C.; Cook, S.P. A concise synthesis of (+)-artemisinin. J. Am. Chem. Soc. 2012, 134, 13577–13579. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.A.; Jennings-White, C.; Chong, W.K.M. The total synthesis of (+)-artemisinin and (+)-9-desmethyltemesinin. Tetrahedron Lett. 1987, 28, 4629–4632. [Google Scholar] [CrossRef]
- Avery, M.A.; Chong, W.K.M.; Jennings-White, C. Stereoselective total synthesis of (+)-artemisinin, the antimalarial constituent of Artemisia annua L. J. Am. Chem. Soc. 1992, 114, 974–979. [Google Scholar] [CrossRef]
- Hao, H.-D.; Li, Y.; Han, W.-B.; Wu, Y. A hydrogen peroxide based access to qinghaosu (artemisinin). Org. Lett. 2011, 13, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.J.; Acton, N. Isolation of arteannuic acid from Artemisia annua. Planta Med. 1987, 53, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Keasling, J.D. Semi-synthetic artemisinin: A model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, H.; Paddon, C.J.; Eng, D.; Lenihan, J.R.; Horning, T.; Anthony, L.C.; Regentin, R.; Keasling, J.D.; Renninger, N.S.; Newman, J.D. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PLoS ONE 2009, 4, e4489. [Google Scholar] [CrossRef] [PubMed]
