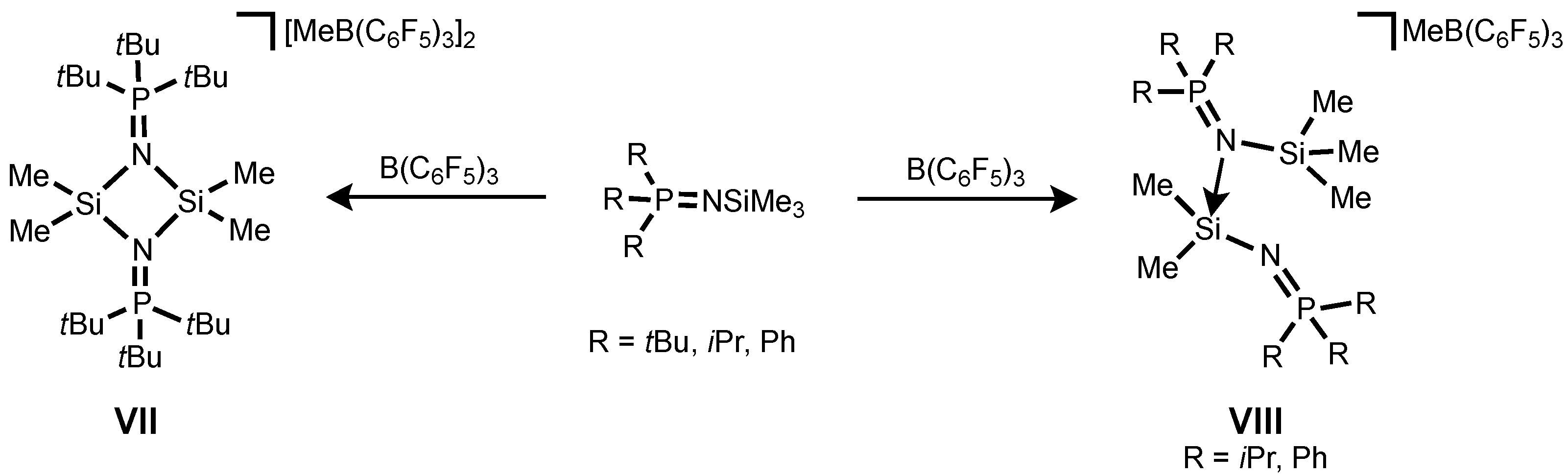1. Introduction
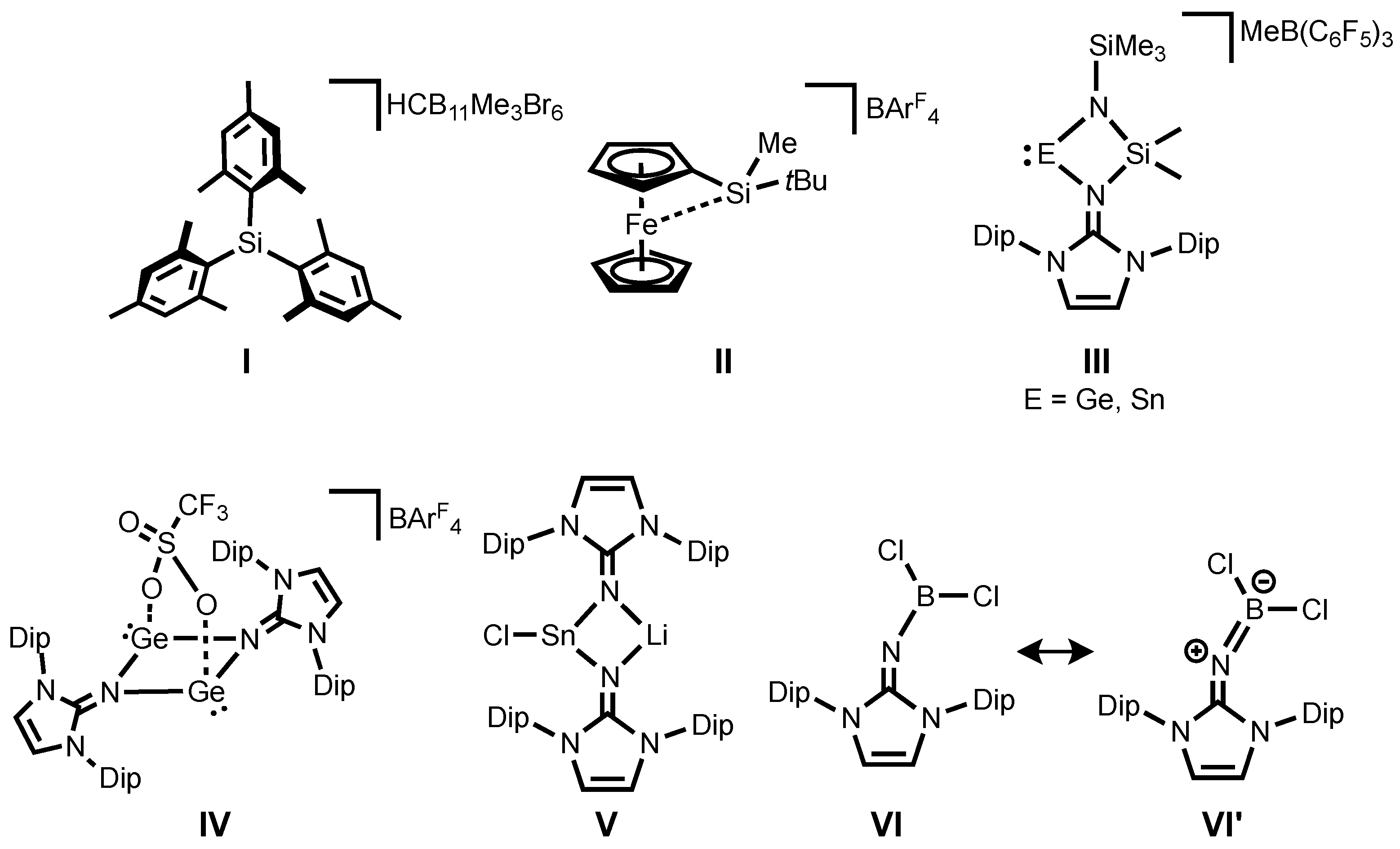
For decades, silicon analogues of carbenium ions, mostly referred to as silylium ions, have been attractive synthetic targets both for fundamental and applied research due to their electronic properties with strong Lewis acidity [
1]. Since the pioneering work on silylium ions via hydride abstraction utilizing trityl cation by Corey and co-workers [
2], several silylium ions were reported, although a weak interaction exists between a pronounced electrophilic silicon center and either solvent molecules or counteranions [
3,
4,
5,
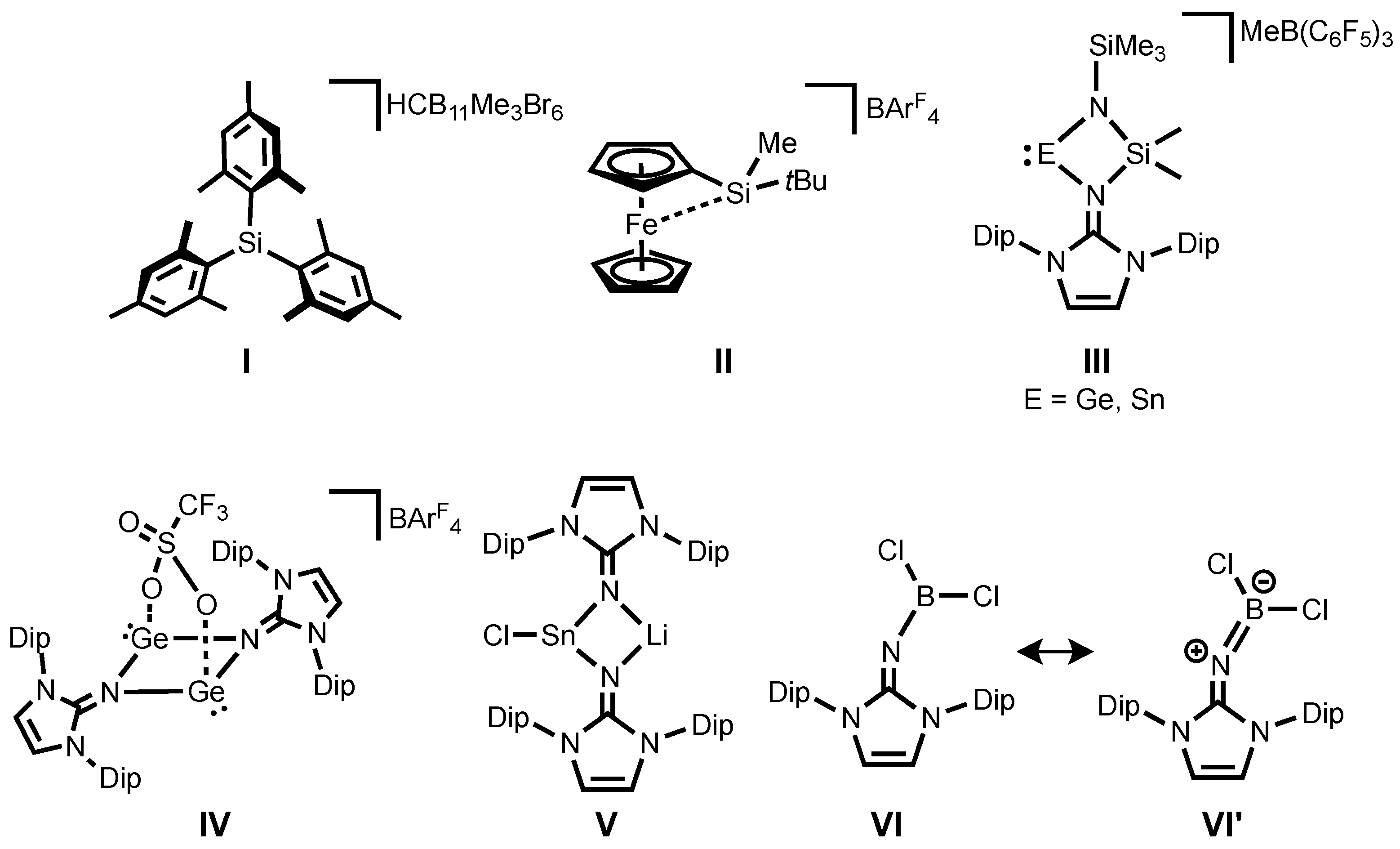
6]. It should be emphasized that the first structurally authenticated free silylium ion
I (
Figure 1) was reported by Lambert, Reed and co-workers in 2002 [
7]. Since then, several further examples of silyl cations have been reported [
8,
9,
10]. On the other hand, the utilization of silylium ions as catalysts has also been an area of intense research interest and their catalytic activity has been examined in various reactions [
11,
12]. For example, the groups of Ozerov as well as Müller have independently reported the defluorination of fluoro- and perfluoro-alkyl groups by using silylium ions [
13,
14,
15]. Moreover, Sawamura implemented a silylium ion as Lewis acid catalyst in Mukaiyama aldol and Diels-Alder reactions [
16]. Catalytic C–C bond forming reactions have also been reported [
17,
18]. Recently, Oestreich and co-workers demonstrated that a ferrocene-based silylium ion
II (
Figure 1) displays catalytic activity in Diels-Alder reactions [
19]. To date, most isolable silylium ions are typically limited to alkyl and aryl substituents and heteroatom-substituted silylium derivatives have remained extremely scarce. However, introduction of a heteroatom such as N, O, or S to the silylium center [
20,
21,
22] produced silylium ions with good catalytic activity [
23].
Tamm and co-workers developed a facile synthetic method for imidazolin-2-imino ligands and reported a number of metal complexes with these ligands [
24,
25,
26,
27]. During the last years,
N-heterocyclic imines (NHIs) have been exploited for the syntheses of various unique main group compounds [
28]. In Group 14, several metallylenes were synthesized by using NHIs as ligand [
29,
30,
31,
32], and two-coordinated metallyliumylidenes
III (
Figure 1) were also successfully isolated owing to stabilization of the NHI ligand by its high σ-donation together with delocalization of a positive charge around the imino fragment [
31,
