3.2. Synthesis
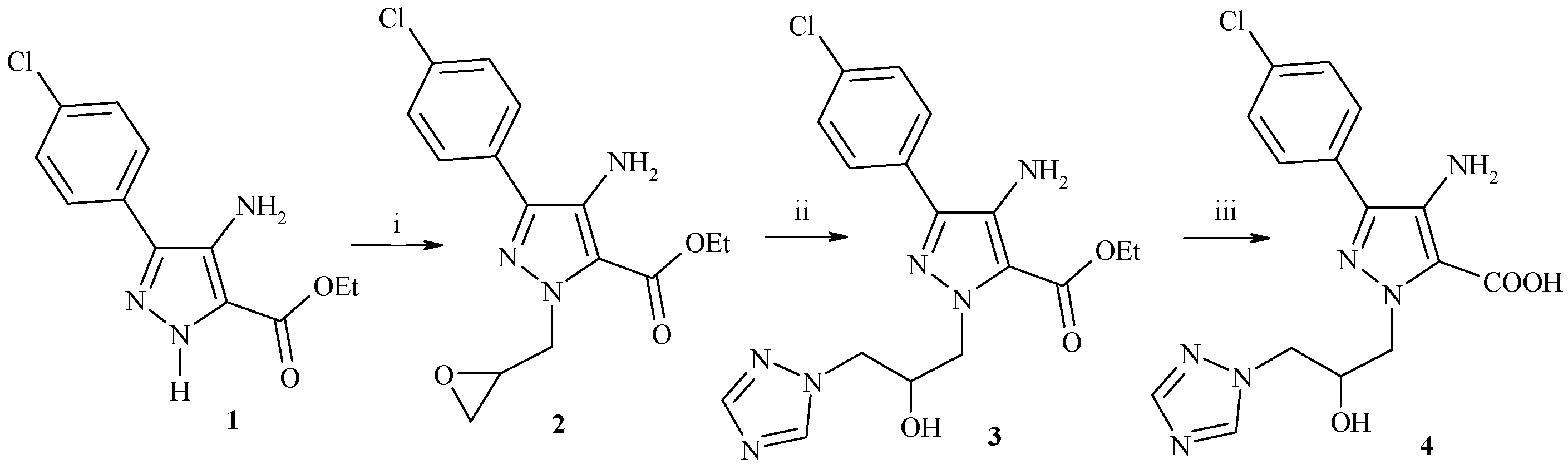
Ethyl 4-amino-3-(4-chlorophenyl)-1H-pyrazol-5-carboxylate (
1) was achieved by a reported method [
19,
20,
21,
22].
Ethyl 4-amino-3-(4-chlorophenyl)-1-(oxiran-2-ylmethyl)-1H-pyrazole-5-carboxylate (2). A mixture of compound 1 (2.65 g, 10 mmol) and potassium carbonate (1.51 g, 12 mmol) was stirred in 2-(chloromethyl)-oxirane (15 mL) under reflux for 4 h. After the reaction was completed, the mixture was cooled to room temperature, the excess 2-(chloromethyl)-oxirane was evaporated under reduced pressure, and water was added. The residue was extracted with chloroform, the combined organic phase was dried over anhydrous sodium sulfate, and then the residue was purified by column chromatography (eluent, chloroform/methanol 70:1, v/v) to give the desired compound 2 as a yellow solid, yield: 70%, m.p. 210–212 °C; IR (KBr, cm−1): ν 3390 (NH2), 3020 (CH-arom.), 2978 (CH-aliph.), 1725(C=O), 1610 (C=N); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.35 (t, 3H, CH3), 2.54–2.55 (m, 2H, OCH2), 3.22–3.40 (m, 2H, N-CH2), 4.25 (q, 2H, OCH2CH3), 4.85–4.90 (m, 1H, O-CH), 7.25 (d, 2H, J = 8.2 Hz, Ar-H), 7.57 (d, 2H, J = 8.2 Hz, Ar-H), 7.80 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 321 (M+); C15H16ClN3O3 (321.7); calcd; % C: 55.99, % H: 5.01, % N: 13.06; found; % C: 55.79, % H: 5.00, % N: 13.03.
Ethyl 4-amino-3-(4-chlorophenyl)-1-[2-hydroxy-3-(1H-1,2,4-triazol-1-yl)-propyl]-1H-pyrazole-5-carboxylate (3) To a stirred suspension of potassium carbonate (1.51 g, 12 mmol) in ethanol was added 1,2,4-triazole (6.91 g, 10 mmol). The mixture was stirred at 60 °C for 1 h. The reaction was cooled to room temperature, compound 2 (3.22 g, 10 mmol) was added at the room temperature and stirred for 2 h under reflux. After the reaction came to an end, the solvent was evaporated and the residue was extracted with chloroform. The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography (eluent, chloroform/methanol 30:1, v/v) to afford compound 3 as a white solid. Yield: 60%; m.p. 250 °C; IR (KBr, cm−1): ν 3443 (broad OH, NH2), 3016 (CH-arom.), 2989 (CH-aliph.), 1730(C=O), 1615 (C=N), 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 1.26 (t, 3H, CH3), 4.19–4.69 (m, 6H, -CH2-), 4.97 (m, 1H, O-CH), 5.66 (m, 1H, OH), 7.30 (d, 2H, J = 8.4 Hz, Ar-H), 7.62 (d, 2H, J = 8.5 Hz, Ar-H), 7.88 (br, 2H, NH2, D2O exchangeable), 8.07 (s, 1H, triazole 5-H), 8.85 (s, 1H, triazole 3-H); its MS (m/z), 390 (M+); C17H19ClN6O3 (390.8): calcd; % C: 52.24, % H: 4.90, % N: 21.50; found; % C: 52.21, % H: 4.90, % N: 21.48.