- Westfall, P.J.; Pitera, D.J.; Lenihan, J.R.; Eng, D.; Woolard, F.X.; Regentin, R.; Horning, T.; Tsuruta, H.; Melis, D.J.; Owens, A.; et al. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc. Natl. Acad. Sci. USA 2012, 109, E111–E118. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Turconi, J.; Griolet, F.; Guevel, R.; Oddon, G.; Villa, R.; Geatti, A.; Hvala, M.; Rossen, K.; Göller, R.; Burgard, A. Semisynthetic artemisinin, the chemical path to industrial production. Org. Process Res. Dev. 2014, 18, 417–422. [Google Scholar] [CrossRef]
- Roth, R.J.; Acton, N. A facile semisynthesis of the antimalarial drug qinghaosu. J. Chem. Ed. 1991, 68, 612. [Google Scholar] [CrossRef]
- Jung, M.; ElSohly, H.N.; Croom, E.M.; McPhail, A.T.; McPhail, D.R. Practical conversion of artemisinic acid in desoxyartemisinin. J. Org. Chem. 1986, 51, 5417–5419. [Google Scholar] [CrossRef]
- Feth, M.P.; Rossen, K.; Burgard, A. Pilot plant pat approach for the diastereoselective diimide reduction of artemisinic acid. Org. Process Res. Dev. 2013, 17, 282–293. [Google Scholar] [CrossRef]
- Pieber, B.; Glasnov, T.; Kappe, C.O. Continuous flow reduction of artemisinic acid utilizing multi-injection strategies—Closing the gap towards a fully continuous synthesis of antimalarial drugs. Chem. A Eur. J. 2015, 21, 4368–4376. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.; Chaudret, R.; Ricci, G.; Kurz, M.; Ochsenbein, P.; Kretzschmar, G.; Kraft, V.; Rossen, K.; Eisenstein, O. Nonclassical CH−π supramolecular interactions in artemisinic acid favor a single conformation, yielding high diastereoselectivity in the reduction with diazene. J. Org. Chem. 2014, 79, 5939–5947. [Google Scholar] [CrossRef] [PubMed]
- Hsing-Jang, L.; Wen-Lung, Y.; Sew, Y.C. A total synthesis of the antimarial natural product (+)-qinghaosu. Tetrahedron Lett. 1993, 34, 4435–4438. [Google Scholar] [CrossRef]