32]. Shortly after, it was also demonstrated the NHIs effectively stabilize a spacer-separated bis(germyliumylidene)
IV (
Figure 1) [
33]. Interestingly, their strong coordination ability enables the synthesis and isolation of a rare halogen-substituted stannylenoid
V (
Figure 1) [
34] by coordinating of the imino-nitrogen to a Li cation, preventing the liberation of LiCl to form divalent tin species. Very recently, several imino-substituted borane compounds, of which the dichloro derivative
VI (
Figure 1) has a significant allenic-type character
VI’ (
Figure 1) were reported [
35]. Note that these iminoboranes act as Frustrated Lewis Pairs (FLPs), showing dehydrogenation of amine-borane. Motivated by this report and from the view point of their isoelectronic nature to silylium ions, iminosilylium ions would be an interesting synthetic target.
It is noteworthy that the syntheses of phosphinoimido complexes of silylium ions were described by Stephan and co-workers and they exist as a dimer
VII or a phosphinoimido-coordinated monomer
VIII depending on the steric crowding of the substituents (
tBu vs.
iPr and Ph) (
Scheme 1) [
36].
Herein we describe the facile synthesis of novel imino-substituted silylium ions by the abstraction of methyl group of the trimethylsilyl ligand. Moreover, base-coordinated silylium ion was also prepared by treatment of Me2(OTf)SiNIPr with DMAP by expelling the OTf anion.
2. Results and Discussion
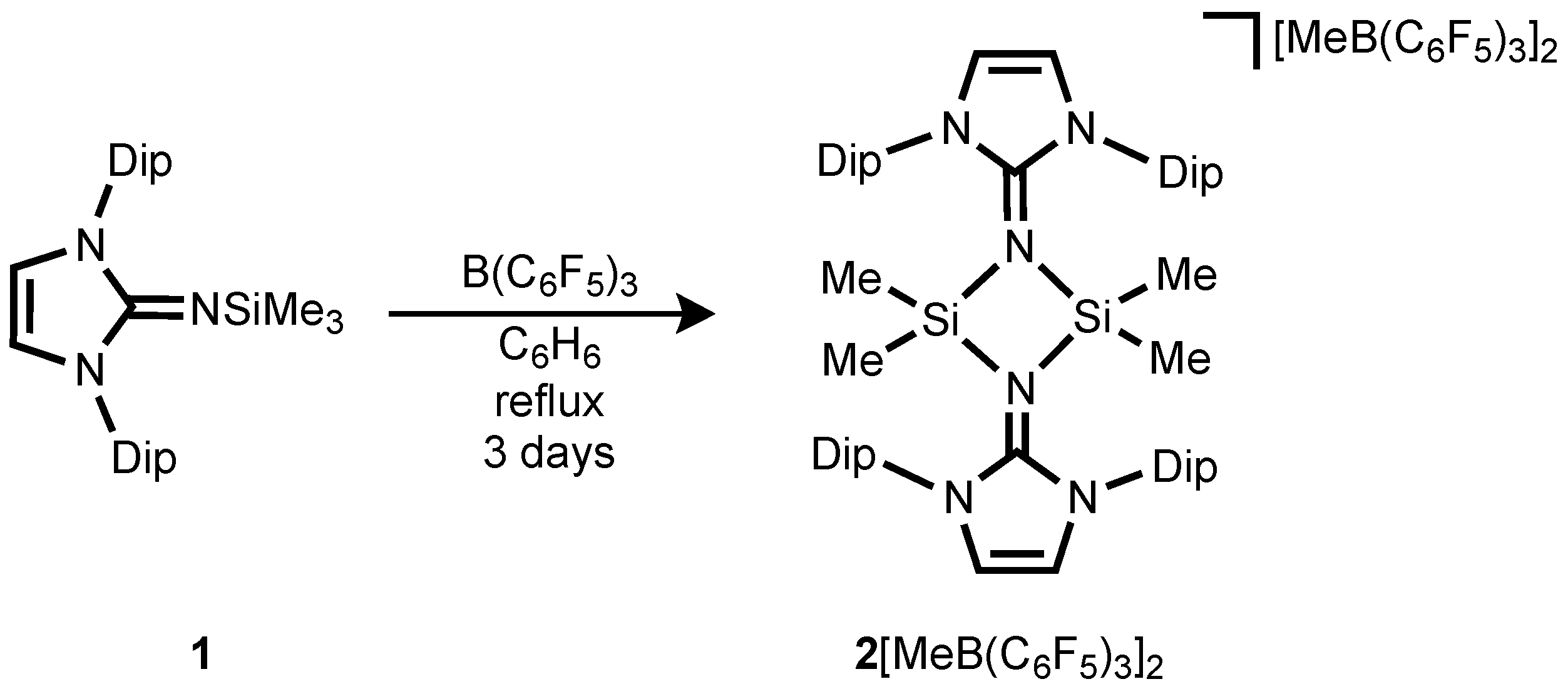
Reaction of Me
3SiNIPr (
1, NIPr = bis(2,6-diisopropylphenyl)imidazolin-2-imino) with B(C
6F
5)
3 in benzene results in the formation of a biphasic mixture, in which the dark-red lower layer was found to contain the silylium ion
2[MeB(C
6F
5)
3]
2 (
Scheme 2). A signal was observed in the
29Si-NMR spectrum at δ = 17.3 ppm, which is significant low-field shifted compared to that of the precursor
1 (−22.3) [
37]. This value is very similar to that of phosphinoimino complex of the silylium ion (δ(
29Si) = 7.9 ppm) [
36], illustrating the existence of dimer
2[MeB(C
6F
5)
3]
2 in solution. The calculated chemical shift of optimized structure of
2 (8.1 ppm) corresponds well with the experimental value, supporting the presence of dimeric structure with a tetracoordinate silicon environment in
2[MeB(C
6F
5)
3]
2. The
19F-NMR spectrum displays three distinct signals characteristic of [MeB(C
6F
5)
3] due to the
ortho,
meta, and
para F atoms of the C
6F
5 groups, which appear at similar shifts to those observed in
III that have [MeB(C
6F
5)
3] as a counterion [
31,
32]. Unfortunately, repeated efforts to grow single crystals of
2[MeB(C
6F
5)
3]
2 suitable for X-ray diffraction analysis were all unsuccessful.