4-Amino-3-(4-chlorophenyl)-1-[2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl]-1H-pyrazole-5-carboxylic acid (4). To a stirred solution of sodium hydroxide (3%, 20 mL) was added compound 3 the mixture was stirred at 100 °C for 3 h. After the reaction was completed, the mixture was treated with formic acid to adjust the pH to 7, and then the suspension was filtered and washed with water three times to give compound 4 as a white solid. Yield: 75%; m.p. 270–272 °C; IR (KBr, cm−1): ν 3420 (broad OH, NH2), 3020 (CH-arom.), 2992 (CH-aliph.), 1730(C=O), 1618 (C=N), 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 4.36–4.84 (m, 4H, 2CH2), 5.05 (m, 1H, O-CH), 5.77 (m, 1H, OH), 7.25 (d, 2H, J = 8.0 Hz, Ar-H), 7.60 (d, 2H, J = 8.1 Hz, Ar-H), 7.85 (br, 2H, NH2, D2O exchangeable), 8.07 (s, 1H, triazole 5-H), 8.95 (s, 1H, triazole 3-H), 14.90 (s, 1H, COOH); its MS (m/z), 362 (M+), C15H15ClN6O3 (362.7): calcd: % C: 49.66, % H: 4.17, % N: 23.17; found; % C: 49.65, % H: 4.15, % N: 23.14.
3.2.1. General Procedure for the Synthesis of 6 and 9
A mixture of compound 1 (2.65 g, 10 mmol) and 1,4-dibromobutane 5 (2.16 g, 10 mmol) and /or (E)-1,4-dibromobut-2-ene 8 (2.14 g, 10 mmol) in dry DMF (10 ml) along with anhydrous potassium carbonate (1.88 g, 15 mmol) was stirred at room temperature for about five hours. The reaction mixture was then poured into ice cold water. The contents were then extracted with diethyl ether and the ether layer was washed with a brine solution and dried over anhydrous sodium sulfate. The solvent was removed and the crude product so obtained was crystallized from ethanol.
Ethyl 4-amino-1-(4-bromobutyl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (6). Pale yellow solid. Yield: 75%, m.p. 190–192 °C; IR (KBr, cm−1): ν 3390, 3360 (NH2), 3018 (CH-arom.), 2983 (CH-aliph.), 1725 (C=O), 1620 (C=N), 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 1.35 (t, 3H, CH3), 2.20–2.25 (m, 4H, 2CH2), 3.40 (t, 2H, CH2), 4.10 (q, 2H, OCH2), 4.28 (t, 2H, CH2), 7.25 (d, 2H, J = 8.2 Hz, Ar-H), 7.57 (d, 2H, J = 8.3 Hz, Ar-H), 7.80 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 400 (M+), C16H19BrClN3O2 (400.6): calcd; % C: 47.96, % H: 4.78, % N: 10.49; found; % C: 47.94, % H: 4.77, % N: 10.47.
Ethyl 4-amino-1-[(2E)-(4-bromobut-2-en-1-yl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (9). Pale yellow solid. Yield: 70%, m.p. 196–198 °C; IR (KBr, cm−1): ν 3400, 3375 (NH2), 3020 (CH-arom.), 2980 (CH-aliph.), 1725 (C=O), 1618 (C=N), 1585 (C=C); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 1.33 (t, 3H, CH3), 3.54 (t, 2H, CH2), 4.14 (t, 2H, CH2), 4.27 (q, 2H, OCH2), 6.09–6.12 (m, 1H, =CH), 6.13–6.19 (m, 1H, =CH), 7.28 (d, 2H, J = 8.3 Hz, Ar-H), 7.60 (d, 2H, J = 8.4 Hz, Ar-H), 7.90 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 398 (M+), C16H17BrClN3O2 (398.6): calcd; % C: 48.20, % H: 4.30, % N: 10.54; found; % C: 48.19, % H: 4.28, % N: 10.51.
3.2.2. General Procedure for Synthesis of 7a–d and 10a–d
N-Nucleophile (10 mmol) was dissolved in DMF (20 mL) along with anhydrous potassium carbonate (15 mmol) The bromo compound (6 and 9) (10 mmol) was added to the solution, and the mixture was stirred at room temperature for 3 h. After completion of the reaction, the mixture was poured into ice cold water. The precipitate obtained was washed with water, dried under a vacuum, and crystallized from ethanol to afford 7a–d and from dioxane to afford 10a–d in good yield.
Ethyl 4-amino-1-(4-(piperidin-1-yl)butyl))-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (7a). White solid, m.p. 160–162 °C; IR (KBr, cm−1): ν 3385, 3360 (NH2), 3023 (CH-arom.), 2978 (CH-aliph.), 1730 (C=O), 1622 (C=N), 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 1.34 (t, 3H, CH3), 1.44–1.55 (m, 2H, piperidine), 1.61–1.63 (m, 4H, piperidine), 1.77–1.82 (m, 2H, piperidine), 2.02 (m, 2H, piperidine), 2.22–2.30 (m, 4H, 2CH2), 3.40 (t, 2H, CH2), 3.68 (t, 2H, CH2), 4.09 (q, 2H, OCH2), 7.20 (d, 2H, J = 8.1 Hz, Ar-H), 7.57 (d, 2H, J = 8.3 Hz, Ar-H), 7.84 (br, 2H, NH2, D2O exchangeable), 13C-NMR (DMSO-d6) δ 18.28 (CH3), 23.86, 24.39, 25.19, 25.44, 25.89, 39.87 (6CH2), 42.58 (OCH2), 48.15, 48.67 (2 NCH2), 117.12, 120.83, 122.37, 124.41, 125.82, 129.92, 130.18, 151.4, 153.07 (nine signals for 9 sp2 carbon), 172.64 ppm (carbonyl) ppm; its MS (m/z), 404 (M+), C21H29ClN4O2 (404.9): calcd; % C: 62.29, % H: 7.22, % N: 13.84; found; % C: 62.29, % H: 7.20, % N: 13.81.