- Constantino, M.G.; Beltrame, M.; da Silva, G.V.J.; Zukerman-Schpector, J. A novel asymmetric total synthesis of (+)-artemisinin. Synth. Commun. 1996, 26, 321–329. [Google Scholar] [CrossRef]
- Yadav, J.S.; Thirupathaiah, B.; Srihari, P. A concise stereoselective total synthesis of (+)-artemisinin. Tetrahedron 2010, 66, 2005–2009. [Google Scholar] [CrossRef]
- Haynes, R.K.; Vonwiller, S.C. Catalysed oxygenation of allylic hydroperoxides derived from qinghao (artemisinic) acid. Conversion of qinghao acid into dehydroginghaosu (artemisitene) and qinghaosu (artemisinin). Chem. Commun. 1990, 451–453. [Google Scholar] [CrossRef]
- Vonwiller, S.C.; Warner, J.A.; Mann, S.T.; Haynes, R.K. Copper(ii) trifluoromethanesulfonate-induced cleavage oxygenation of allylic hydroperoxides derived from qinghao acid in the synthesis of qinghaosu derivatives: Evidence for the intermediacy of enols. J. Am. Chem. Soc. 1995, 117, 11098–11105. [Google Scholar] [CrossRef]
- Sy, L.-K.; Zhu, N.-Y.; Brown, G.D. Syntheses of dihydroartemisinic acid and dihydro-epi-deoxyarteannuin b incorporating a stable isotope label at the 15-position for studies into the biosynthesis of artemisinin. Tetrahedron 2001, 57, 8495–8510. [Google Scholar] [CrossRef]
