
Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
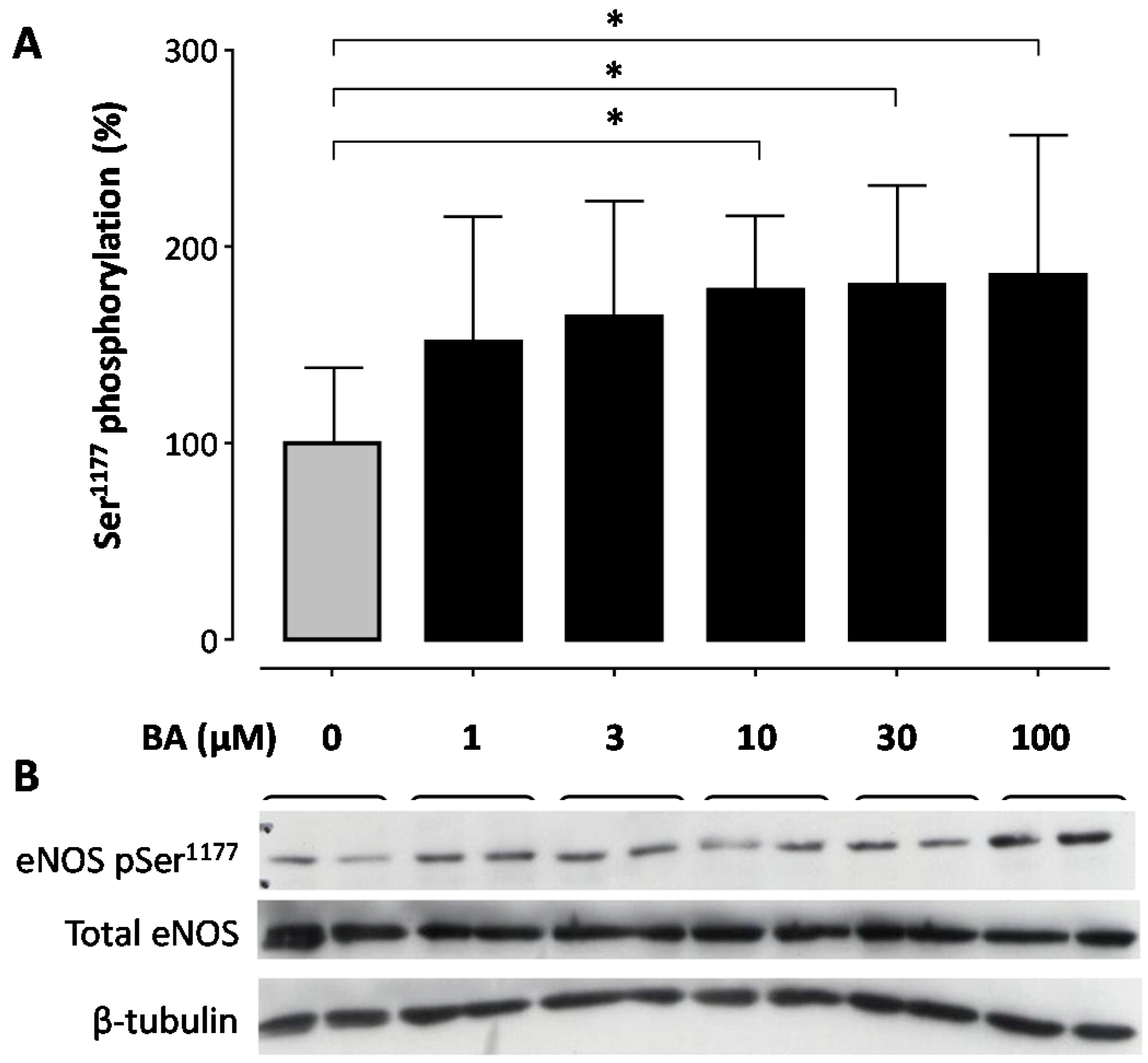
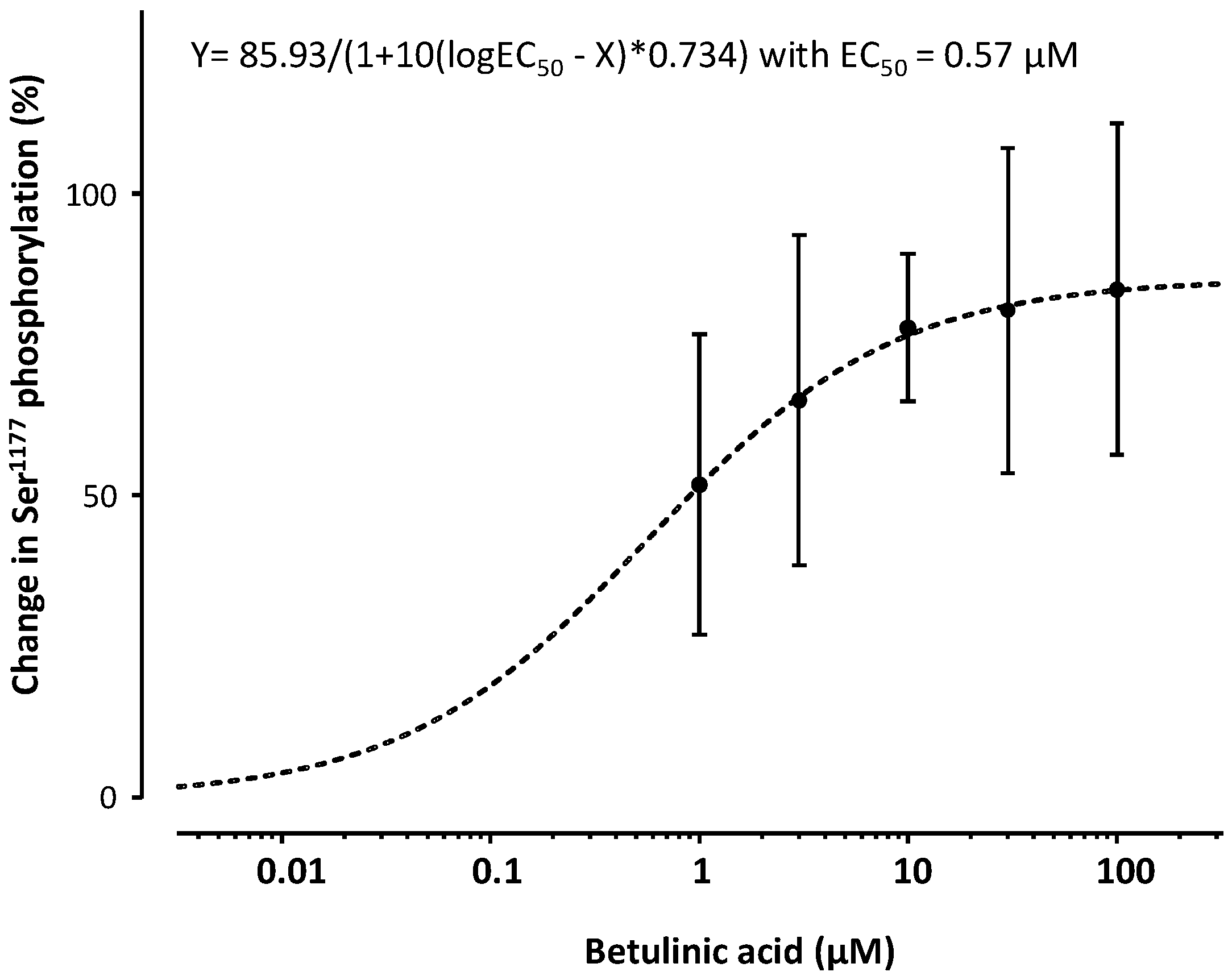
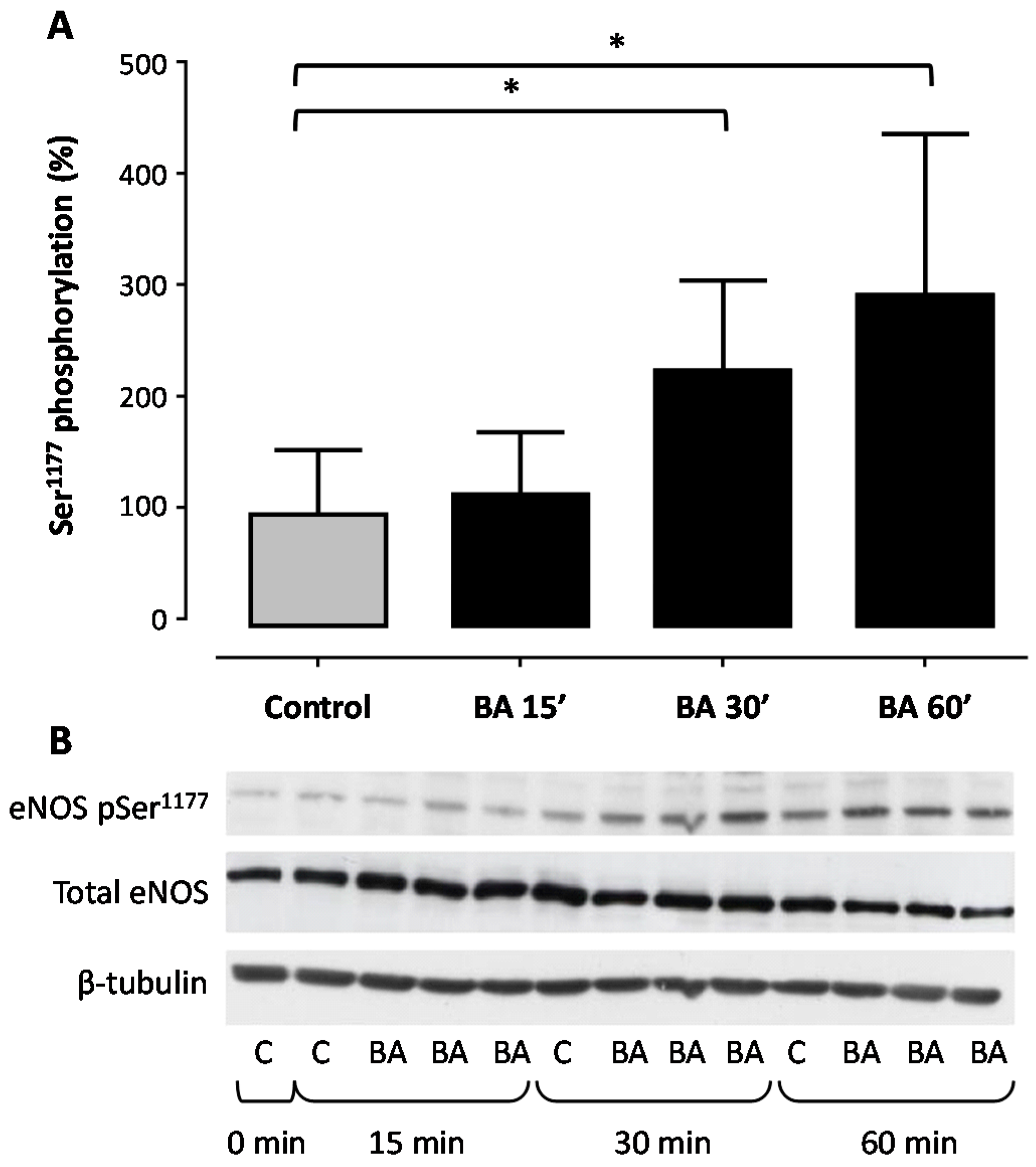
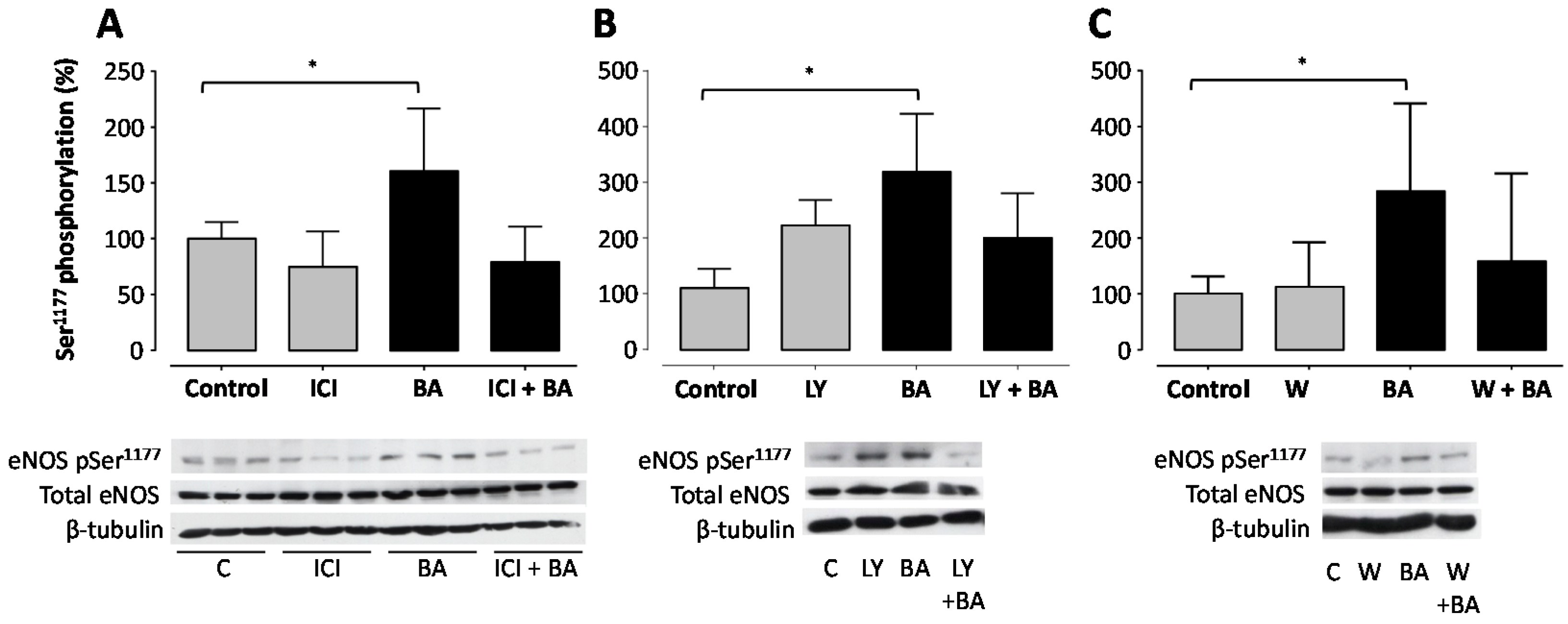
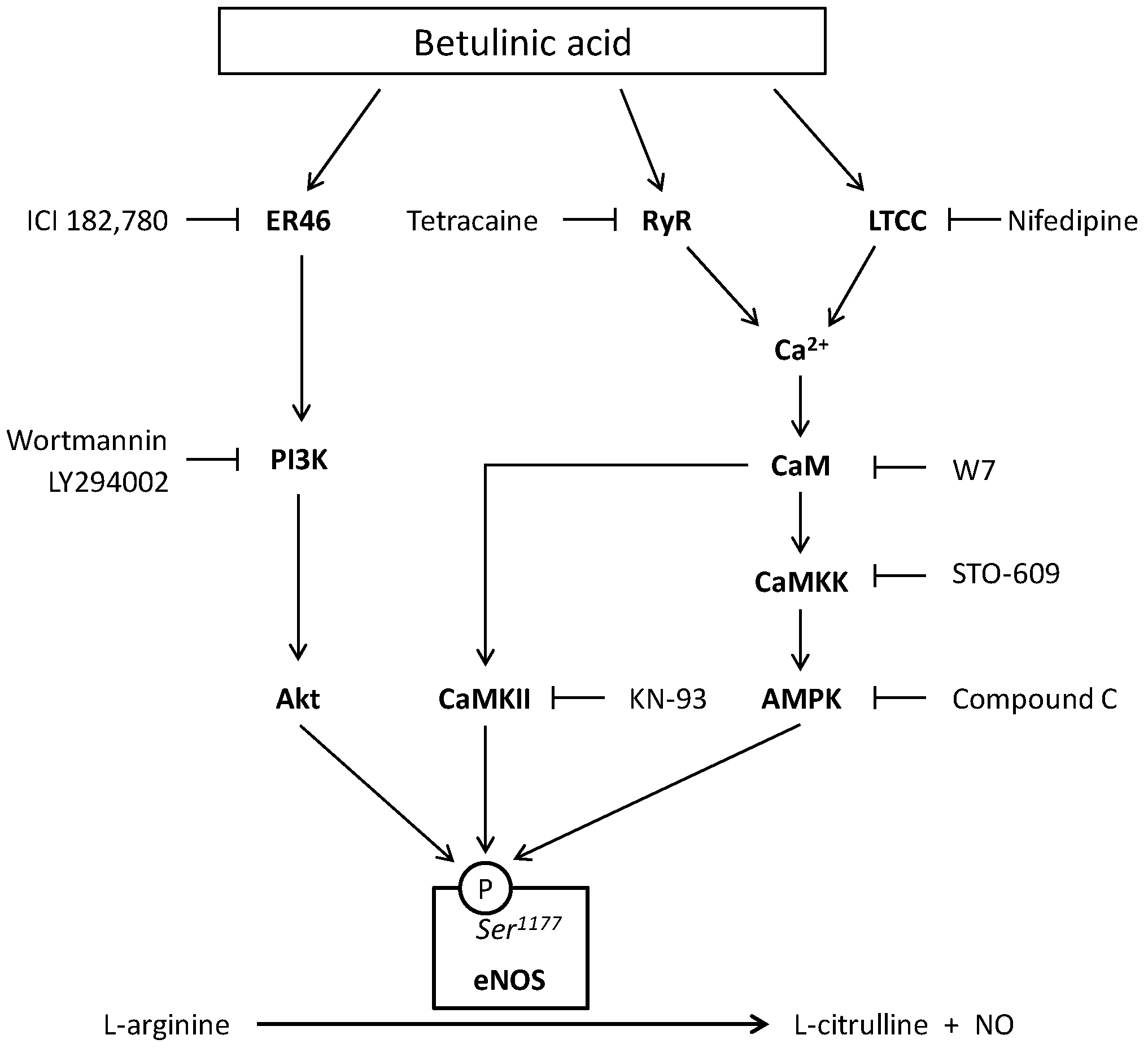
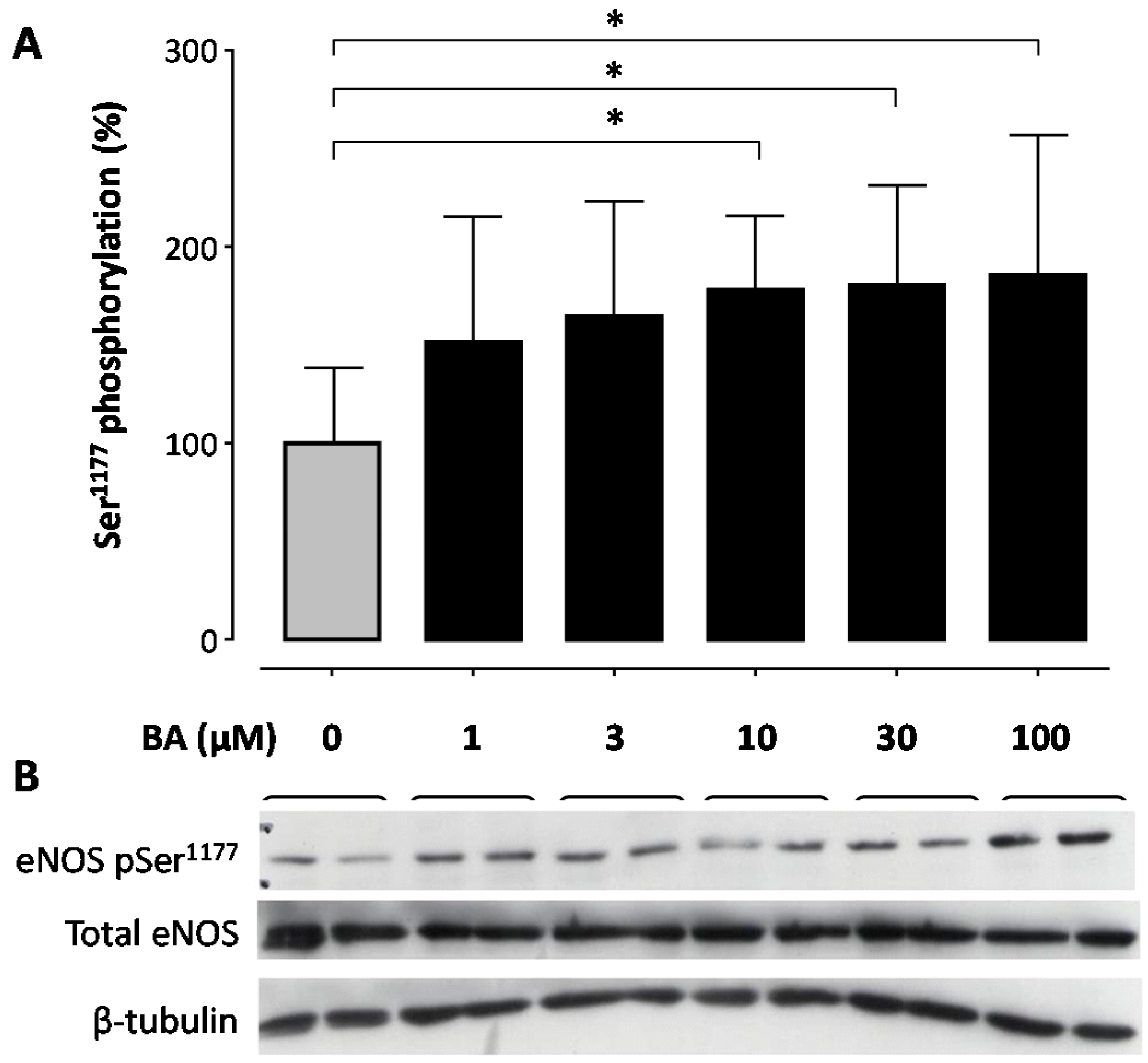
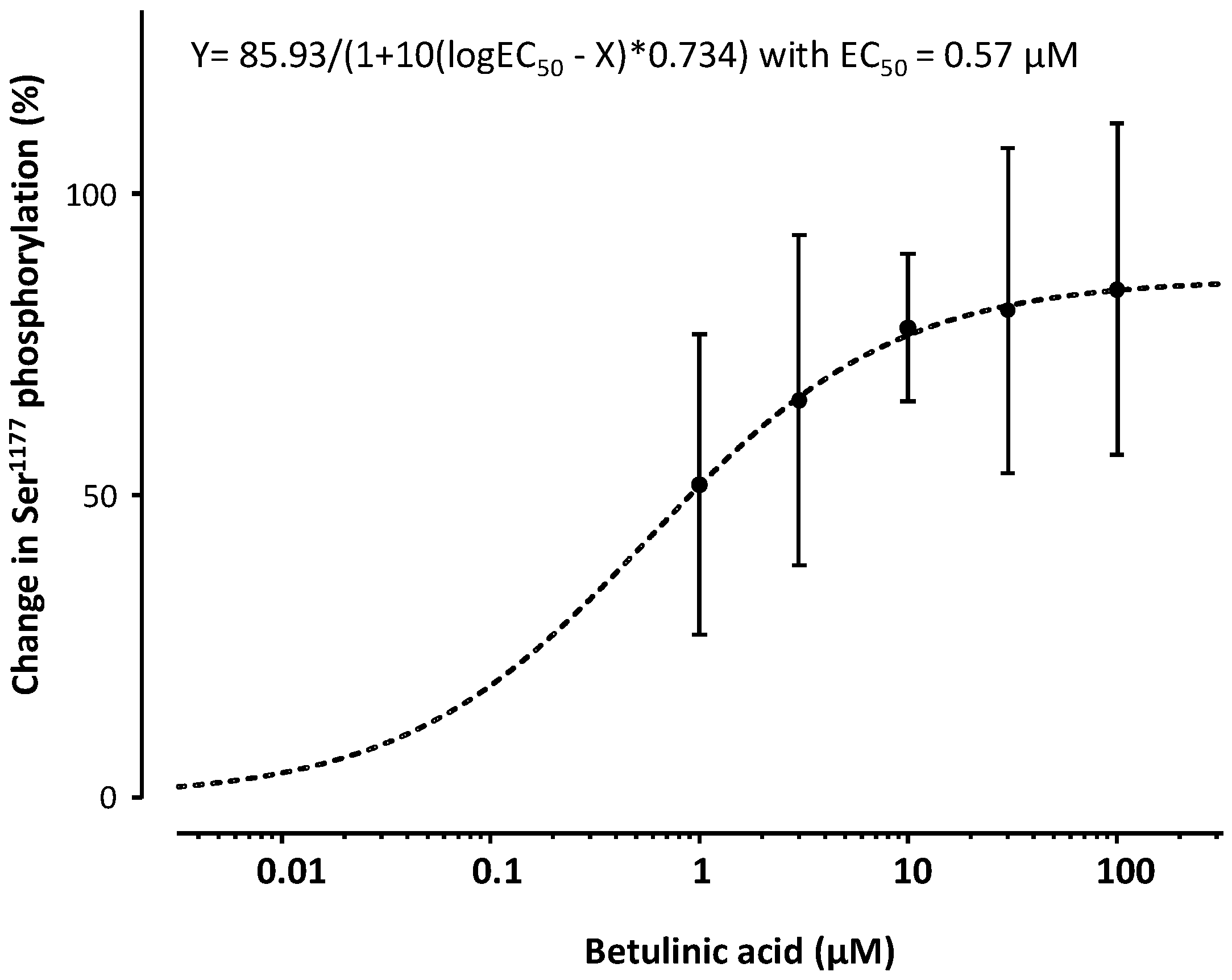
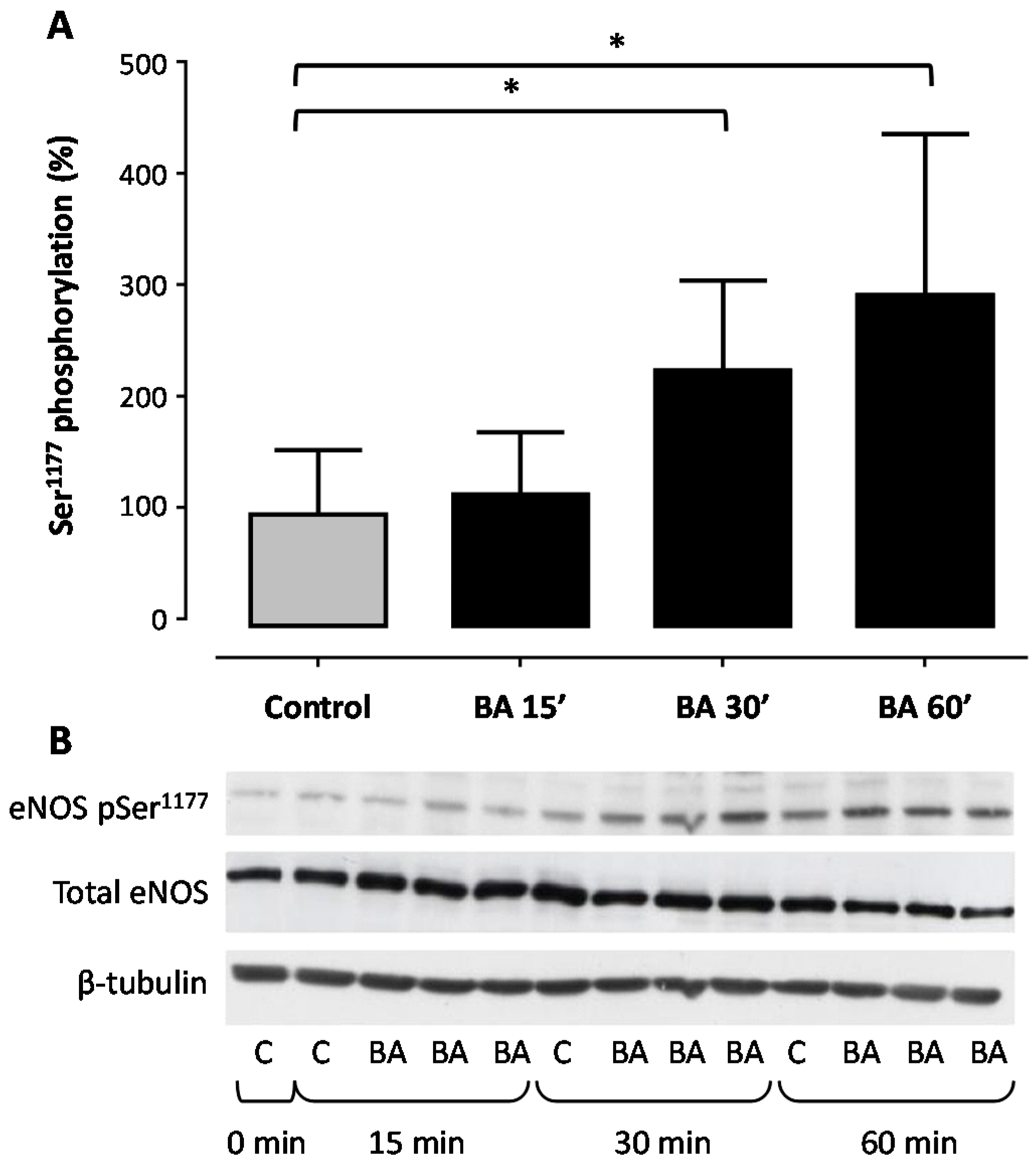
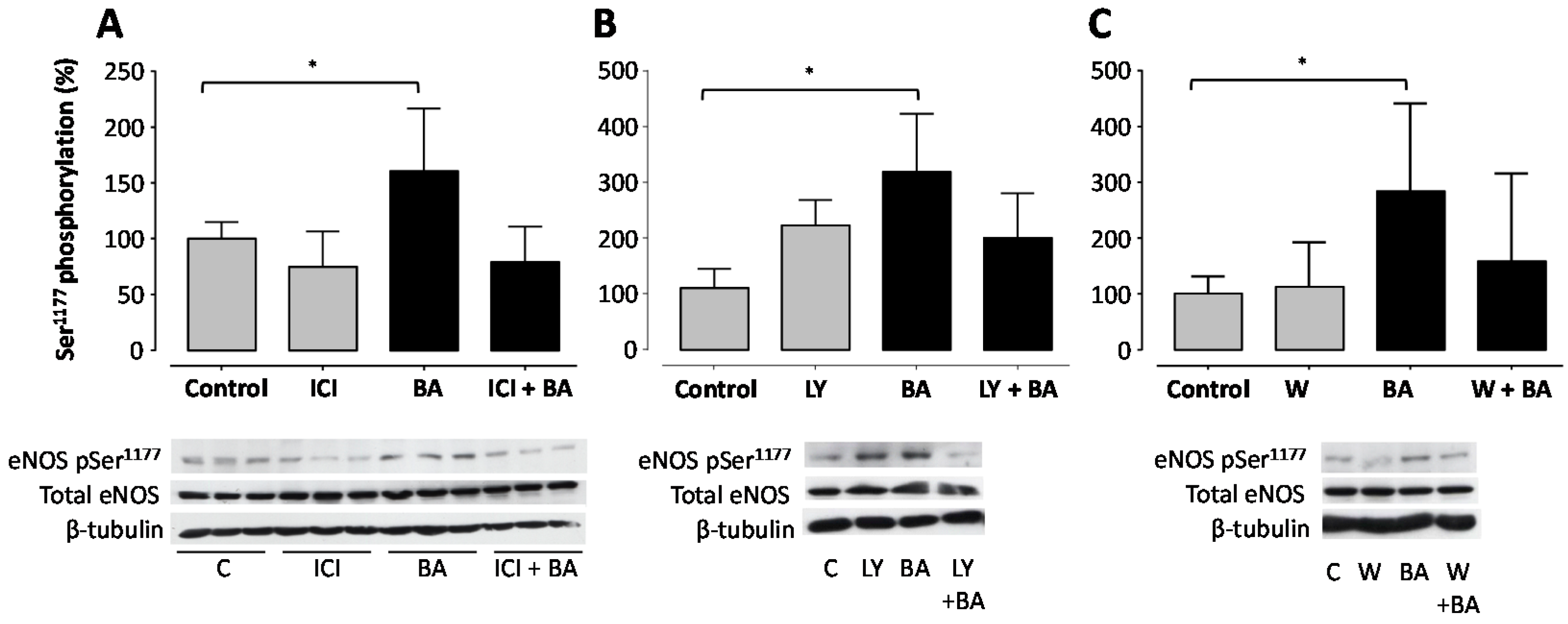
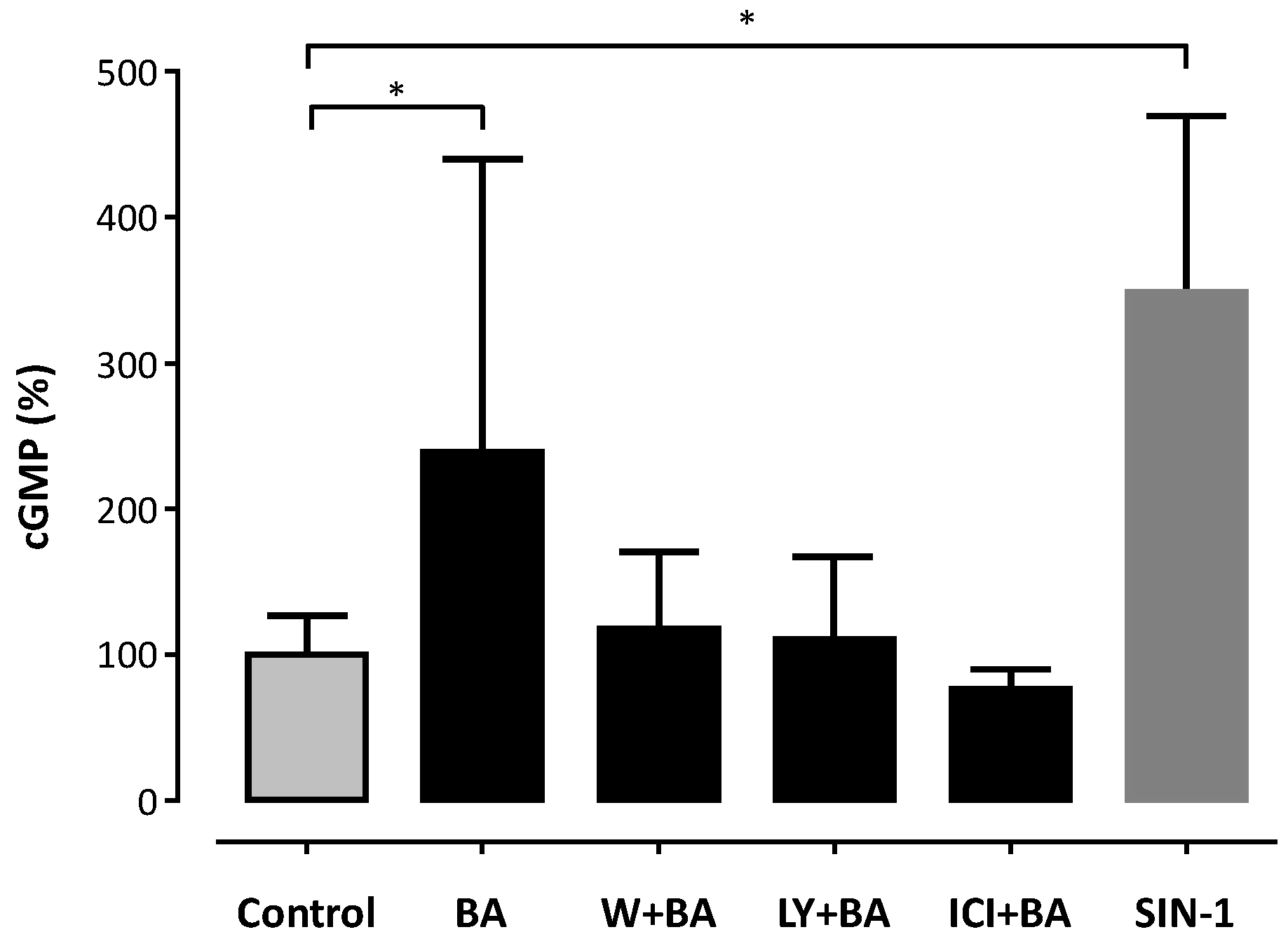