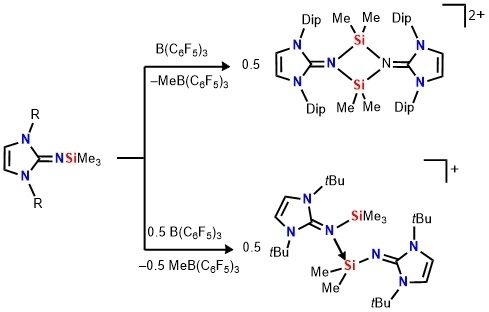
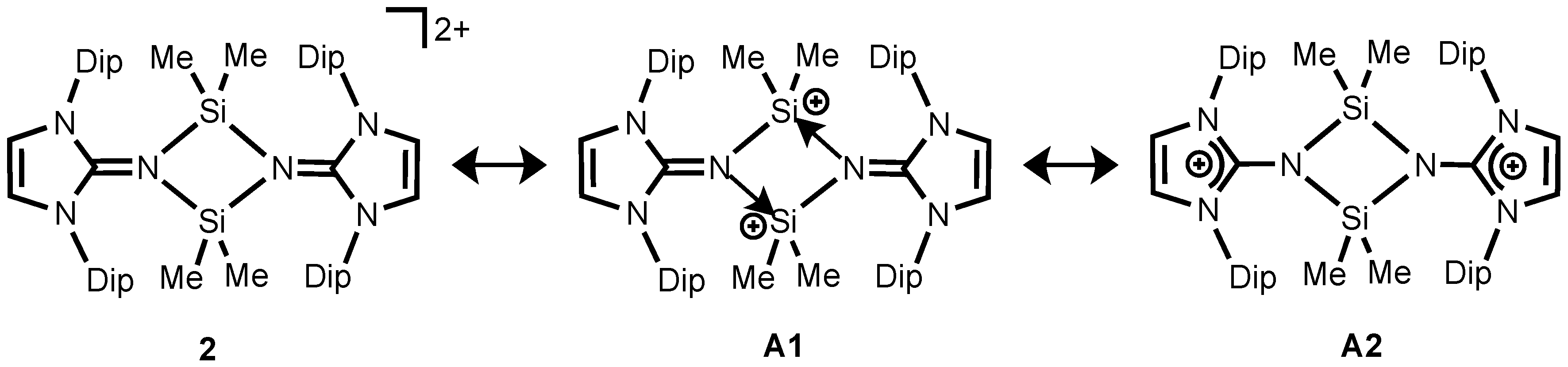
In order to gain deeper insight into the electronic nature of the structure of
2, we performed a Natural Resonance Theory (NRT) analysis. Evaluation of the relative contributions of all important canonical forms for compound
2 shows that the imino-coordinated silylium ion formulation
A1 (78.9%) is dominant (
Scheme 3). Additionally, compound
2 possesses significant imidazolium cation character
A2 (21.1%), where the formal positive charges reside on the both imidazoline rings. Consequently, the optimized structure of
2 has relatively long C–N
imino bonds (1.321 Å) which are comparable to those found in bis(germyliumylidene)
IV (1.331(3), 1.335(2) Å) [
33]. The calculated Si–N
imino bond length (1.820 Å) is significantly longer than that in the starting material
1 (1.677(2) Å) [
26], indicating the high dative bond interaction between the silicon and the imino-nitrogen (
Figure 2). The computed HOMO for
2 shows major π contributions located on the Dip unit (
Figure S2). The LUMO shows N–C and C–C antibonding interactions on the imidazolium ring (
Figure S2).
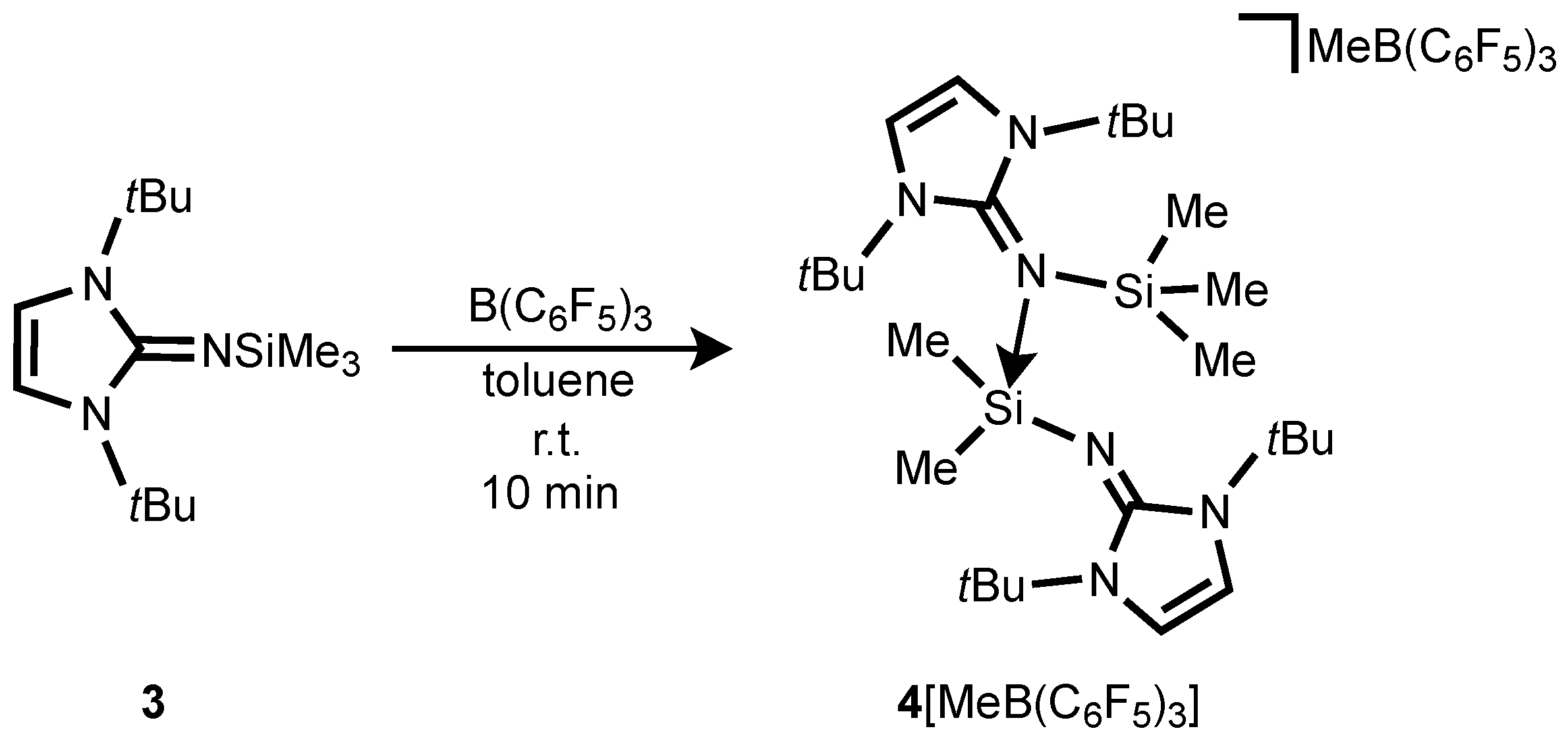
The treatment of Me
3SiNI
tBu
3 (NI
tBu = bis(
tert-butyl)imidazolin-2-imino) with B(C
6F
5) in a 2:1 to form a two-layer reaction mixture (
Scheme 4), accompanied by a color change from colorless to dark-red akin to
2[MeB(C
6F
5)
3]
2. Although our efforts to obtain X-ray-quality crystals of this species have been unsuccessful, the NMR data as well as the ESI mass spectroscopy results are all consistent with the formulation of silylium salt [NI
tBuSiMe
2(NI
tBuSiMe
3)][MeB(C
6F
5)
3] (
4[MeB(C
6F
5)
3]) (
Scheme 4). This salt is formed by abstraction of a Si-bound methyl group, affording a silylium cation which is stabilized by intermolecular coordination of a second Me
3SiNI
tBu molecule. The
1H-NMR spectrum shows two sets of signal for the imidazoline ligands. The two signals at δ = −0.01 and 0.34 in an integral ratio of 2:3 can be attributed to the Si-bound methyl groups. The
29Si-NMR spectrum reveals two resonances for
4[MeB(C
6F
5)
3] (δ(
29Si) = −32.0, 13.1 ppm), in which the latter is assigned to the silicon atom of the silylium ion because this value is comparable to that of
2[MeB(C
6F
5)
3]
2. The signals at −32.0 and 13.1 ppm are well reproduced by gauge-independent atomic orbitals (GIAO) calculation of the optimized structure of