- Sy, L.-K.; Brown, G.D. The mechanism of the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron 2002, 58, 897–908. [Google Scholar] [CrossRef]
- Sy, L.-K.; Brown, G.D. The role of the 12-carboxylic acid group in the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron 2002, 58, 909–923. [Google Scholar] [CrossRef]
- Brown, G.D.; Sy, L.-K. Synthesis of labelled dihydroartemisinic acid. Tetrahedron 2004, 60, 1125–1138. [Google Scholar] [CrossRef]
- Brown, G.D.; Sy, L.-K. In vivo transformations of artemisinic acid in Artemisia annua plants. Tetrahedron 2007, 63, 9548–9566. [Google Scholar] [CrossRef]
- Acton, N.; Roth, R.J. On the conversion of dihydroartemisinic acid into artemisinin. J. Org. Chem. 1992, 57, 3610–3614. [Google Scholar] [CrossRef]
- Kopetzki, D.; Lévesque, F.; Seeberger, P.H. A continuous-flow process for the synthesis of artemisinin. Chem. Eur. J. 2013, 19, 5450–5456. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, F.; Seeberger, P.H. Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem. Int. Ed. 2012, 51, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.; Kopetzki, D.; Lee, J.W.; Horvath, Z.; McQuade, D.T.; Seidel-Morgenstern, A.; Seeberger, P.H. Continuous synthesis of artemisinin-derived medicines. Chem. Commun. 2014, 50, 12652–12655. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Han, W.-B.; Hao, H.-D.; Wu, Y. A facile and scalable synthesis of qinghaosu (artemisinin). Tetrahedron 2013, 69, 1112–1114. [Google Scholar] [CrossRef]
- Amara, Z.; BellamyJessica, F.B.; Horvath, R.; Miller, S.J.; Beeby, A.; Burgard, A.; Rossen, K.; Poliakoff, M.; George, M.W. Applying green chemistry to the photochemical route to artemisinin. Nat. Chem. 2015, 7, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.D.P.; Vil’, V.; Terent’ev, A.; Alabugin, I.V. Stereoelectronic source of the anomalous stability of bis-peroxides. Chem. Sci. 2015, 6, 6783–6791. [Google Scholar] [CrossRef]

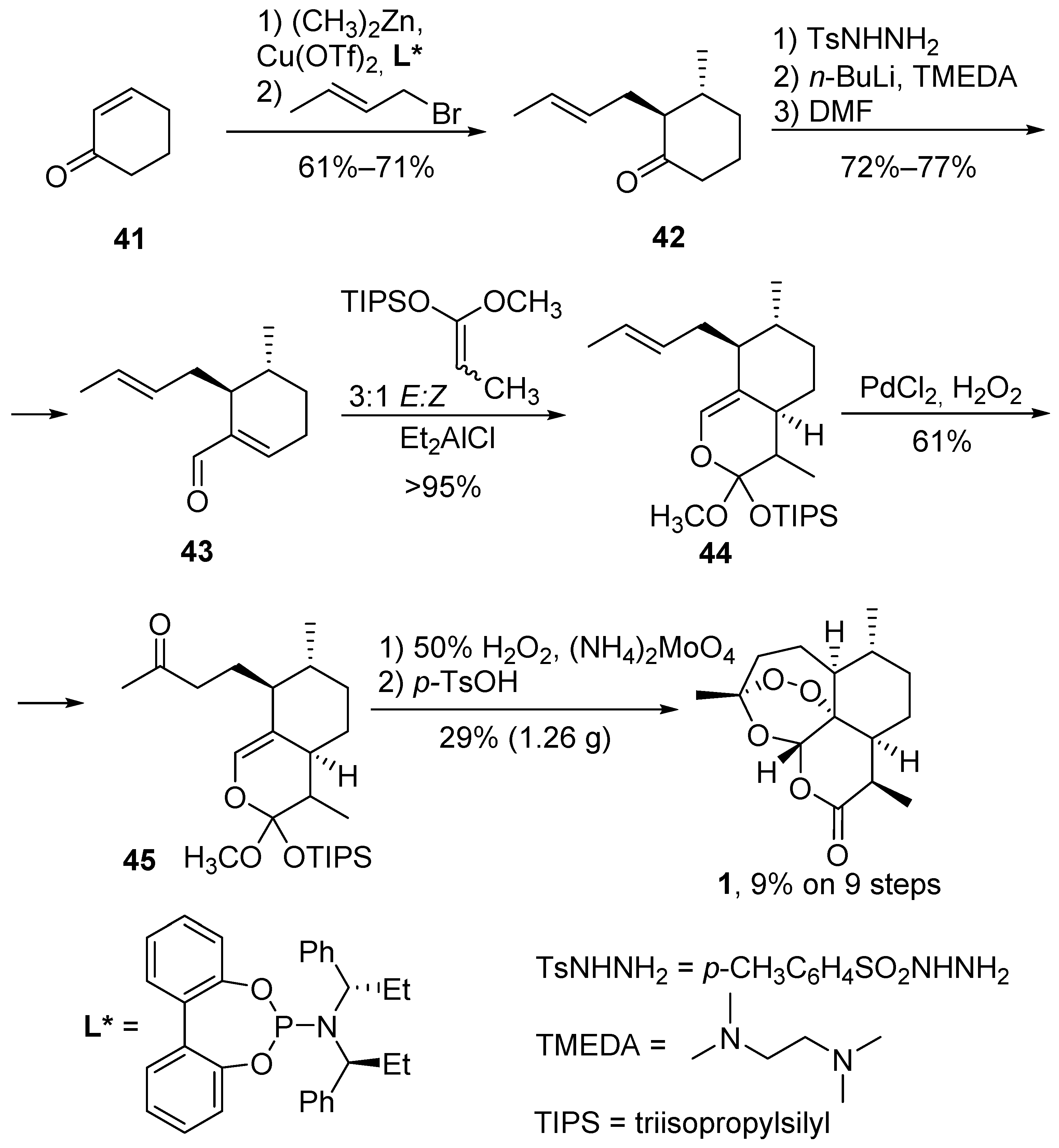
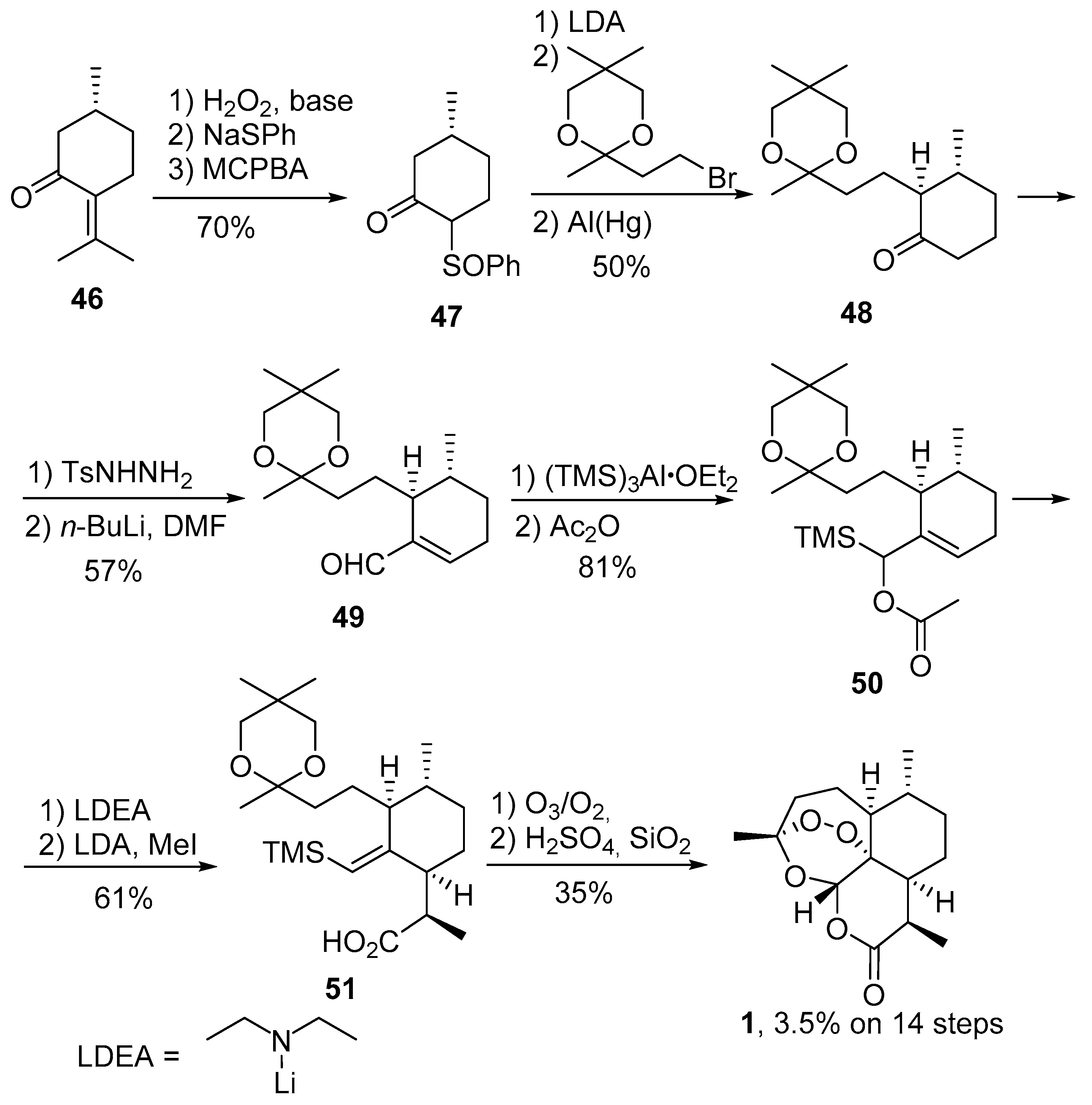