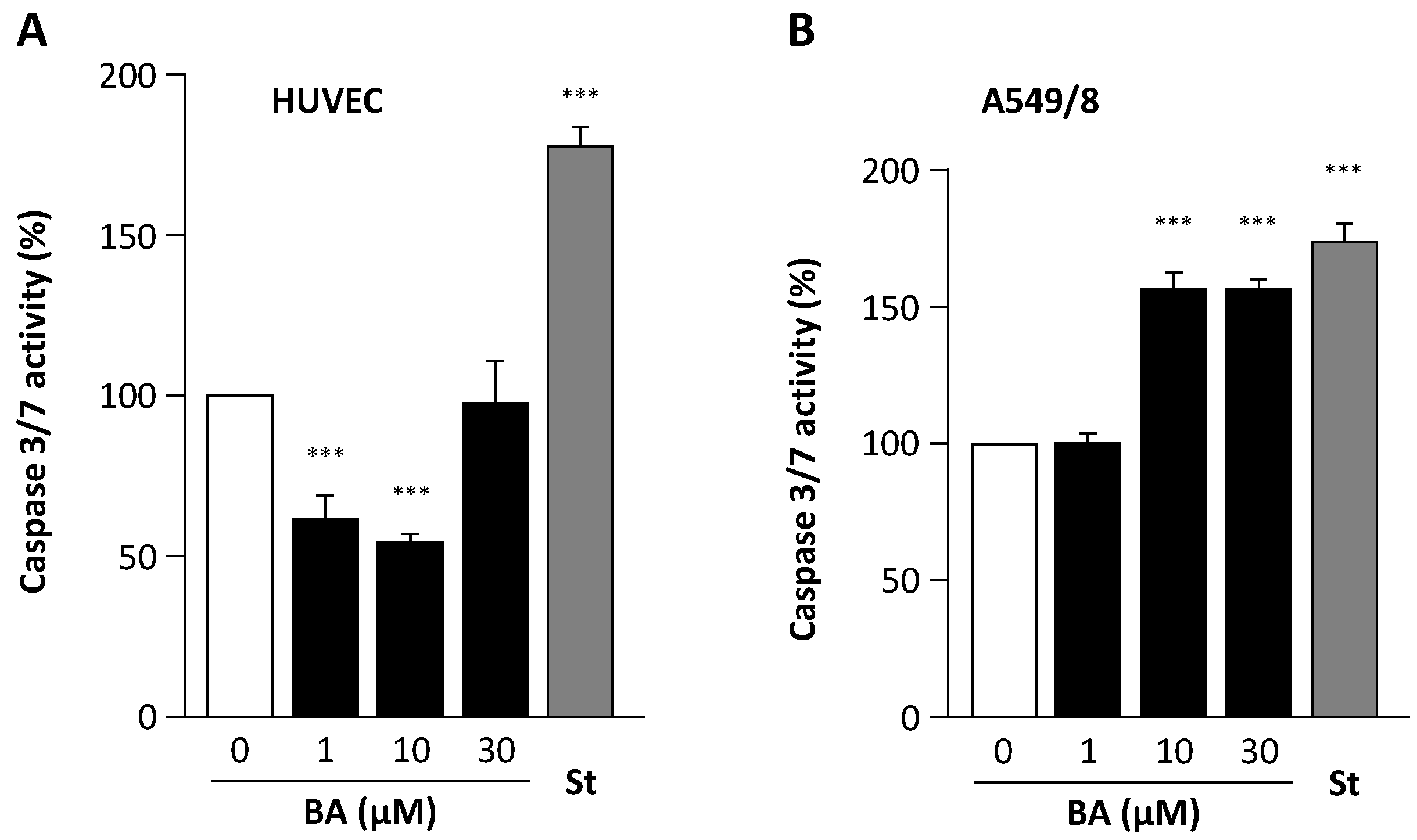
2. Results
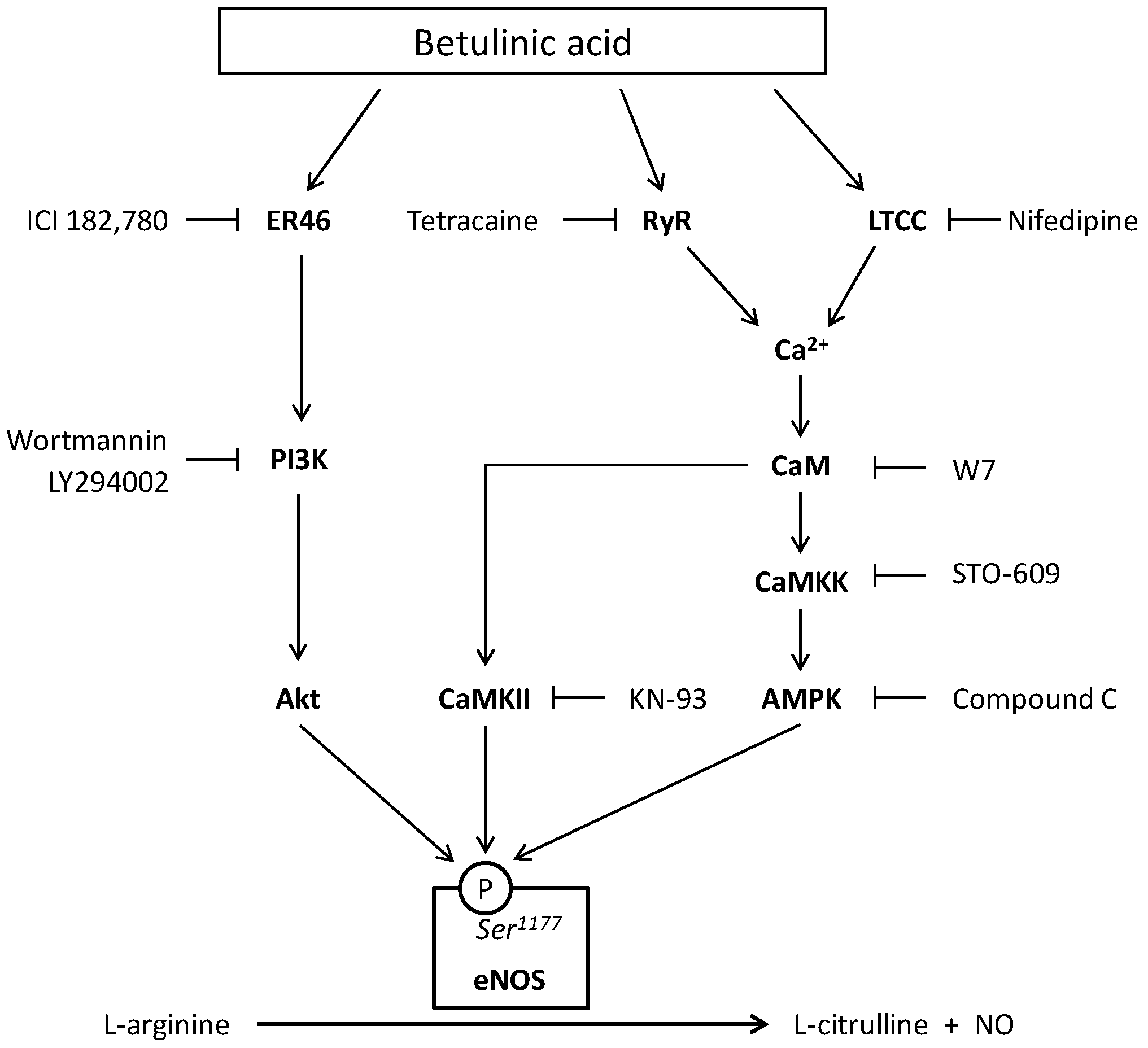
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2 Cell Lines
4.3. Cell Culture
4.4. Western Blots
4.5. NO Measurement through cGMP-Reporter Cell Assay
4.6. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, H.; Förstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Förstermann, U.; Mülsch, A.; Böhme, E.; Busse, R. Stimulation of soluble guanylate