Ethyl 4-amino-1-(4-(4-methylpiperazin-1-yl)butyl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxy-late (7b). White solid, m.p. 156–158 °C; IR (KBr, cm−1): ν 3395, 3363 (NH2), 3020 (CH-arom.), 2982 (CH-aliph.), 1725 (C=O), 1622 (C=N), 1H-NMR (500 MHz, DMSO-d6, δ, ppm), 1.33 (t, 3H, CH3), 1.75–1.80 (m, 4H, 2CH2), 2.03 (t, J = 6.8 Hz, 2H, CH2), 2.29 (s, 3H, piperazinyl NCH3), 2.59 (brs, 4H, piperazinyl, CH3N(CH2)2), 3.36 (brs, 4H, piperazinyl, N(CH2)2), 4.02 (t, J = 6.0 Hz, 2H, CH2), 4.15 (q, 2H, OCH2), 7.23 (d, 2H, J = 8.0 Hz, Ar-H), 7.58 (d, 2H, J = 8.2 Hz, Ar-H), 7.90 (br, 2H, NH2, D2O exchangeable), its MS (m/z), 419 (M+), C21H30ClN5O2 (419.9): calcd; % C: 60.06, % H: 7.20, % N: 16.68; found; % C: 60.04, % H: 7.20, % N: 16.66.
Ethyl 4-amino-1-(4-morpholinobutyl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (7c). White solid, m.p. 175–178 °C; IR (KBr, cm−1): ν 3400, 3370 (NH2), 3022 (CH-arom.), 2978 (CH-aliph.), 1730 (C=O), 1618 (C=N); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.33 (t, 3H, CH3), 1.78–1.80 (m, 4H, 2CH2), 2.03–2.05 (m, 2H, CH2), 2.20 (t, 2H, CH2), 3.29 (t, 4H, morpholinyl, N(CH2)2, J = 5.0 Hz), 3.90 (t, 4H, morpholinyl, O(CH2)2), J = 5.0 Hz), 4.05 (t, 2H, CH2), 4.15 (q, 2H, OCH2), 7.20 (d, 2H, J = 8.3 Hz, Ar-H), 7.58 (d, 2H, J = 8.4 Hz, Ar-H), 7.89 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 406 (M+); C20H27ClN4O3 (406.9); calcd; % C: 59.03, % H: 6.69, % N: 13.77; found; % C: 59.02, % H: 6.67, % N: 13.75.
Ethyl 4-amino-1-(4-(pyrrolidin-1-yl)butyl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (7d). White solid, m.p. 173–175 °C; IR (KBr, cm−1): ν 3410, 3380 (NH2), 3022 (CH-arom.), 2978 (CH-aliph.), 1726 (C=O), 1625 (C=N); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.35 (t, 3H, CH3), 1.75–1.79 (m, 2H, CH2), 2.05–2.08 (m, 2H, CH2), 2.20 (t, 2H, CH2), 2.53 (brs, 4H, 2CH2 pyrrole), 2.76–2.89 (br, 2H, 2CH2), 4.09 (t, 2H, CH2), 4.22 (q, 2H, OCH2), 7.25 (d, 2H, Ar-H), 7.64 (d, 2H, J = 8.3 Hz, Ar-H), 7.89 (br, 2H, J = 8.5 Hz, NH2, D2O exchangeable); its MS (m/z), 390 (M+); C20H27ClN4O2 (390.9); calcd; % C: 61.45, % H: 6.69, % N: 14.33; found; % C: 61.43, % H: 6.67, % N: 14.30.
(E)-Ethyl 4-amino-1-(4-(piperidin-1-yl)but-2-en-1-yl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (10a). White solid, m.p. 223–225 °C; IR (KBr, cm−1): ν 3380 (NH2), 3020 (CH-arom.), 2980 (CH-aliph.), 1726 (C=O), 1628 (C=N), 1595(C=C), 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 1.35 (t, 3H, CH3), 2.72 (brs, 4H, piperidine), 2.39 (brs, 4H, piperidine), 1.59 (m, 2H, piperidine), 3.04 (d, 2H, CH2), 4.15 (d, 2H, CH2), 4.30 (q, 2H, OCH2), 5.89–6.00 (m, 2H, 2CH), 7.30 (d, 2H, J = 8.0 Hz, Ar-H), 7.65 (d, 2H, J = 8.2 Hz, Ar-H), 7.93 (br, 2H, NH2, D2O exchangeable); 13C-NMR (DMSO-d6) δ 19.14 (CH3), 24.76, 25.56, 25.95, 40.11 (4CH2), 42.58 (OCH2), 48.15, 48.67 (2NCH2), 118.19, 119.38, 120.87, 123.27, 124.78, 125.90, 129.25, 130.18, 131.21, 151.81, 153.11 (11 signals for 11 sp2 carbon), 173.21 ppm (carbonyl) ppm; its MS (m/z), 402 (M+); C21H27ClN4O2 (402.9): calcd: % C: 62.60, % H: 6.75, % N: 13.91; found; % C: 62.59, % H: 6.73, % N: 13.90.
(E)-Ethyl-4-amino-1-(4-(4-methylpiperazin-1-yl)but-2-en-1-yl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxy-late (10b). White solid, m.p. 185–187 °C; IR (KBr, cm−1): ν 3387 (NH2), 3022 (CH-arom.), 2980 (CH-aliph.), 1727 (C=O), 1630 (C=N), 1590 (C=C); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.30 (t, 3H, CH3), 2.03 (d, 2H, CH2), 2.20 (s, 3H, -NCH3), 2.54 (brs, 4H, piperazinyl, -N(CH2)2), 3.40 (brs, 4H, piperazinyl, N(CH2)2), 4.09 (d, 2H, CH2), 4.20 (q, 2H, OCH2), 5.88–5.90 (m, 1H, CH), 5.97–5.99 (m, 1H, CH), 7.25 (d, 2H, J = 8.2 Hz, Ar-H), 7.60 (d, 2H, J = 8.2 Hz, Ar-H), 7.90 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 417 (M+); C21H28ClN5O2 (417.9); calcd; % C: 60.35, % H: 6.75, % N: 16.76; found; % C: 60.32, % H: 6.73, % N: 16.75.