4 and confirmed the assignment (−33.1 and 12.6 ppm). We also employed DFT calculations to assess the likely structure of the complex. The Me
2Si–N
imino distance of 1.686 Å is comparable to that in
3 1.655(2) Å) [
24], meaning the covalent bond character. Alternatively, the contact between the coordinating imino ligand and the silylium center of 1.855 Å is significantly longer owing to the high dative bond character. The extent of the positive charge delocalization can be evaluated by the bond length of C
NHC–N bond in the imino moiety. In this context, the bond distance of C
NHC–N (1.374 Å) in the coordinating imine Me
3SiNI
tBu is relatively longer than corresponding value of the main framework of [Me
2SiNI
tBu]
+ (1.313 Å), indicating the positive charge density is mainly delocalized into the coordinating silylimine ligand. Moreover, the bond angle of C
NHC–N–Si (139.63°) shows a remarkably bent structure. This value is much larger than that in the iminosilylene-borane adduct having a significant silaimine character [
29], indicating the π-electron interaction between the imino-nitrogen towards the silylium center can be neglected.
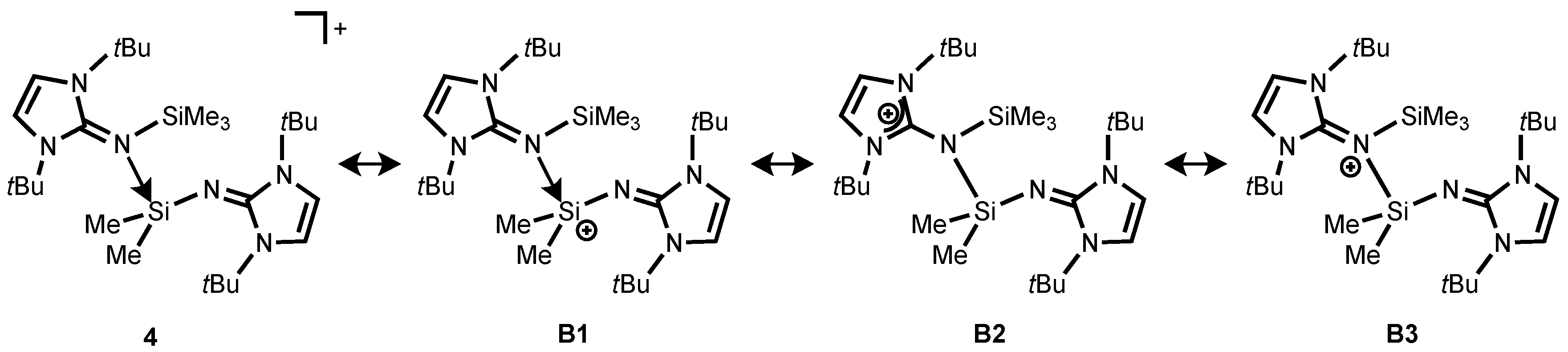
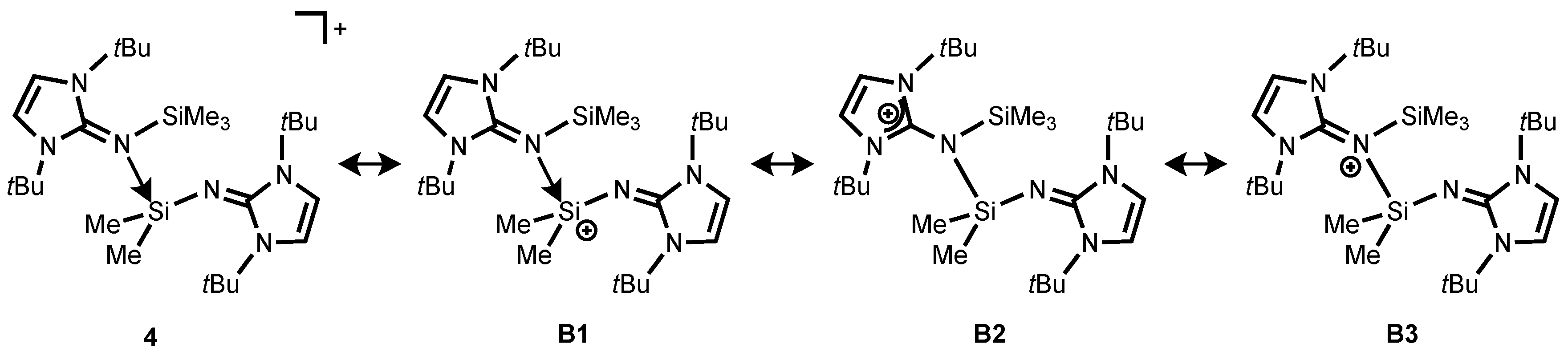
Moreover, the bonding situation of
4 were analyzed by means of NRT analysis (
Scheme 5). The study shows that the dominant resonance form is
B1 (56.6%), in which the iminosilane (Me
3SiNI
tBu) coordinates the vacant orbital on the silicon atom in the silylium ion (
Scheme 5). As expected for the calculated structure of
4, the resonance structure
B2 also has considerable weight (35.8%), with a positive charge being located on one of the imidazoline rings. In addition, compound
4 possesses a small contribution of the iminium ion
B3 (8.1%) (
Scheme 5). Akin to
2, HOMO is localized on imidazolin-2-imine fragment while LUMO is mainly on the π-contribution of the coordinated imidazolium ring (
Figure S3).
When an excess amount of B(C
6F
5)
3 is utilized, dimeric silylium [Me
2SiNI
tBu]
2[MeB(C
6F
5)
3]
2 is not formed. This different reactivity of Me
3SiNIPr
1 and Me
3SiNI
tBu
3 towards B(C
6F
5)
3 can be caused by the steric bulk of the imidazoline fragment. This trend of the reactivity with B(C
6F
5)
3 is very similar to phosphinoiminosilanes, where the bulky Me
3SiNP(
tBu)
3 formed the dimeric silylium ion
VII while the less bulky silanes (Me
3SiNP(R)
3, R =
iPr, Ph) led to the formations of phosphinoimine-coordinated silylium ions
VIII (
Scheme 1) [
36].