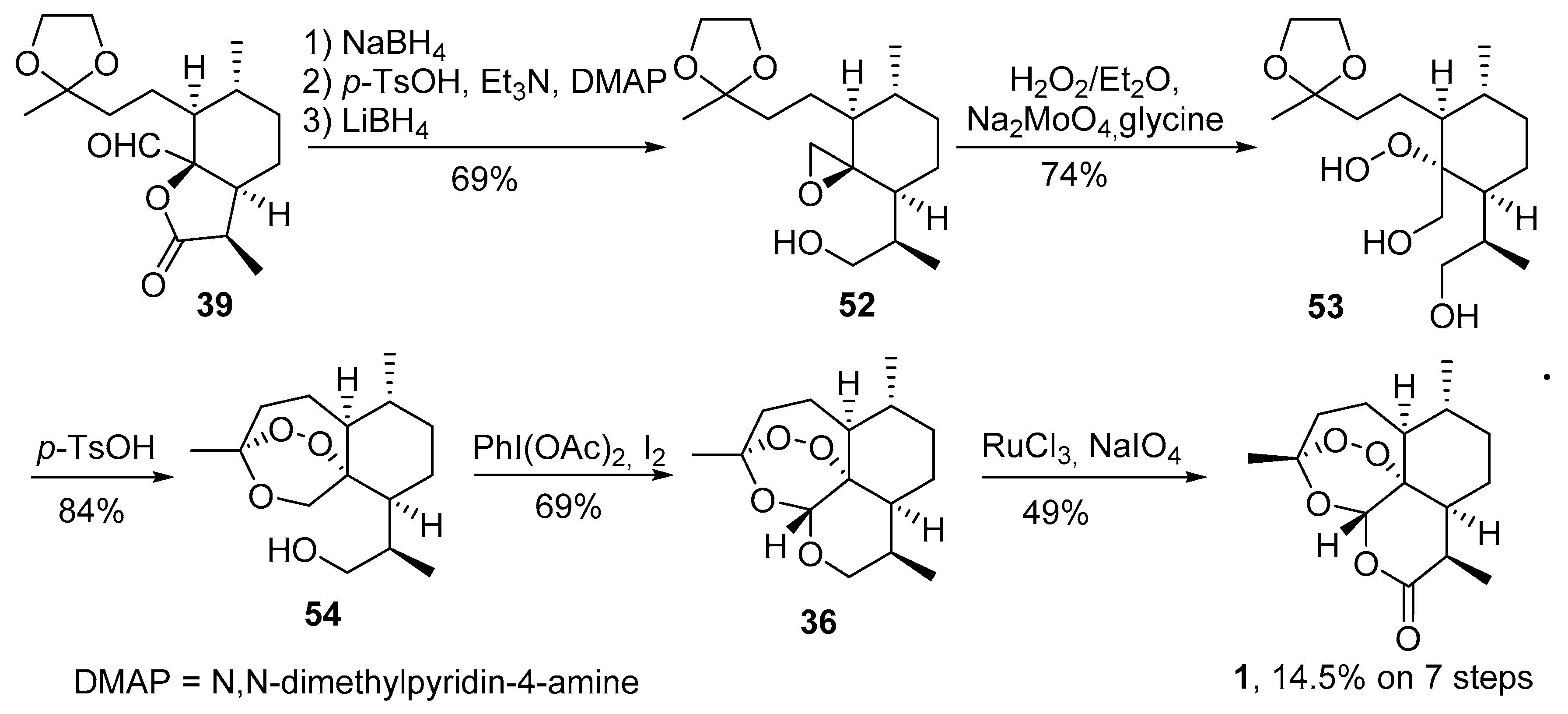
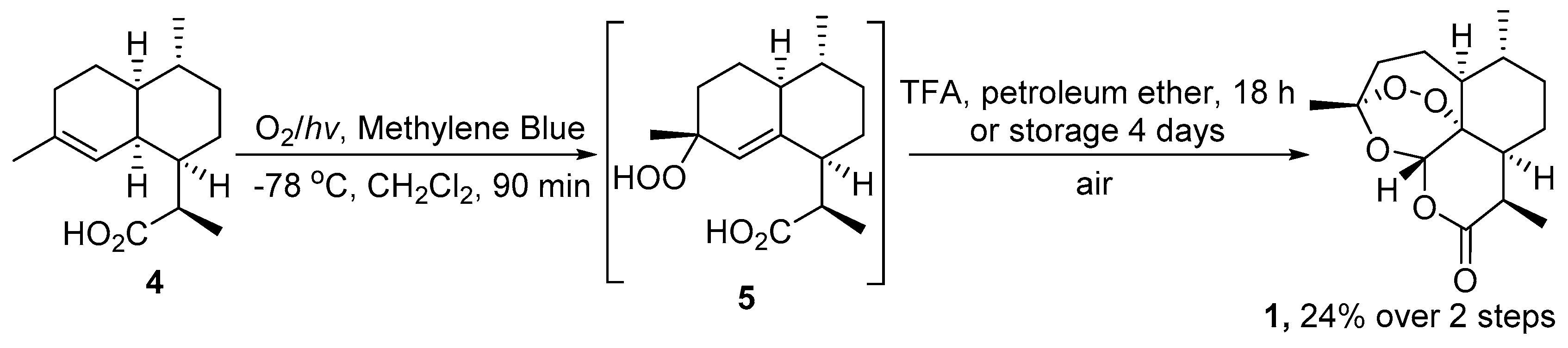
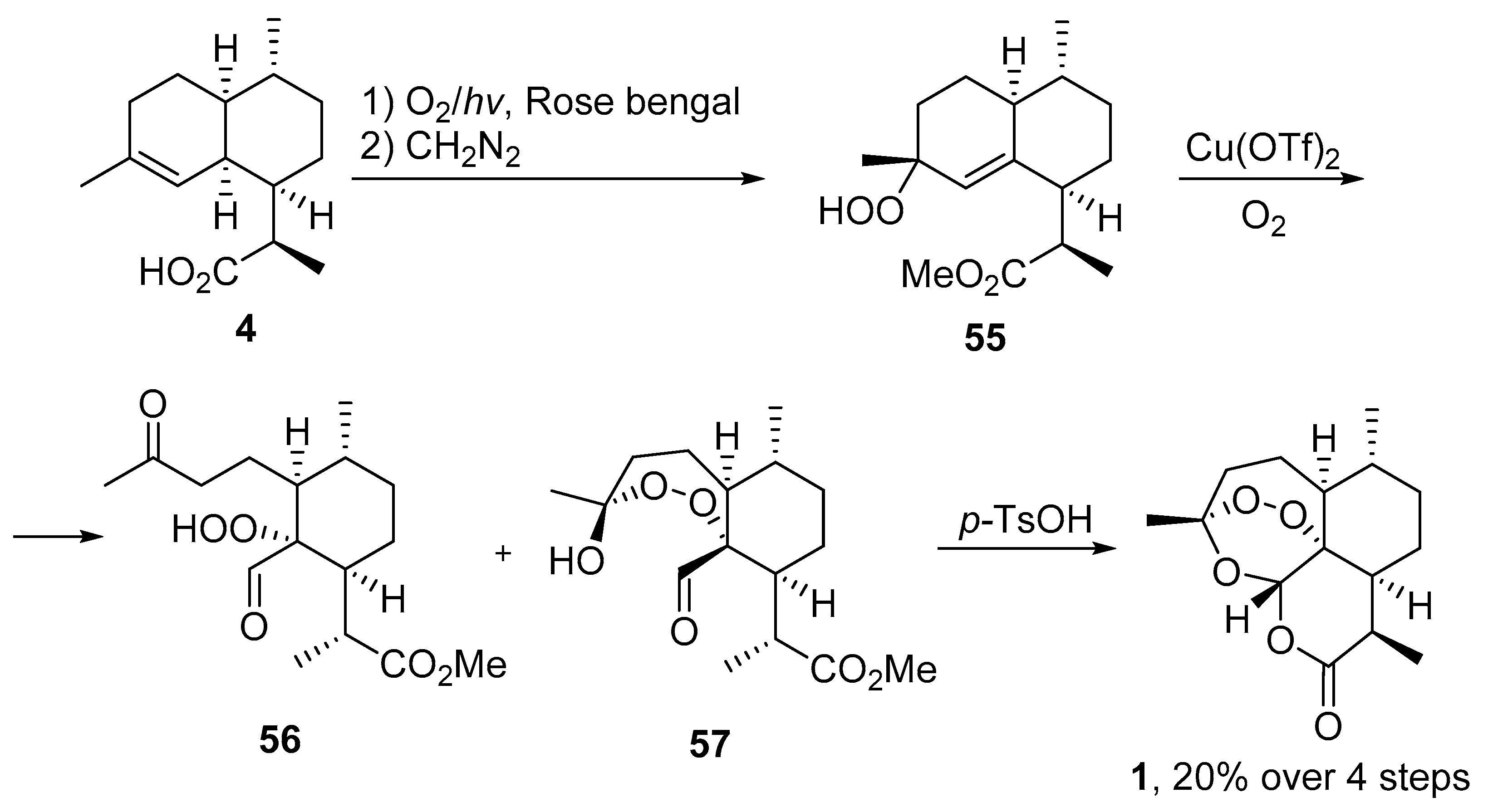
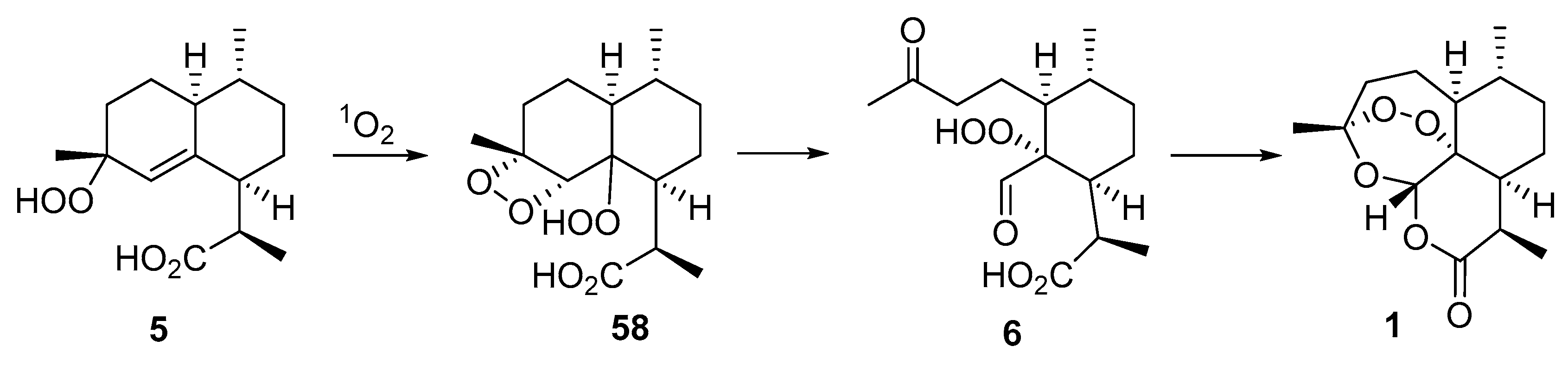
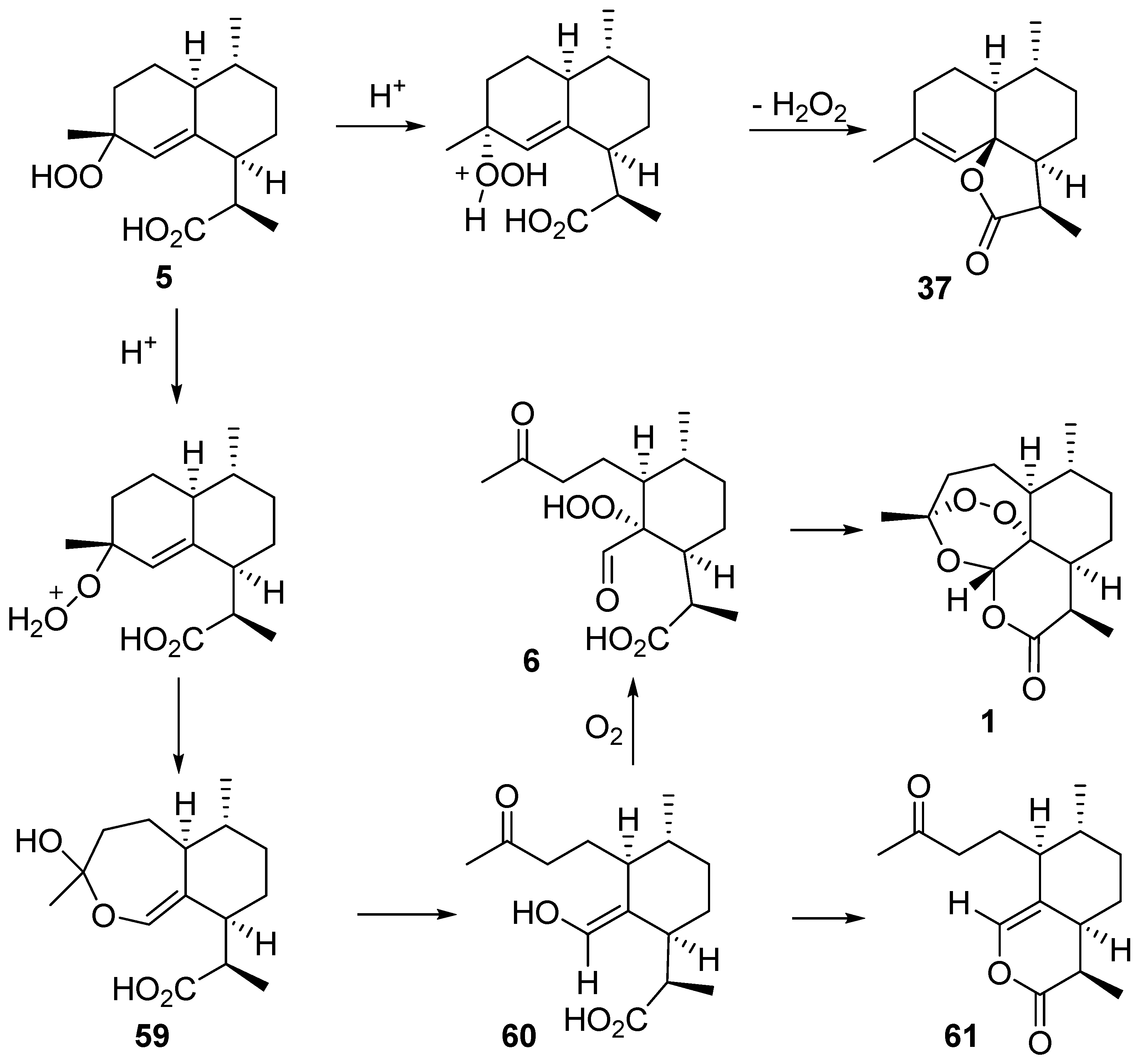

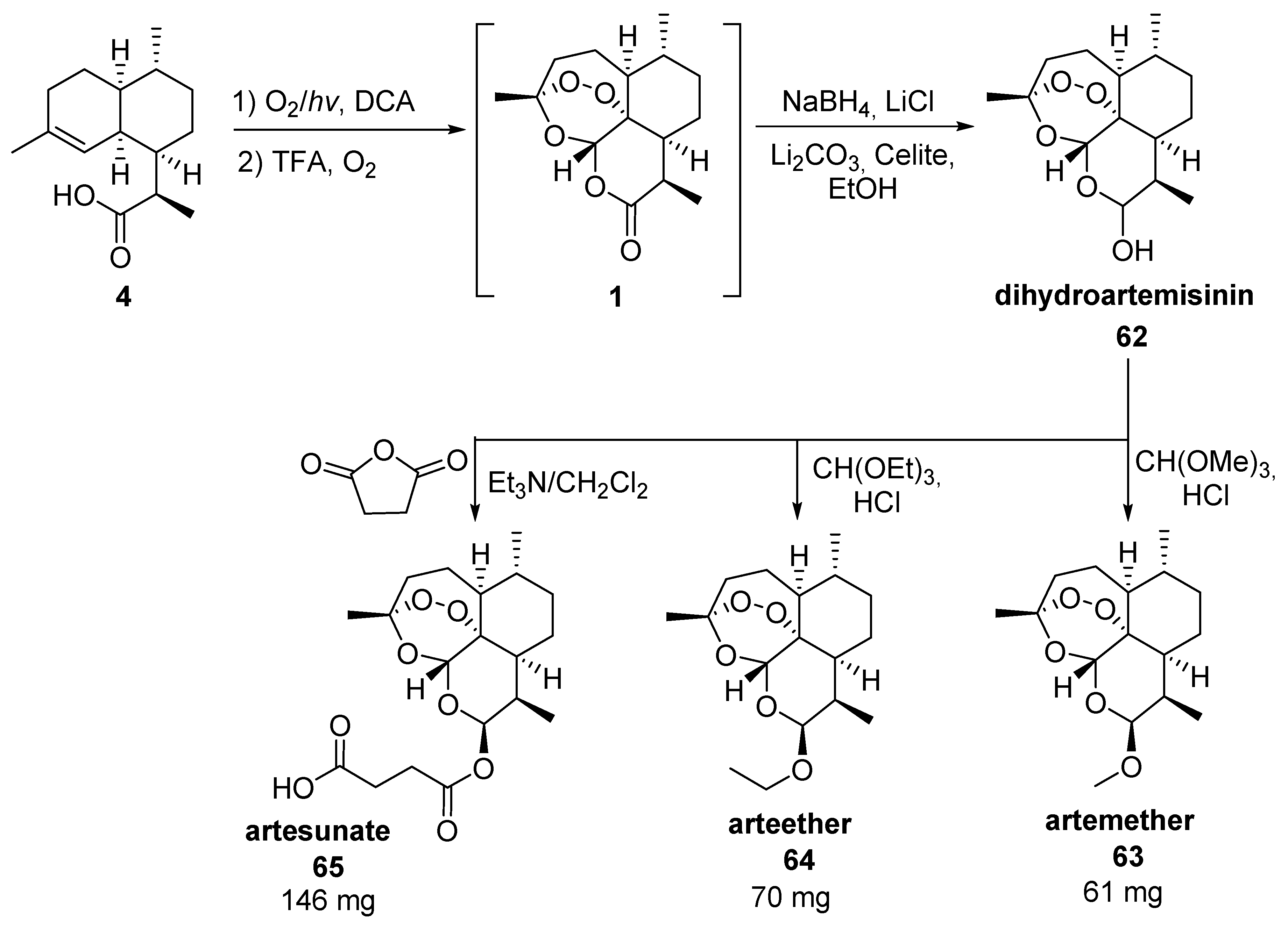
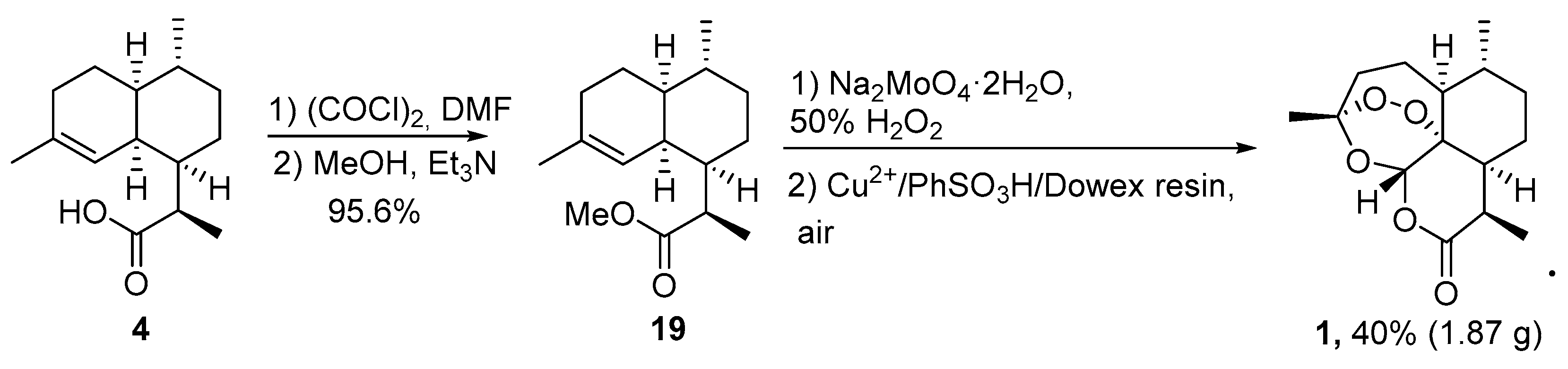
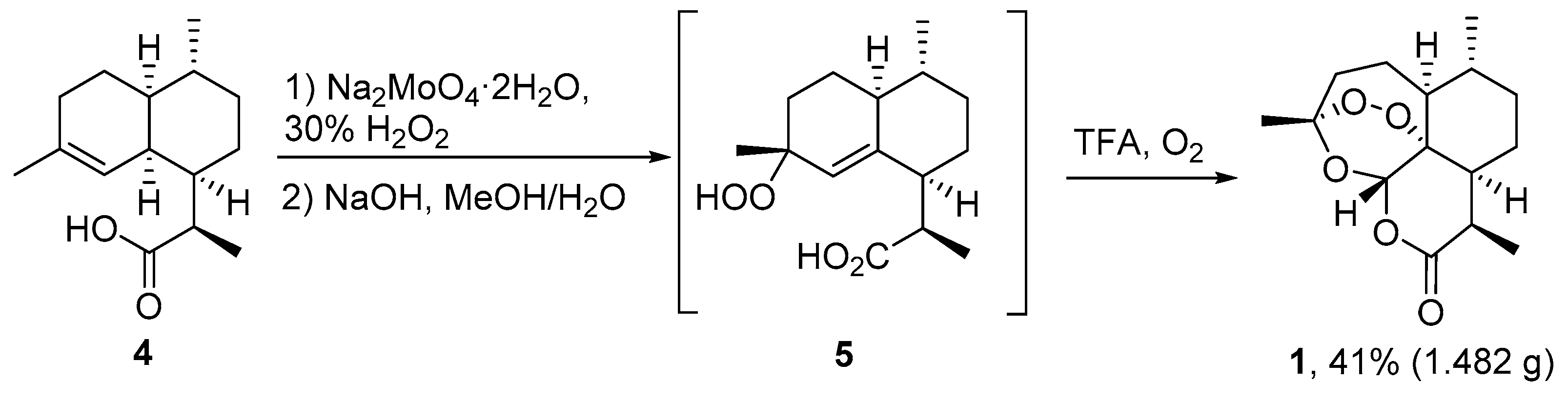

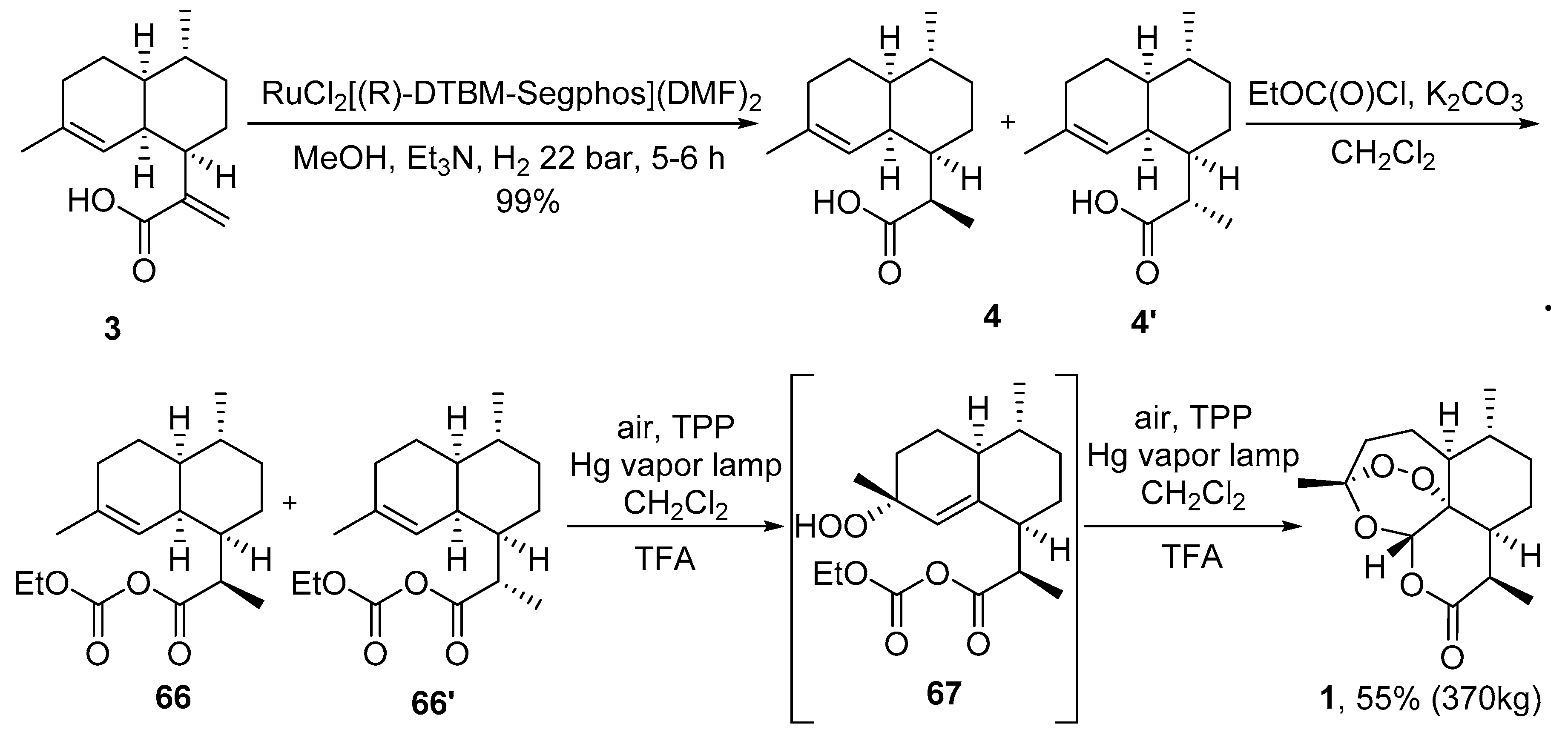
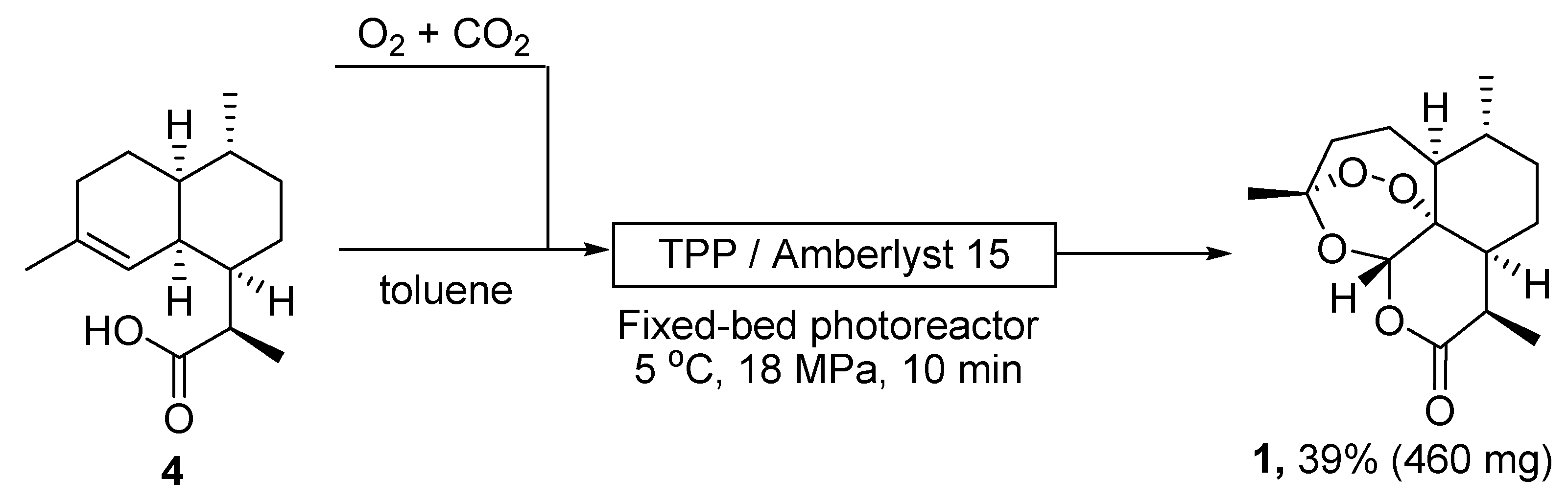
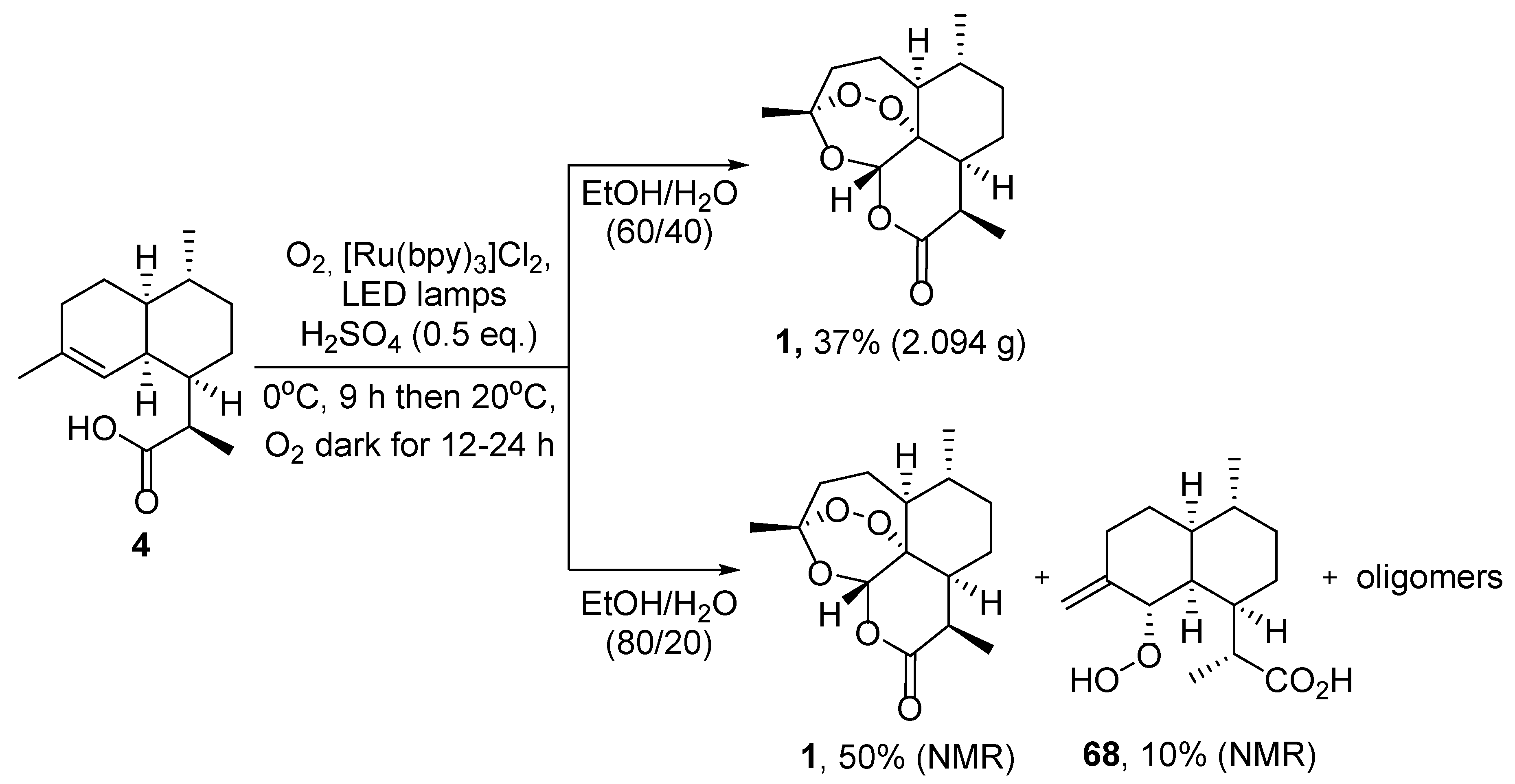

| No. | Substrate | Condition of Peroxidation Step | Condition of Rearrangement/Cyclization Step | Yield of 1, % | Reference |
|---|---|---|---|---|---|
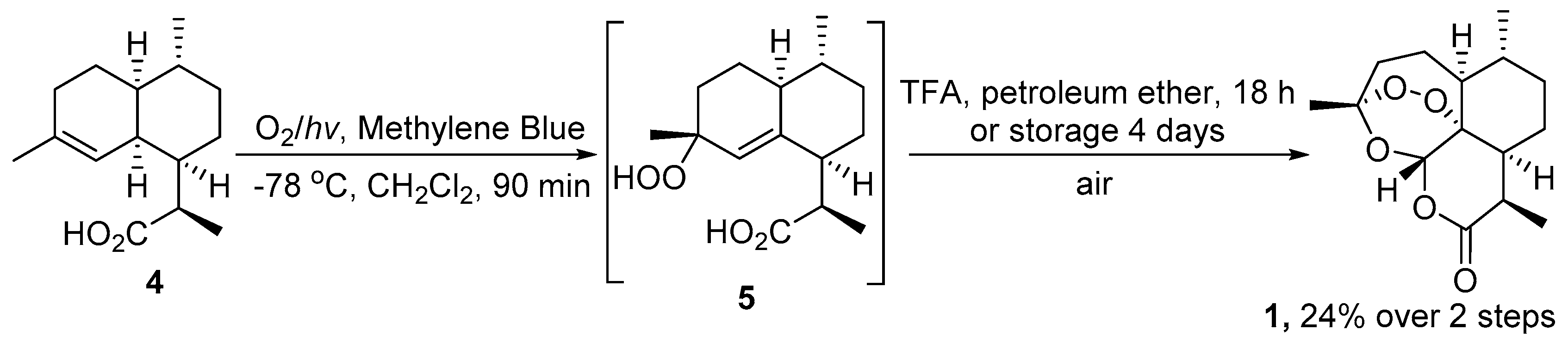
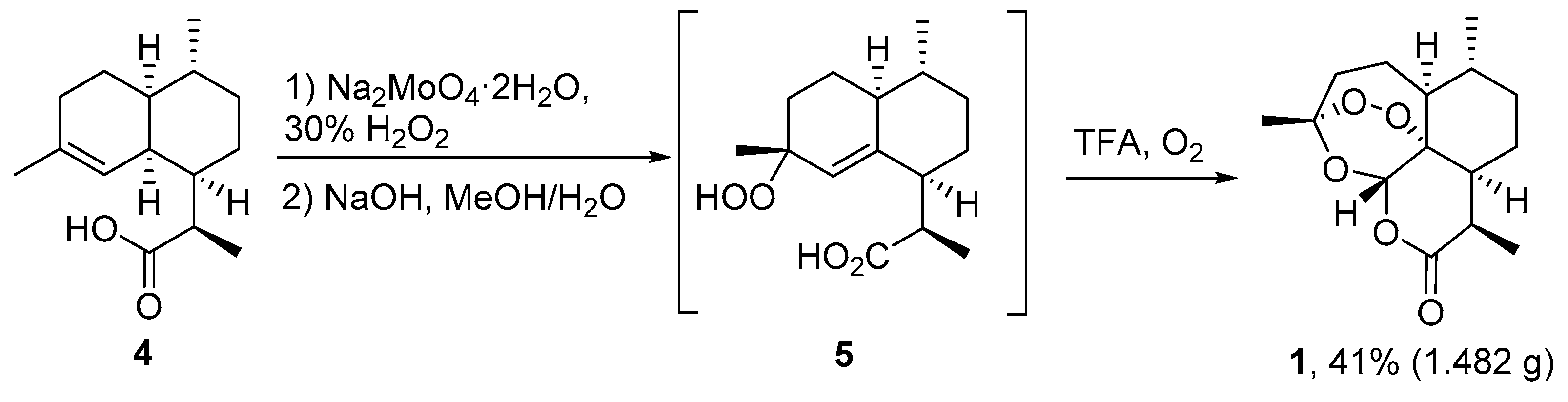
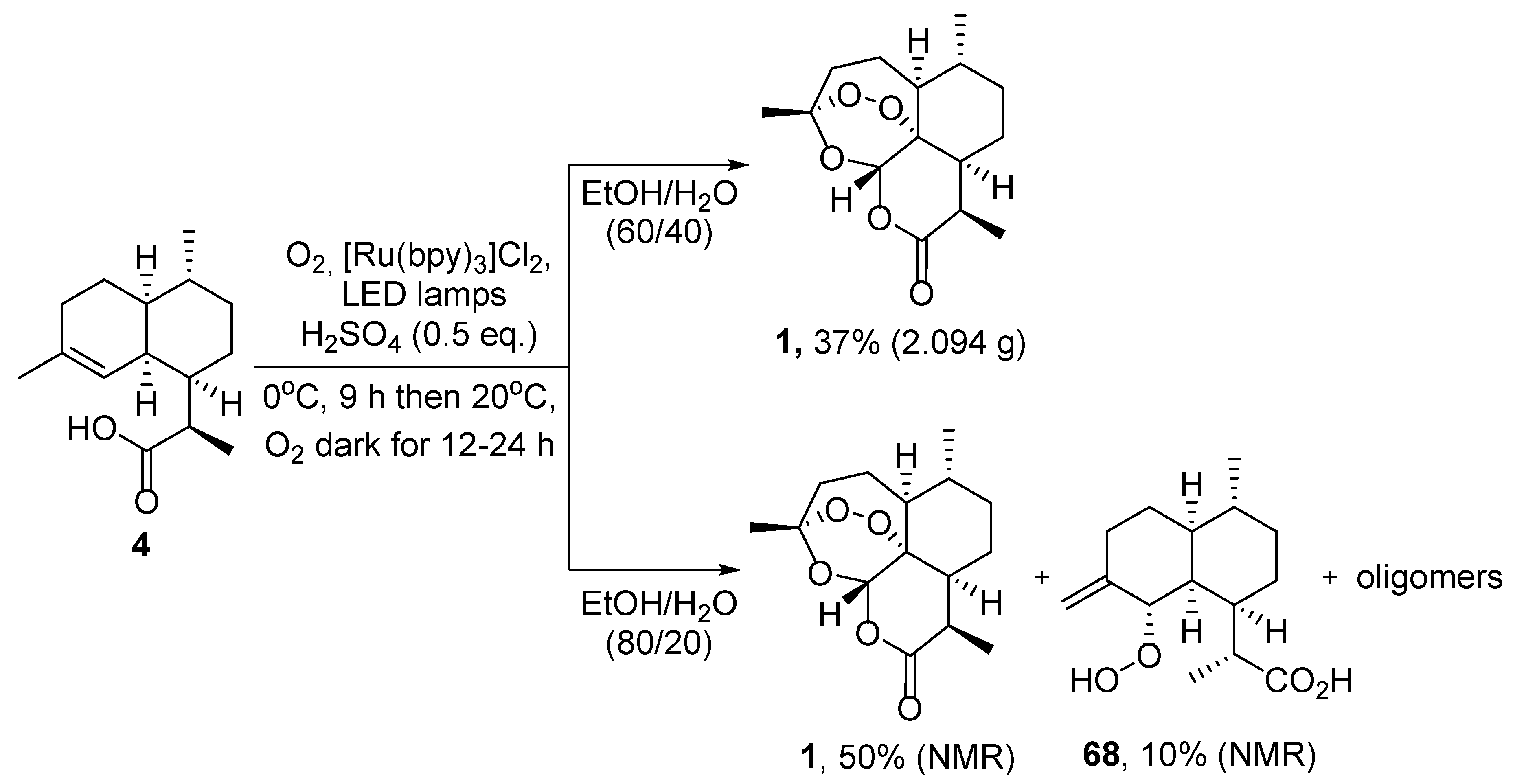
| 1 | 4 | O2/hv, Methylene Blue, acetone, 0 °C, 30 min | TFA (cat.), petroleum ether, air, r.t., 4 days | 30 | [59] |
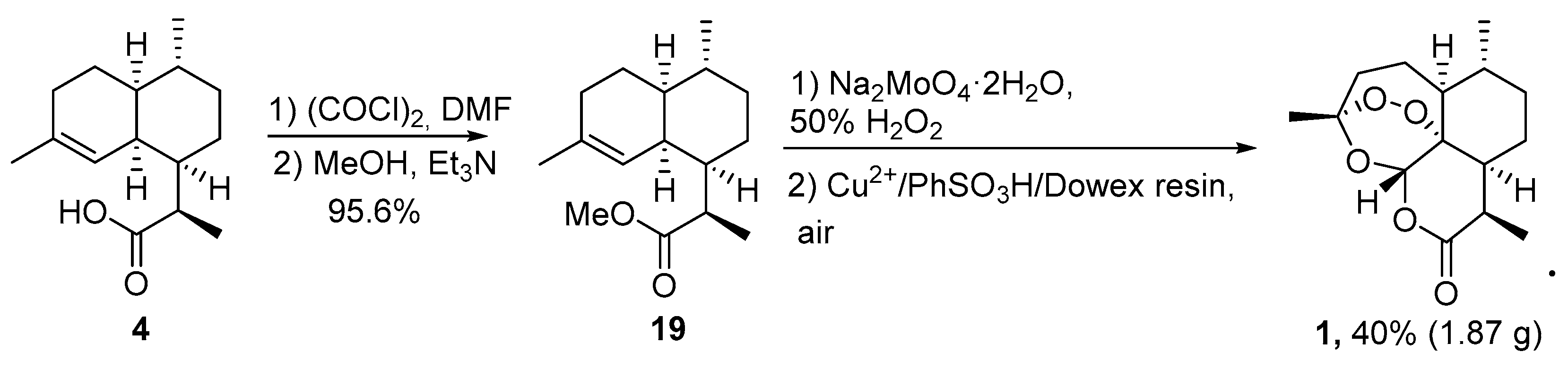
| 2 | crude mixture of 19 and isomer with double bond C6-C7 position | O2/hv, Methylene Blue, CH2Cl2, 20 °C, 90 min | TFA (cat.), petroleum ether, air, 20 °C, 4 days | 30 | [64] |