(E)-Ethyl 4-amino-1-(4-morpholin-1-yl)but-2-en-1-yl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (10c). White solid, m.p. 215–217 °C; IR (KBr, cm−1): ν 3383 (NH2), 3028 (CH-arom.), 2978 (CH-aliph.), 1728 (C=O), 1625 (C=N), 1595 (C=C); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.30 (t, 3H, CH3), 2.20 (d, 2H, CH2), 3.30 (t, 4H, morpholinyl, N(CH2)2), 3.95 (t, 4H, morpholinyl, O(CH2)2), 4.05 (d, 2H, CH2), 4.20 (q, 2H, OCH2), 5.98–6.12 (m, 2H, 2CH), 7.23 (d, 2H, J = 8.4 Hz, Ar-H), 7.58 (d, 2H, J = 8.5 Hz, Ar-H), 7.89 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 404 (M+); C20H25ClN4O3 (404.8); calcd; % C: 59.33, % H: 6.22, % N: 13.84; found; % C: 59.32, % H: 6.22, % N: 13.82.
(E)-Ethyl 4-amino-1-(4-(pyrrolidin-1-yl)but-2-en-1-yl)-3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (10d). White solid, m.p. 180–182 °C; IR (KBr, cm−1): ν 3380 (NH2), 3022 (CH-arom.), 2978 (CH-aliph.), 1730 (C=O), 1628 (C=N),1590 (C=C); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.29 (t, 3H, CH3), 2.25 (d, 2H, CH2), 2.55 (brs, 4H, 2CH2 pyrrole), 2.69–2.78 (brs, 4H, 2CH2 pyrrole), 4.10 (d, 2H, CH2), 4.22 (q, 2H, OCH2), 5.92–6.12 (m, 2H, 2CH), 7.28 (d, 2H, J = 8.2 Hz, Ar-H), 7.66 (d, 2H, J = 8.3 Hz, Ar-H), 7.92 (br, 2H, NH2, D2O exchangeable); its MS (m/z), 388 (M+); C20H25ClN4O2 (388.8); calcd; % C: 61.77, % H: 6.48, % N: 14.41; found; % C: 61. 75, % H: 6. 47, % N: 14. 39.
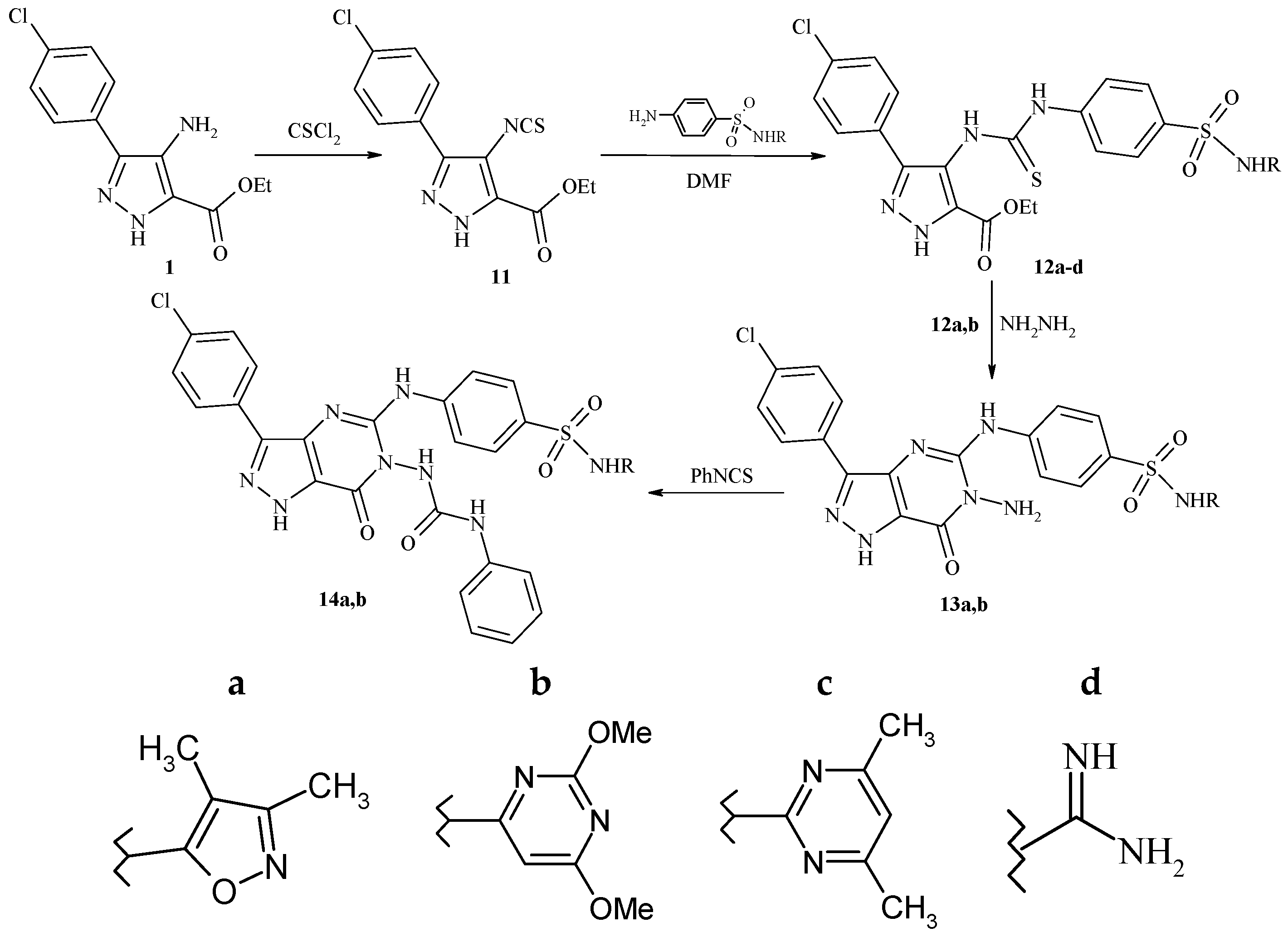
Ethyl 3-(4-chlorophenyl)-4-isothiocyanato-1H-pyrazol-5-carboxylate (11). A mixture of compound 1 (2.65 g, 10 mmol) and thiophosgene (1.14 mL, 10 mmol) in dry chloroform (25 mL) was stirred under reflux for 5 h. The solvent was evaporated and the solid obtained was recrystallized from ethanol to give 11 as a deep yellow solid, yield: 70%. m.p. 190–192 °C; IR (KBr, cm−1): ν 3386 (NH), 3019 (CH-arom.), 2980 (CH-aliph.), 1728 (C=O), 1604 (C=N), 1265 (C=S); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.35 (t, 3H, CH3), 4.20 (q, 2H, OCH2), 7.25 (d, 2H, J = 8.0 Hz, Ar-H), 7.58 (d, 2H, J = 8.1, Hz Ar-H), 9.05 (br, 1H, NH, D2O exchangeable); its MS (m/z), 307 (M+); C13H10ClN3O2S (307.7); calcd; % C: 50.73, % H: 3.28, % N: 13.65; found; % C: 50.71, % H: 3.26, % N: 13.62.