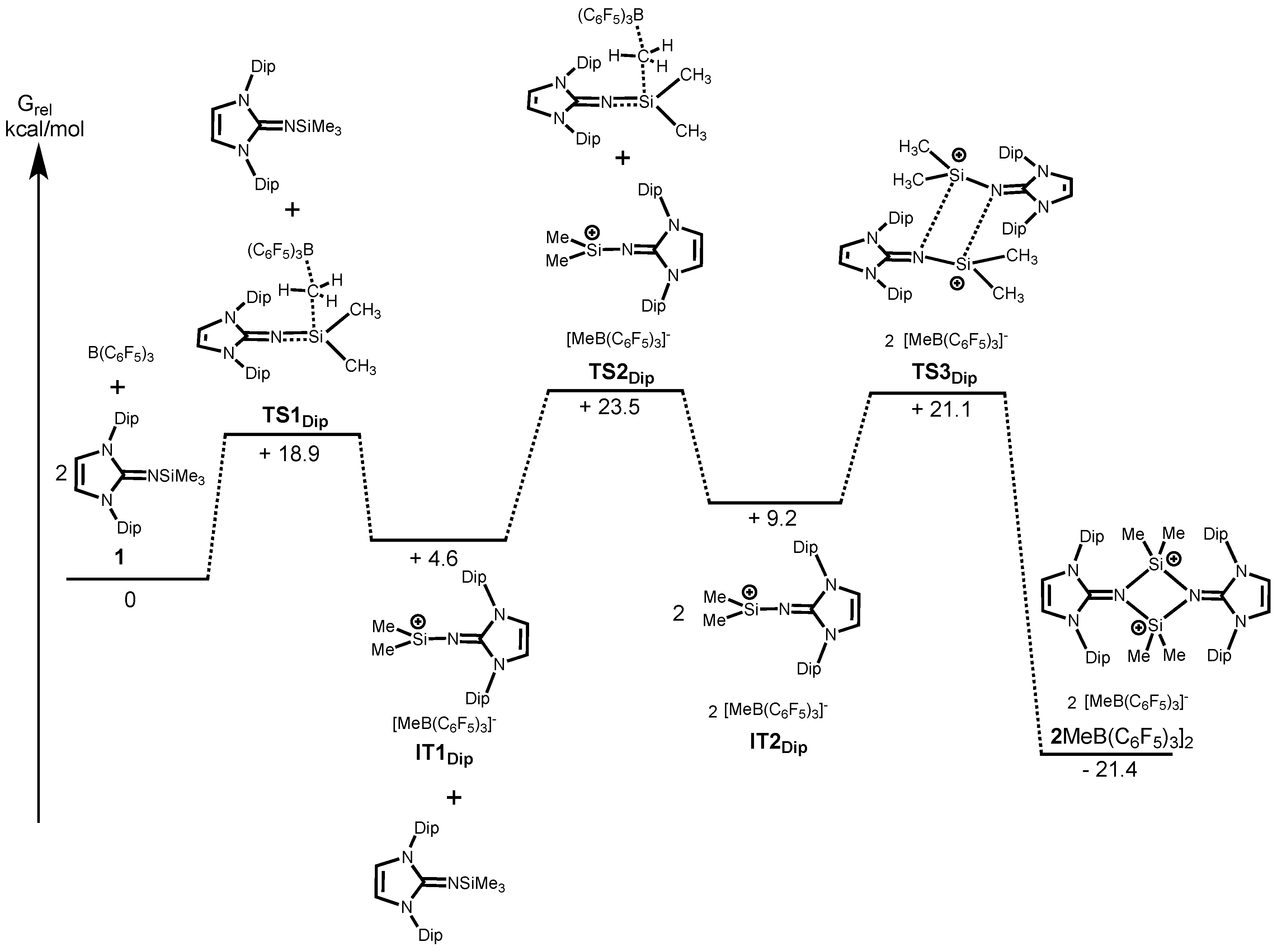
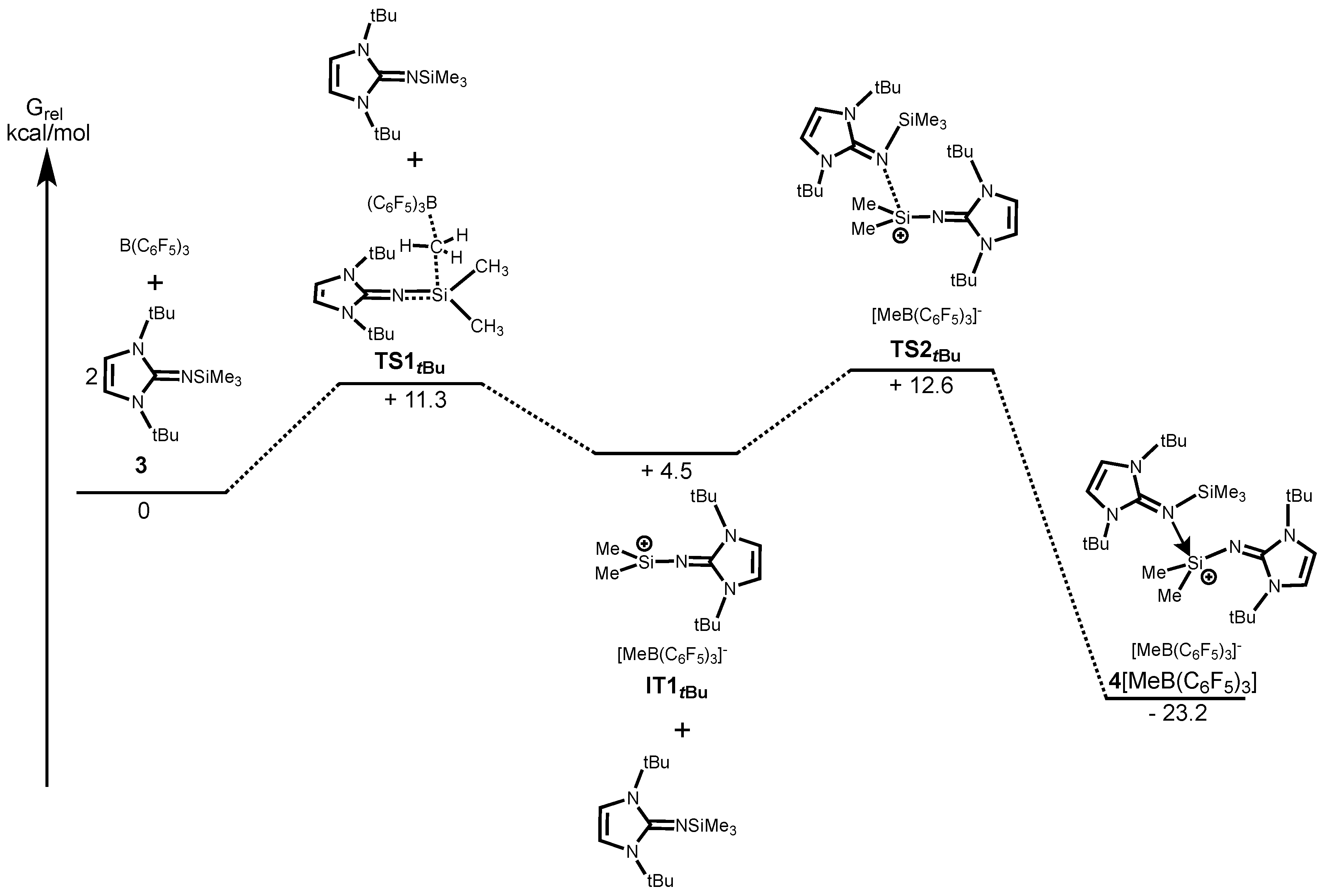
We next carried out the DFT calculations for elucidation of the mechanisms of formation of silylium ions
2[MeB(C
6F
5)
3]
2 and
4[MeB(C
6F
5)
3], respectively. The difference in the steric bulk of the precursors
1 and
3 leads to different products based on the reaction kinetics.