| 3 | crude mixture of 4 and isomer with double bond C6-C7 position | O2/hv, Methylene Blue, acetone, 0 °C, 30 min | TFA (cat.), petroleum ether, air, 20 °C, 24 h | 26 | [65] |
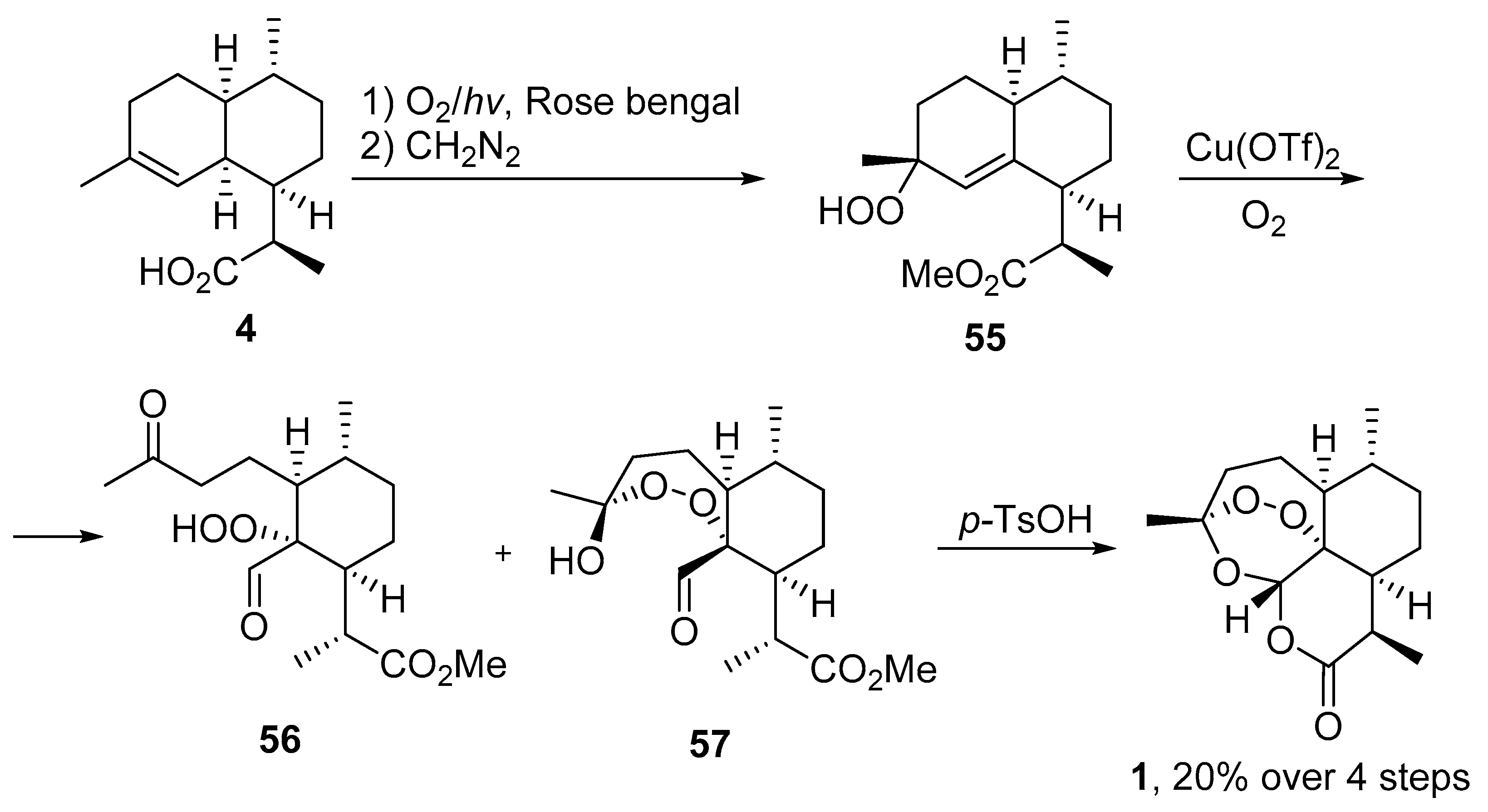
| 4 | 19 | O2/hv, Rose bengal, CH3CN, −30 °C, 6 h | (1) O2, Cu(OTf)2, CH3CN, −20 °C; | 25 | [66] |
| (2) p-TsOH (cat.), CH2Cl2, 20 °C, 4 h |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vil’, V.A.; Yaremenko, I.A.; Ilovaisky, A.I.; Terent’ev, A.O. Synthetic Strategies for Peroxide Ring Construction in Artemisinin. Molecules 2017, 22, 117. https://doi.org/10.3390/molecules22010117
Vil’ VA, Yaremenko IA, Ilovaisky AI, Terent’ev AO. Synthetic Strategies for Peroxide Ring Construction in Artemisinin. Molecules. 2017; 22(1):117. https://doi.org/10.3390/molecules22010117
Chicago/Turabian StyleVil’, Vera A., Ivan A. Yaremenko, Alexey I. Ilovaisky, and Alexander O. Terent’ev. 2017. "Synthetic Strategies for Peroxide Ring Construction in Artemisinin" Molecules 22, no. 1: 117. https://doi.org/10.3390/molecules22010117