3.2.3. General Procedure for Synthesis of 12a–d
A mixture of compound 11 (3.07 g, 10 mmol) and sulfa drugs (10 mmol) in DMF (30 mL) was stirred under reflux for 4 h. The reaction mixture was poured onto ice water and the obtained product was recrystallized from dioxane to give compounds 12a–d, respectively.
Ethyl-3-(4-chlorophenyl)-3-(4-(N-(3,4-dimethyl[1,2]isooxazolo)sulfamoyl)phenyl)thioureido-1H-pyrazole-5-carboxylate (12a). Pale brown solid, yield: 75%, m.p. 250–252 °C; IR (KBr, cm−1): ν 3386, 3350, 3201 (NH), 3019 (CH-arom.), 2989, 2850 (CH-aliph.), 1725 (C=O),, 1612 (C=N), 1260 (C=S), 1325, 1153 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.29 (t, 3H, CH3), 2.20 (s, 3H, CH3), 2.28 (s, 3H, CH3), 4.18 (q, 2H, OCH2), 7.20 (d, 2H, J = 8.1 Hz, Ar-H), 7.55 (d, 2H, J = 8.2 Hz, Ar-H), 7.88 (d, 2H, J = 8.2 Hz, Ar-H), 8.10 (d, 2H, J = 8.2 Hz, Ar-H), 9.00 (s, 1H, NH-Ph, exchangeable with D2O), 9.15 (br, 2H, 2NH, D2O exchangeable), 10.90 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 575 (M+); C24H23ClN6O5S2 (575); calcd; % C: 50.13, % H: 4.03, % N: 14.61; found; % C: 50.11, % H: 4.00, % N: 14.60.
Ethyl-3-(4-chlorophenyl)-3-(4-(N-(2,6-dimethoxypyrimidin-4-yl)sulfamoyl)phenyl)thioureido-1H-pyrazole-5-carboxylate (12b). Brown solid, yield: 65%, m.p. 279–281 °C; IR (KBr, cm−1): ν 3398, 3274, 3178 (NH), 3070 (CH-arom.), 2970, 2860 (CH-aliph.), 1732 (C=O), 1620 (C=N), 1265 (C=S), 1388, 1168 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.29 (t, 3H, CH3), 3.80, 3.90 (2s, 6H, 2OCH3), 4.15 (q, 2H, OCH2), 6.90 (d, 2H, J = 8.0 Hz, Ar-H), 7.20 (d, 2H, J = 8.1 Hz, Ar-H), 7.54 (d, 2H, J = 8.3 Hz, Ar-H), 7.90 (d, 2H, J = 8.3 Hz, Ar-H), 8.60 (s, 1H, CH pyrimidine), 11.10 (s, 3H, 3NH, exchangeable with D2O), 12.40 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 618 (M+); C25H24ClN7O6S2 (618.08); calcd; % C: 48.58, % H: 3.91, % N: 15.86; found; % C: 48.55, % H: 3.90, % N: 15.84.
Ethyl-3-(4-chlorophenyl)-3-(4-(N-(4,6-dimethylypyrimidin-2-yl)sulfamoyl)phenyl)thioureido-1H-pyrazole-5-carboxylate (12c). Brown solid, yield: 65%, m.p. 290–292 °C; IR (KBr, cm−1): ν 3390, 3251 (NH), 3035 (CH-arom.), 2950, 2865 (CH-aliph.), 1710 (C=O), 1600 (C=N), 1265 (C=S), 1345, 1161 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.30 (t, 3H, CH3), 2.30 (s, 6H, 2CH3), 4.10 (q, 2H, OCH2), 6.85 (d, 2H, J = 8.1 Hz, Ar-H), 7.19 (d, 2H, J = 8.3 Hz, Ar-H), 7.50 (d, 2H, J = 8.1 Hz, Ar-H), 7.80 (d, 2H, J = 8.4 Hz, Ar-H), 8.05 (s, 1H, CH pyrimidine), 9.00 (br, 1H, NH, D2O exchangeable), 11.40 (s, 2H, 2NH, exchangeable with D2O), 12.10 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 586 (M+); C25H24ClN7O4S2 (586.08); calcd; % C: 51.23, % H: 4.13, % N: 16.73; found; % C: 51.22, % H: 4.11, % N: 16.71.
Ethyl-3-(4-chlorophenyl)-N-carbamimidoylbenzenesulfonamido-4-thioureido-1H-pyrazole-5-carboxylate (12d). Orange solid, yield: 60%, m.p. >300 °C; IR (KBr, cm−1): ν 3250, 3125 (NH, NH2), 3050 (CH-arom.), 2970, 2850 (CH-aliph.), 1720 (C=O), 1604 (C=N), 1265 (C=S), 1350, 1113 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.23 (t, 3H, CH3), 3.54 (s, 1H, NH, exchangeable with D2O), 4.18 (q, 2H, OCH2), 6.80 (s, 2H, NH2, exchangeable with D2O), 7.25 (d, 2H, J = 8.2 Hz, Ar-H), 7.49 (d, 2H, J = 8.3 Hz, Ar-H), 7.77 (d, 2H, J = 8.2 Hz, Ar-H), 7.90 (d, 2H, J = 8.2 Hz, Ar-H), 9.05 (br, 1H, NH, D2O exchangeable), 11.30 (s, 2H, 2NH thiourea, exchangeable with D2O), 13.10 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 522 (M+); C20H20ClN7O4S2 (522.0); calcd; % C: 46.02, % H: 3.86, % N: 18.78; found; % C: 46.00, % H: 3.84, % N: 18.75.