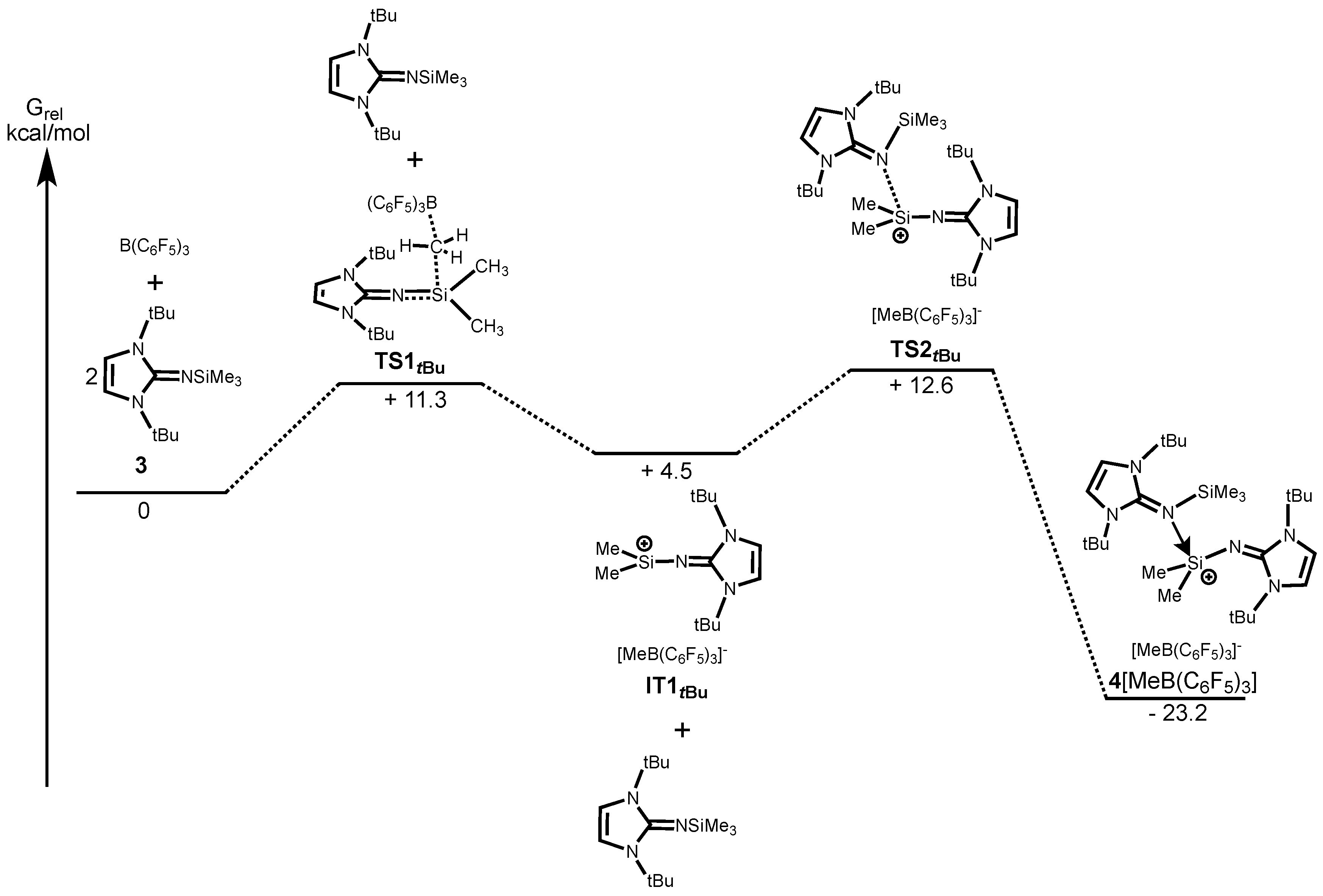
Figure 3 shows the energy profile along the reaction path calculated at the B97-D/6-31G(d) level of theory which has been successfully applied in low-valent silicon compounds [
38,
39,
40,
41,
42,
43,
44,
45,
46,
47]. The first step in the reaction is the abstraction of a Me group of a Me
3Si moiety by the B(C
6F
5)
3 (
TS1Dip) with the associated energy of +18.9 kcal/mol for
2[MeB(C
6F
5)
3]
2. On the other hand, the calculation revealed that a methyl-abstraction of
3 requires less energy (
TS1tBu, +11.3 kcal/mol) than that of
1 mainly due to the less sterically congested environment around the Si center of
3 (
Figure 4). This finding is consistent with the experimental results, in which the reaction of
1 with B(C
6F
5)
3 only proceeds at elevated temperature with prolonged reaction time, whereas
3 reacts with B(C
6F
5)
3 instantly at room temperature. After abstracting of the first methyl group, a methyl-abstraction from the second Me
3SiNIPr (
TS2Dipp) with associated energy of +23.5 kcal/mol followed by the dimerization (
TS3Dip) to form dicationic
2[MeB(C
6F
5)
3]
2. Alternatively, coordination of the second Me
3SiNI
tBu (
TS2tBu) with the associated energy of +12.6 kcal/mol leads to the imine-adduct
4[MeB(C
6F
5)
3]. We also calculated the reaction pathway with a concerted reaction that a precursor molecule supports a Me-group abstraction with B(C
6F
5)
3 via its coordination to the back side of the forming silylium ion. However, it turned out that the corresponding transition state was not found. This is probably because the severe steric hindrance makes coordination of the second imine Me
3SiNI
tBu to the silicon center highly unfavorable (
Scheme S1).
To clarify the difference in the reactivity between Me
3SiNIPr
1 and Me
3SiNI
tBu
3, the mechanism of the formation of [NIPrSiMe
2(NIPrSiMe
3)][MeB(C
6F
5)
3] (
5[MeB(C
6F
5)
3]) and [Me
2SiN
tBu]
2[MeB(C
6F
5)
3]
2 (
6[MeB(C
6F
5)
3]
2) were also carried out (see
Supporting Materials). The associated energy of the coordination of the second Me
3SiNIPr is relatively high (
TS2Dip’, +26.7 kcal/mol) and the net energy gain of the formation
5[MeB(C
6F
5)
3] is small (−15.8 kcal/mol) mainly due to the steric repulsion between the bulky Dip groups (
Figure S7). On the other hand, the net energy gain of the formation of the dication
6[MeB(C
6F
5)
3]
2 (−18.9 kcal/mol) is smaller than that of
4[MeB(C
6F
5)
3] (−23.2 kcal/mol) (
Figure S8).
We also considered the possibility of the direct Me abstraction from
4[MeB(C
6F
5)
3] to lead to
6[MeB(C
6F
5)
3]
2. However, the transition state in this reaction is relatively high (25.2 kcal/mol) and it is an uphill process (+5.3 kcal/mol). Thus, this pathway to dimeric silylium ion
6[MeB(C
6F
5)
3]
2 is both kinetic and thermodynamic unfavorable (
Figure S9). Consequently, these theoretical results are also in accordance with the experimental results.
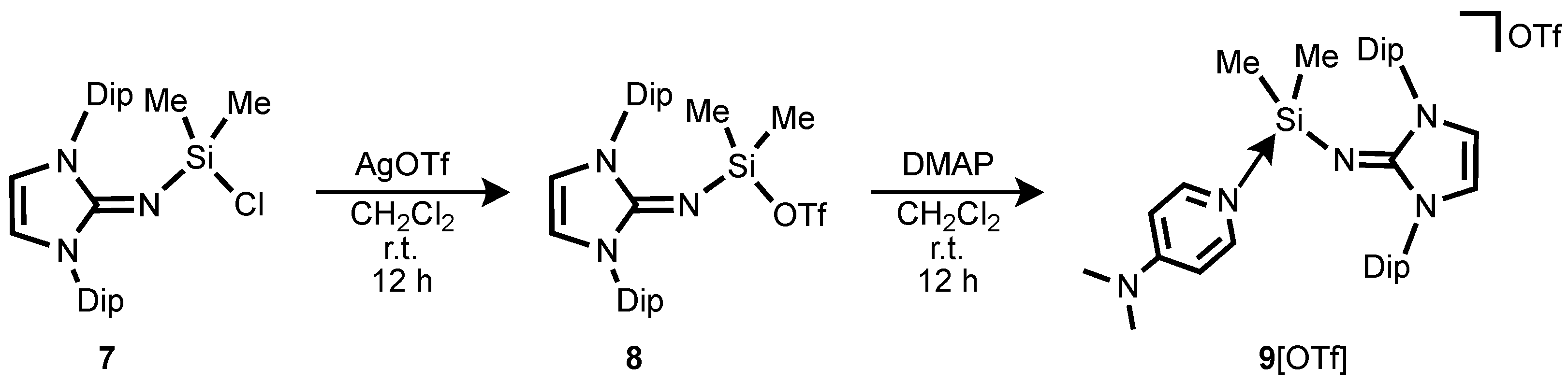
To further explore the scope of iminosilylium ions, we have investigated different synthetic approaches. Treatment of Me
3SiNIPr with one equivalent of Me
2SiCl
2 affords the Me
2(Cl)SiNIPr
7 in high yield. The reaction of Me
2(Cl)SiNIPr
7 with one equivalent of AgOTf results in the formation of iminotriflatosilane Me
2(OTf)SiNIPr
8 by substitution of the chloride with a triflate group (
Scheme 6). The observed
29Si resonance value for
8 (−18.5 ppm) was comparable with that of Me
2Si(Cl)NIPr
7 (−19.2 ppm). Single crystal X-ray diffraction analysis of
8 reveals that it crystallizes as a contact ion pair with the triflate anion in the solid state to form a tetrahedral silicon center with an Si–O bond distance of 1.7696(17) Å (
Figure 5). The bond angle of Si–N–C is 158.77(17)°, which falls in the range of the mono-imino-substituted tetravalent silanes [
27,
29]. Furthermore, the Si–N
imino bond distance (1.6231(18) Å) is comparable to those reported for tetravalent silane bearing an imino ligand and indicative of a negligible Si=N double bond character.
The reaction of
8 in toluene at 25 °C with one equivalent of DMAP affords a Lewis base adduct
9[OTf] (
Scheme 6). This complexation leads to displacement of the TfO–Si interaction in
8. The
29Si-NMR spectrum of
9[OTf] (in CD
2Cl
2) displays a singlet resonance at −16.4 ppm, which is slightly downfield shifted compared to those in
7 (−19.2 ppm) and
8 (−18.5 ppm). Compound
9[OTf] crystallizes in monoclinic space group
P2
1/
c as separated ion pairs (closest Si–anion separation: 4.699 Å). The molecular structure of
9[OTf] is depicted in
Figure 5. The Si–N
imino distance (1.6350(19) Å) is significantly shorter than the Si–N
DMAP separation (1.8470(18) Å), but is very close to the Si–N
imino bond length in
6 (1.6231(18) Å). Also, the N–C
NHC bond distance of 1.275(3) Å is a typical value for carbon-nitrogen double bonds in neutral imidazolin-2-imino compounds [
26,
27], indicating the delocalization of positive charge density into the imidazoline ring is negligible. Accordingly, instead of observing a linear Si–N–C angle in
9, the Si–N–C angle is significantly bent (155.86(17)°). These structural features suggest little electron donation from the imino nitrogen to the silicon core, exhibiting no allenic-type bonding character.
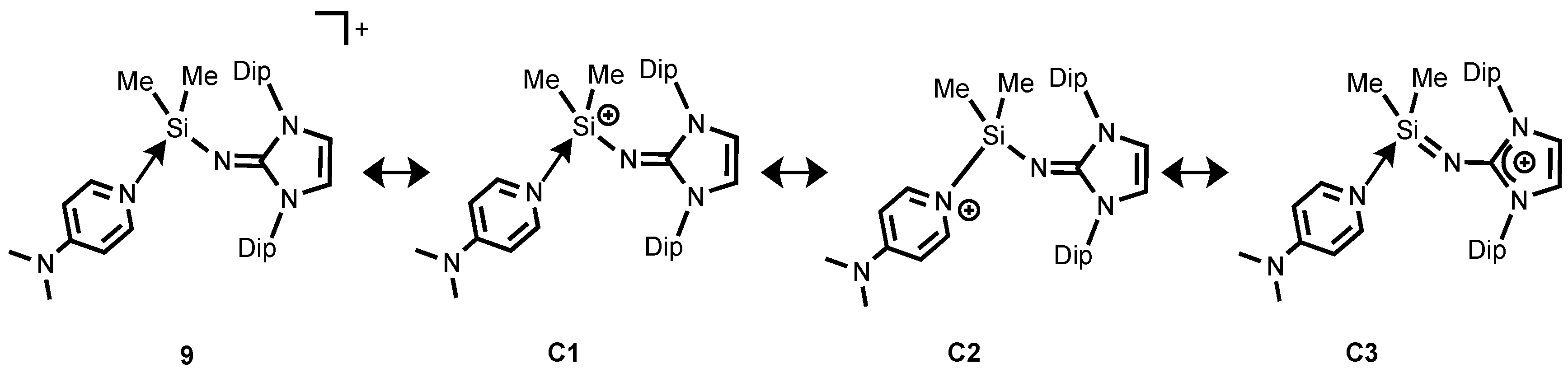
Judging from our NRT analysis, the relative contributions of resonance structures for compound
9 denotes that the DMAP-coordinated silylium ion
C1 is dominant (51.2%) (
Scheme 7). Furthermore,
9 possesses significant pyridinium character represented by
C2. Interestingly, the resonance form
C3 bearing the Si–N
imino double bond character, with formal positive charge being located at the imidazolium ring, has a non-negligible role in the description of
9 (18.5%,
Scheme 7) despite the long Si–N
imino bond and the short N
imino–C distance that observed in the X-ray structure.
3. Experimental Section
3.1. General Information
All experiments and manipulations were conducted under dry oxygen-free nitrogen using standard Schlenk techniques or in an MBraun glovebox workstation containing an atmosphere of purified nitrogen. Solvents were dried by standard methods. NMR solvents were degassed by multiple freeze-pump-thaw cycles and stored over molecular sieves (3 Å). Reagents were purchased from commercial suppliers and processed as received if not stated otherwise. The starting material
1 and
3 was prepared according to the reported procedure [
25,
26].
1H-,
11B-,
13C{
1H}-, and
19F-spectra were recorded on an Avance II 200 MHz or 400 MHz spectrometer (Bruker) and referenced to residual solvent signals as internal standards (
1H and
13C) or an external standard (Et
2O·BF
3 for
11B, and CFCl
3 for
19F). Values for the chemical shift (δ) are given in parts per million. Abbreviations: s = singlet; d = doublet; t = triplet; sept = septet. Elemental analyses and ESI-HRMS were carried out by the microanalytical laboratory and the MS-Service of the Institut für Chemie, Technische Universität Berlin, Germany, respectively.
3.2. Synthesis of 2[MeB(C6F5)3]2
A Schlenk tube equipped with a PTFE-coated magnetic stirring bar was charged with Me3SiNIPr 1 (157 mg, 0.330 mmol) and B(C6F5)3 (175 mg, 0.342 mmol). Benzene (5 mL) was transferred to the reaction vessel by cannula and the resulting mixture was stirred under reflux condition for 72 h, resulting in a biphasic reaction mixture. The upper phase was removed, and the lower oily substance was washed with benzene. The oily residue was subjected to prolonged drying in high vacuum (6 h) to afford 2[MeB(C6F5)3]2 as a pale red oil (144 mg, 44%); 1H-NMR (400 MHz, CD2Cl2): δ −0.19 (s, superimposed, 6H, Si(CH3)2), −0.19 (s, superimposed, 3H MeB(C6F5)3), 1.23 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 1.35 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 2.56 (sept, 3JHH = 7 Hz, 4H, CH(CH3)2), 7.01 (s, 2H, NCH), 7.47 (d, 3JHH = 8 Hz, 4H, Ar-H), 7.67 (t, 3JHH = 8 Hz, 2H, Ar-H); 11B-NMR (64.2 MHz, CD2Cl2): δ = −16.6. 13C{1H}-NMR (100.6 MHz, CD2Cl2): δ = 0.3 (Si(CH3)2), 23.1 (CH(CH3)2), 25.3 (CH(CH3)2), 29.7 (CH(CH3)2), 120.2 (NCH), 126.4 (Ar-C), 128.5 (Ar-C), 133.6 (Ar-C), 146.9 (Ar-C), 148.0 (NCN). 19F-NMR (188.3 MHz, CD2Cl2): δ = −133.0 (s, 6F), −163.7 (t, J = 20 Hz, 3F), −167.6 (t, J = 20 Hz, 6F). 29Si{1H}-NMR (79.5 MHz, CD2Cl2): δ = 17.3.