3.2.4. General Procedure for Synthesis of 13a,b
A mixture of 12a,b (10 mmol) and hydrazine hydrate (80%, 7mL) in butanol (30 mL) was refluxed for 5 h. After cooling, the reaction mixture was poured onto ice water and the solid obtained was recrystallized from dioxane to give 13a,b, respectively.
4-{[6-Amino-(N-(3,4-dimethyl[1,2]isooxazolo)sulfamoyl)phenyl)amino}-3-(4-chlorophenyl)-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidine (13a). Yellow solid, yield: 73%, m.p. >300 °C; IR (KBr, cm−1): ν 3350, 3201 (NH, NH2), 3055 (CH-arom.), 2989, 2850 (CH-aliph.), 1698 (C=O), 1612 (C=N), 1315, 1153 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 2.20 (s, 3H, CH3), 2.26 (s, 3H, CH3), 5.60 (s, 2H, N-NH2, exchangeable with D2O), 7.00 (d, 2H, J = 8.3 Hz, Ar-H), 7.30 (d, 2H, J = 8.1 Hz, Ar-H), 7.89 (d, 2H, J = 8.2 Hz, Ar-H), 8.10 (d, 2H, J = 8.2 Hz, Ar-H), 9.00, 9.15, 10.90 (3br, 3NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ 20.47, 21.32 (2 CH3), 118.23, 120.81, 121.27, 122.81, 123.29, 125.65, 126.91, 130.55, 135.12, 137.31, 151.80, 153.17, 154.12, 154.28, 155.23, 155.87, 156.31, 156.92, 157.81, 168.21 ppm (20 signals for 20 sp2 carbon); its MS (m/z), 526 (M+); C22H19ClN8O4S (526.9); calcd; % C: 50.14, % H: 3.63, % N: 21.26; found; % C: 50.12, % H: 3.61, % N: 21.23.
4-{[6-Amino-(N-(2,6-dimethoxypyrimidin-4-yl)sulfamoyl)phenyl)amino]}-3-(4-chlorophenyl)-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidine (13b). Pale yellow solid, yield: 65%, m.p. >300 °C; IR (KBr, cm−1): ν 3301, 3205 (NH, NH2), 3035 (CH-arom.), 2993, 2889 (CH-aliph.), 1685 (C=O), 1627 (C=N), 1315, 1157 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 3.82 (s, 6H, 2OCH3), 5.50 (s, 2H, N-NH2,exchangeable with D2O), 6.90 (d, 2H, J = 8.2 Hz, Ar-H), 7.20 (d, 2H, J = 8.1 Hz, Ar-H), 7.58 (d, 2H, J = 8.3 Hz, Ar-H), 7.90 (d, 2H, J = 8.2 Hz, Ar-H), 8.01 (s, 1H, CH pyrimidine), 8.80 (s, 1H, NH, exchangeable with D2O), 9.15 (br, 1H, 1NH, D2O exchangeable), 10.80 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 569 (M+); C23H20ClN9O5S (569.9); calcd; % C: 48.47, % H: 3.54, % N: 22.12; found; % C: 48.46, % H: 3.54, % N: 22.10.
3.2.5. General Procedure for Synthesis of 14a,b
A mixture of compound (13a,b) (10 mmol) and phenyl isothiocyanate (1.35 g, 10 mmol) in ethanol (50 mL) was refluxed for 10 h. The solvent was then concentrated and the residue was recrystallized from ethanol to give 14a,b, respectively.
N-(3,4-Dimethyl[1,2]isooxazol-5-yl)-4-3-(4-chlorophenyl)-7-oxo-6-[3-(3-phenylthioureido)-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidine-5-ylamino)benzenesulfonamide (14a). Brown solid, yield: 60%, m.p. >300 °C; IR (KBr, cm−1): ν 3205 (NH), 3035 (CH-arom.), 2927, 2865 (CH-aliph.), 1668 (C=O), 1550 (C=N), 1242 (C=S), 1315, 1168 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 2.15 (s, 3H, CH3), 2.21 (s, 3H, CH3), 6.90–7.02 (m, 5H, Ar-H), 7.23 (d, 2H, J = 8.0 Hz, Ar-H), 7.54 (d, 2H, J = 8.0 Hz, Ar-H), 7.89 (d, 2H, J = 8.2 Hz, Ar-H), 8.11 (d, 2H, J = 8.2 Hz, Ar-H), 8.51, 8.62 (2s, 2H, 2NH, exchangeable with D2O), 9.15 (s, 1H, NH exchangeable with D2O), 9.70 (s, 1H, NH-Ph, exchangeable with D2O), 10.72 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 662 (M+); C29H24ClN9O4S2 (662.1); calcd; % C: 52.60, % H: 3.65, % N: 19.04; found; % C: 52.59, % H: 3.63, % N: 19.02.
N-(2,6-Dimethoxypyrimidin-4-yl)-4-3-(4-chlorophenyl)-7-oxo-6-[3-(3-phenylthioureido)-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidine-5-ylamino)benzenesulfonamide (14b). Brown solid, yield: 63%, m.p. >300 °C; IR (KBr, cm−1): ν 3210 (NH), 3030 (CH-arom.), 2928, 2865 (CH-aliph.), 1669 (C=O), 1550 (C=N), 1240 (C=S), 1317, 1168 (SO2); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 3.86 (s, 6H, 2OCH3), 6.92–7.05 (m, 5H, Ar-H), 7.25 (d, 2H, J = 8.3 Hz, Ar-H), 7.54 (d, 2H, J = 8.1 Hz, Ar-H), 7.90 (d, 2H, J = 8.2 Hz, Ar-H), 8.14 (d, 2H, J = 8.2 Hz, Ar-H), 8.19 (s, 1H, CH pyrimidine), 8.50, 8.60 (2s, 2H, 2NH, exchangeable with D2O), 9.15 (s, 1H, NH exchangeable with D2O), 9.73 (s, 1H, NH, exchangeable with D2O), 10.90 (s, 1H, SO2NH, exchangeable with D2O); its MS (m/z), 705 (M+); C30H25ClN10O5S2 (705.1); calcd; % C: 51.10, % H: 3.57, % N: 19.86; found; % C: 51.10, % H: 3. 55, % N: 19. 84.