3.3. Synthesis of 4[MeB(C6F5)3]
A Schlenk tube equipped with a PTFE-coated magnetic stirring bar was charged with Me3SiNItBu 2 (355 mg, 1.33 mmol) and B(C6F5)3 (340 mg, 0.67 mmol). Toluene (10 mL) was transferred to the reaction vessel by cannula and the resulting mixture was stirred at room temperature for 1 h, resulting in a biphasic reaction mixture. The upper phase was removed, and the lower oily substance was washed with toluene. The oily residue was subjected to prolonged drying in high vacuum (6 h) to afford 4[MeB(C6F5)3] as a pale red oil (672 mg, 48%); 1H-NMR (200 MHz, C6D6): δ −0.01 (s, 9H, Si(CH3)3), 0.34 (s, 6H, Si(CH3)2), 1.00 (s, 18H, N(CH3)3), 1.20 (s, 18H, N(CH3)3), 5.96 (s, 2H, NCH), 6.34 (s, 2H, NCH); 11B-NMR (64.2 MHz, C6D6): δ = −14.3; 13C{1H}-NMR (50.3 MHz, C6D6): δ = 4.3 (Si(CH3)3), 8.8 (Si(CH3)2), 28.7 (NC(CH3)3), 31.1 (NC(CH3)3), 55.2 (NC(CH3)3), 64.0 (NC(CH3)3), 108.6 (NCH), 117.7 (NCH), 141.9 (NCN), 147.0 (NCH). 19F-NMR (188.3 MHz, C6D6): δ = −131.8 (d, J = 20 Hz, 6F), −164.6 (t, J = 20 Hz, 3F), −167.0 (t, J = 20 Hz, 6F). 29Si{1H}-NMR (79.5 MHz, CD2Cl2): δ = 13.1, −32.0; ESI-HRMS: m/z: 519.4031 (calcd. 519.4021 for [M − MeB(C6F5)3]+).
3.4. Synthesis of Me2(Cl)SiNIPr (7)
To a stirring mixture of Me3SiNIPr (1.88 g, 3.95 mmol) in toluene (10 mL) was added Me2SiCl2 (0.65 mL, 5.39 mmol) via syringe at −78 °C. The reaction mixture was stirred for 12 h while allowed to slowly warm to room temperature. The solvent was removed under vacuum and washed with hexane to give Me2(Cl)SiNIPr as a yellow solid (1.55 g, 79%). Recrystallization from hexane (3 mL) afforded Me2(Cl)SiNIPr in the form of colorless crystals; 1H-NMR (200 MHz, C6D6): δ 0.10 (s, 6H, SiMe2), 1.16 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 1.39 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 3.07 (sept, 3JHH = 7 Hz, 4H, CH(CH3)2), 5.94 (s, 2H, NCH), 7.10–7.26 ppm (m, 6 H, Ar-H); 13C{1H}-NMR (50.3 MHz, C6D6): δ 6.3 (Si(CH3)2), 23.5 (CH(CH3)2), 24.5 (CH(CH3)2), 29.0 (CH(CH3)2), 114.4 (NCH), 124.1 (Ar-C), 129.9 (Ar-C), 134.2 (Ar-C), 143.0 ppm (NCN), 147.7 (Ar-C); 29Si{1H}-NMR (79.5 MHz, C6D6): δ −19.2; Elemental analysis calcd. (%) for C29H42ClN3Si: C 70.20, H 8.53, N 8.47; found: C 69.69, H 8.33, N 9.10. m.p. at 155–158 °C (dec.).
3.5. Synthesis of Me2(OTf)SiNIPr (8)
A Schlenk tube equipped with a PTFE-coated magnetic stirring bar was charged with Me2ClSiNIPr 7 (133 mg, 0.268 mmol) and Ag[OTf] (69 mg, 0.268 mmol). CH2Cl2 (5 mL) was transferred to the reaction vessel by cannula at room temperature and the resulting mixture was stirred for 12 h. The volatiles were removed under reduced pressure and the residue was recrystallized from CH2Cl2 (5 mL) at −30 °C to afford Me2(OTf)SiNIPr (8) as colorless crystals (107 mg, 65%). The batch contained crystals suitable for single crystal X-ray diffraction analysis. 1H-NMR (200 MHz, C6D6): δ 0.00 (s, 6H, SiMe2), 1.10 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 1.30 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 2.91 (sept, 3JHH = 7 Hz, 4H, CH(CH3)2), 5.89 (s, 2H, NCH), 7.07–7.25 ppm (m, 6 H, Ar-H); 13C{1H}-NMR (100.6 MHz, C6D6): δ 2.10 (Si(CH3)2), 23.3 (CH(CH3)2), 24.4 (CH(CH3)2), 29.0 (CH(CH3)2), 114.7 (NCH), 124.3 (Ar-C), 130.2 (Ar-C), 133.4 (Ar-C), 143.5 ppm (NCN), 147.3 (Ar-C); 29Si{1H}-NMR (79.5 MHz, C6D6): δ −18.5; ESI-HRMS: m/z: 462.3480 (calcd. 462.3299 for [M − OTf + 2 H]+). 208–210 °C (dec.).
3.6. Synthesis of [Me2(DMAP)SiNIPr][OTf] 9[OTf]
A Schlenk tube equipped with a PTFE-coated magnetic stirring bar was charged with Me2(OTf)SiNIPr 8 (106 mg, 0.174 mmol) and DMAP (26 mg, 0.213 mmol). CH2Cl2 (5 mL) was transferred to the reaction vessel by cannula at room temperature and the resulting mixture was stirred for 12 h. The volatiles were removed under reduced pressure and the residue was recrystallized from CH2Cl2 (5 mL) at −30 °C to afford [Me2(DMAP)SiNIPr][OTf] (9[OTf]) as colorless crystals (53 mg, 42%). The batch contained crystals suitable for single crystal X-ray diffraction analysis. 1H-NMR (400.1 MHz, CD3CN): δ = −0.21 (s, 6H, Si(CH3)2), 1.17 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 1.21 (d, 3JHH = 7 Hz, 12H, CH(CH3)2), 2.87 (sept, 3JHH = 7 Hz, 4 H, CH(CH3)2), 3.08 (s, 6H, N(CH3)2), 6.36 (d, 3JHH = 7 Hz, 2 H, DMAP-H-2,6 or -3,5), 6.82 (s, 2 H, NCH), 7.31 ppm (d, 3JHH = 7 Hz, 2H, DMAP-H-2,6 or -3,5), 7.40 (d, 3JHH = 8 Hz, 4H, Ar-H), 7.56 (t, 3JHH = 8 Hz, 2H, Ar-H); 13C{1H}-NMR (100.6 MHz, CD2Cl2): δ 1.6 (Si(CH3)2), 23.1 (CH(CH3)2), 24.7 (CH(CH3)2), 29.1 (CH(CH3)2), 40.1(N(CH3)2), 107.4 (DMAP-C-2,6 or -3,5), 115.5 (NCH), 124.7 (Ar-C), 130.6 (Ar-C), 133.3 (Ar-C), 142.8 (DMAP-C-2,6 or -3,5), 147.9 (Ar-C), 155.1 (DMAP-C-4), 156.8 ppm (NCN); 19F-NMR (188.3 MHz, CD2Cl2): δ = −78.9; 29Si{1H}-NMR (79.5 MHz, CD2Cl2): δ = −16.4 ppm; ESI-HRMS: m/z: 592.3985 (calcd. 592.3586 for [M − OTf]+). 173–176 °C (dec.).
3.7. X-ray Crystallography
X-ray data collection and structural refinement. Intensity data for compounds
7,
8 and
9[OTf] collected on an Agilent SuperNova diffractometer, equipped with a CCDC area Atlas detector and a mirror monochromator utilizing Cu
Kα radiation (λ = 1.54184 Å). CCDC deposition numbers: 1491068 for
7, and 1491070 for
8, 1491069 for
9[OTf]. The data can be obtained free of charge from the Cambridge Crystallography Data Center via
http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail:
[email protected]).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}