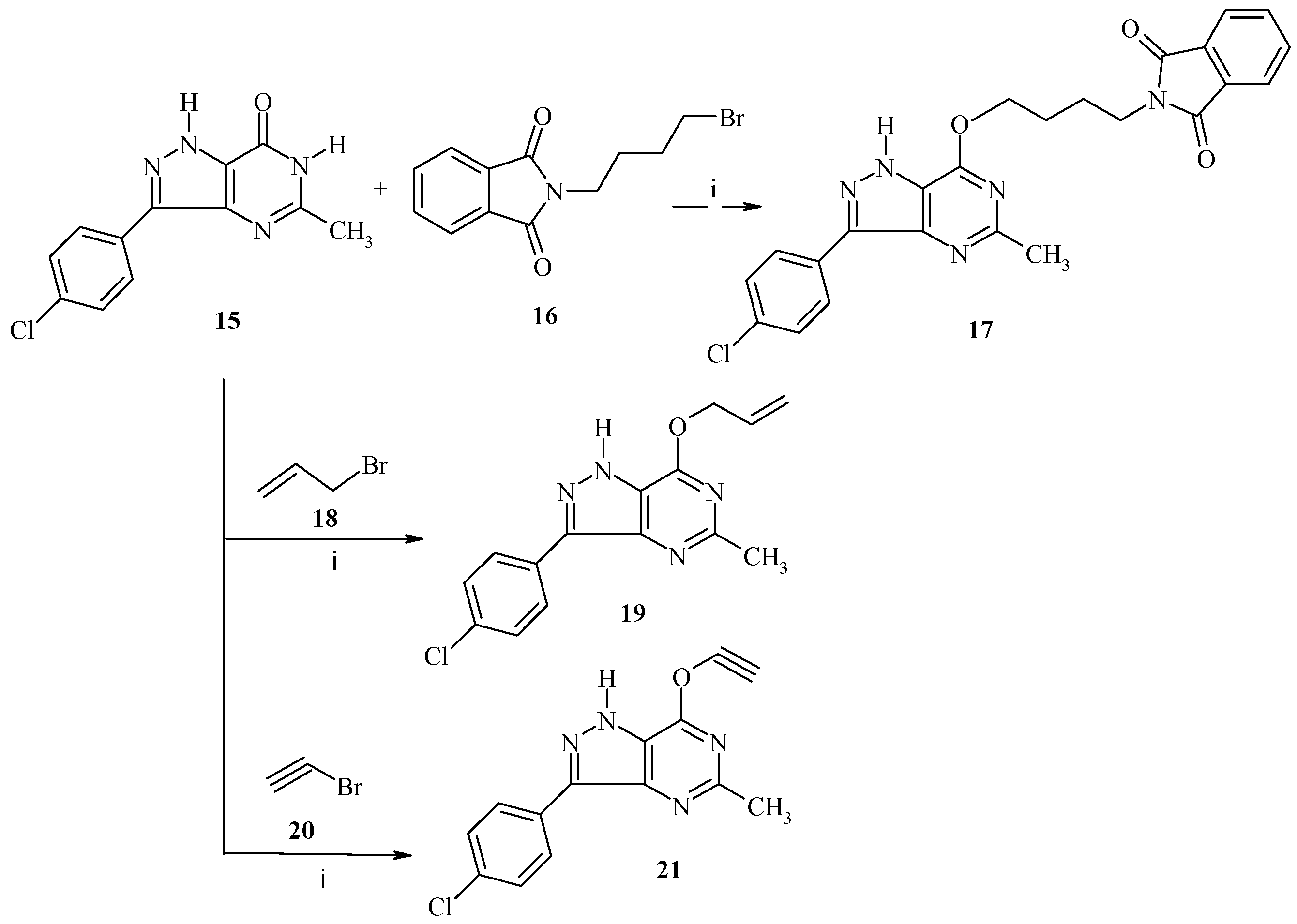
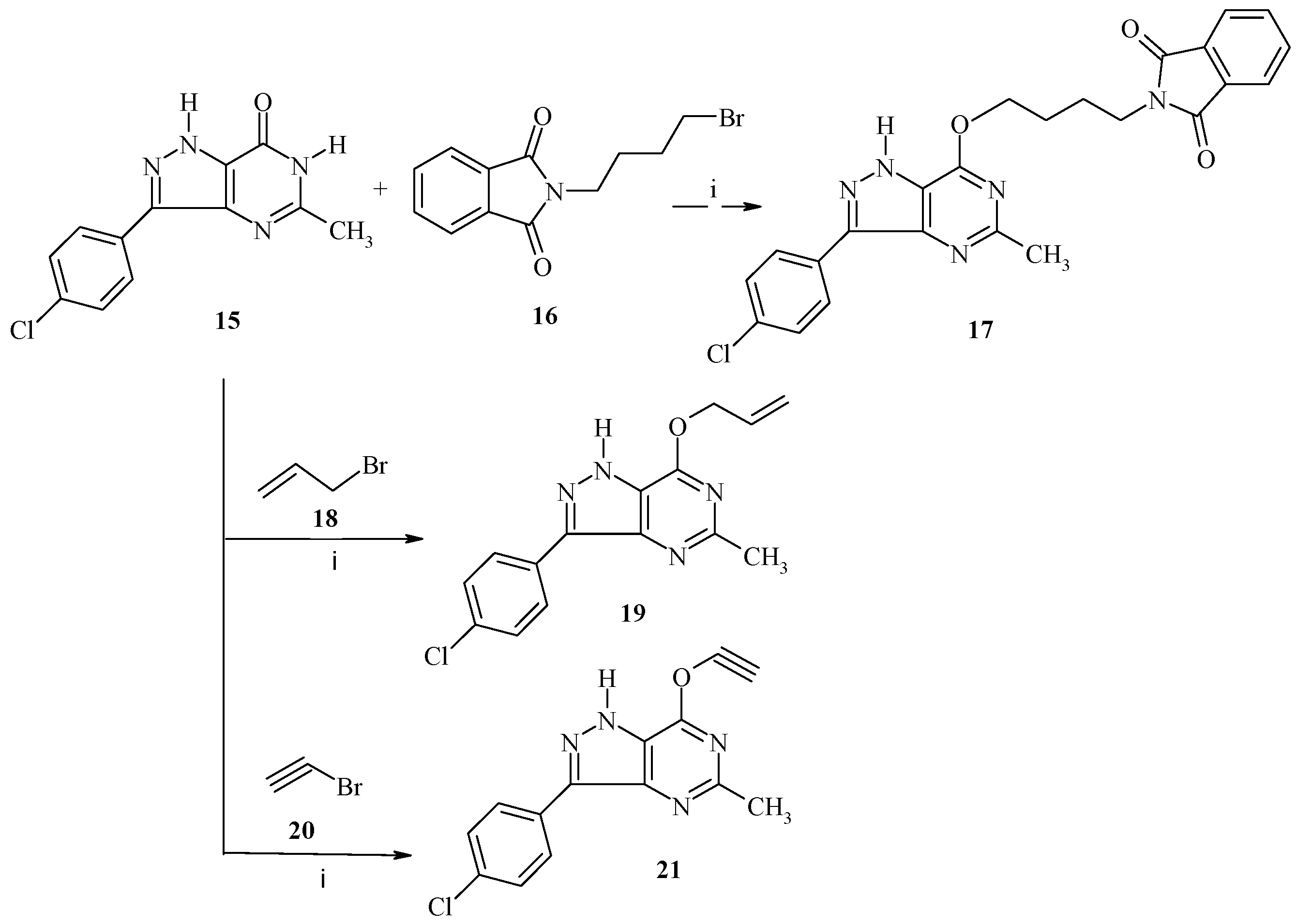
3-(4-Chlorophenyl)-5-methyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (15) was achieved by a reported method [
9].
3-(4-Chlorophenyl)-5-methyl-7-oxybutyl)-isoindoline-1,3-dioxo-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin (17). A mixture of 15 (2.60 g, 10 mmol), N-(4-bromobutyl)-phthalimide 16 (2.82 g, 10 mmol) and anhydrous K2CO3 (1.8, 10 mmol) in 5 mL DMF was stirred in a round bottom flask for 6–8 h at room temperature. The reaction mixture was poured onto ice water and the obtained product was recrystallized from dioxane to give compound 17 as colorless solid, yield: 72%, m.p. 290–292 °C; IR (KBr, cm−1): ν 3210 (NH), 3020 (CH-arom.), 2922, 2865 (CH-aliph.), 1680, 1668 (C=O), 1550 (C=N), 1390 (CH3), 1080 (C-O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.20 (s, 3H, CH3), 1.90 (brs, 4H, 2CH2), 3.72 (t, 2H, CH2, J = 6.6 Hz, 2H), 4.54 (t, J = 5.6 Hz, 2H, CH2), 7.02 (s, 1H, Ar-H), 7.30 (d, 2H, J = 8.1 Hz, Ar-H), 7.60 (d, 2H, J = 8.2 Hz, Ar-H), 7.70 (m, 1H-Ar), 7.78 (m, 1H, Ar-H), 7.91 (m, 1H, Ar-H), 9.30 (br, 1H, NH, D2O exchangeable); its MS (m/z), 461 (M+); C24H20ClN5O3 (461.9); calcd; % C: 62.41, % H: 4.36, % N: 15.16; found; % C: 62.39, % H: 4.34, % N: 15.15.
3.2.6. General Procedure for Synthesis of 19 and 21
A mixture of 15 (2.60 g, 10 mmol), was stirred with potassium carbonate (1.38 g, 10 mmol) in dry DMF (20 mL) for 1 h, followed by the addition of the appropriate alkenyl halide (10 mmol) in portions. The reaction mixture was stirred at room temperature overnight, then refluxed for 4 h. After cooling it was then filtered to remove insoluble materials, and the filtrate was poured into ice water to give the crude product as a precipitate, which in turn was filtered off and dried. The crude product obtained in all cases was purified by recrystallization from ethanol.
3-(4-Chlorophenyl)-5-methyl-7-(prop-2-en-1-yloxy)-1H-pyrazolo[4,3-d]pyrimidine (19). This compound was prepared from 15 (2.60 g, 10 mmol) and allyl bromide (1.2 g, 10 mmol) was obtained as a white powder, yield: 75%, m.p. 280–282 °C; IR (KBr, cm−1): ν 3220 (NH), 3080 (CH-arom.), 2930, 2870 (CH-aliph.), 1640 (C=C), 1550 (C=N), 1380 (CH3), 1150 (C-O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.23 (s, 3H, CH3), 4.12 (d, 2H, OCH2), 6.08–6.14 (m, 1H, =CH), 6.15–6.20 (m, 1H, CH2), 7.28 (d, 2H, J = 8.2 Hz, Ar-H), 7.58 (d, 2H, J = 8.2 Hz, Ar-H), 9.28 (br, 1H, NH, D2O exchangeable); its MS (m/z), 300 (M+); C15H13ClN4O (300.7); calcd; % C: 59.91, % H: 4.36, % N: 18.63; found; % C: 59.90, % H: 4.34, % N: 18.60.
3-(4-Chlorophenyl)-7-(ethynyloxy)-5-methyl-1H-pyrazolo[4,3-d]pyrimidine (21). This compound was prepared from 15 (2.60 g, 10 mmol) and bromoethyne (1.05 g, 10 mmol) was obtained as a white powder, yield: 80%, m.p. 268–270 °C; IR (KBr, cm−1): ν 3200 (NH), 3085 (CH-arom.), 2922, 2870 (CH-aliph.), 2150 (C≡C), 1600 (Aryl), 1550 (C=N), 1350 (CH3), 1153 (C-O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm); 1.28 (s, 3H, CH3), 3.68 (s, 1H, CH), 7.30 (d, 2H, J = 8.0 Hz, Ar-H), 7.68 (d, 2H, J = 8.2 Hz, Ar-H), 9.25 (br, 1H, NH, D2O exchangeable); its MS (m/z), 284 (M+); C14H9ClN4O (284.7); calcd; % C: 59.06, % H: 3.19, % N: 19.68; found; % C: 59.04, % H: 3.17, % N: 19.65.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}