cyclase by an acetylcholine-induced endothelium-derived factor from rabbit and canine arteries. Circ. Res. 1986, 58, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Harbison, R.G.; Wood, K.S.; Kadowitz, P.J. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: Stimulation by acetylcholine, bradykinin and arachidonic acid. J. Pharmacol. Exp. Ther. 1986, 237, 893–900. [Google Scholar] [PubMed]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Halbrügge, T.; Lütsch, K.; Thyen, A.; Graefe, K.H. Role of nitric oxide formation in the regulation of haemodynamics and the release of noradrenaline and adrenaline. Naunyn-Schmied. Arch. Pharmacol. 1991, 344, 720–727. [Google Scholar] [CrossRef]
- Van Vliet, B.N.; Chafe, L.L.; Montani, J.P. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J. Physiol. 2003, 549, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.W.; Scalia, R.; Murohara, T.; Guo, J.P.; Lefer, A.M. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Cayatte, A.J.; Palacino, J.J.; Horten, K.; Cohen, R.A. Chronic inhibition of nitric oxide production accelerates neointima formation and impairs endothelial function in hypercholesterolemic rabbits. Arterioscler. Thromb. 1994, 14, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Kauser, K.; da Cunha, V.; Fitch, R.; Mallari, C.; Rubanyi, G.M. Role of endogenous nitric oxide in progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1679–H1685. [Google Scholar] [PubMed]
- Alheid, U.; Frolich, J.C.; Forstermann, U. Endothelium-derived relaxing factor from cultured human endothelial cells inhibits aggregation of human platelets. Thromb. Res. 1987, 47, 561–571. [Google Scholar] [CrossRef]
- Li, H.; Förstermann, U. Prevention of atherosclerosis by interference with the vascular nitric oxide system. Curr. Pharm. Des. 2009, 15, 3133–3145. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Dirsch, V. Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 2014, 20, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Förstermann, U. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar]
- Dudzinski, D.M.; Igarashi, J.; Greif, D.; Michel, T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 235–276. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. Eur. J. Physiol. 2010, 459, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.J.; Colditz, G.A. Estrogen replacement therapy and coronary heart disease: A quantitative assessment of the epidemiologic evidence. Prev. Med. 1991, 20, 47–63. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Green, S.; Kumar, V.; Krust, A.; Walter, P.; Chambon, P. Structural and functional domains of the estrogen receptor. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Greene, G.L.; Press, M.F. Structure and dynamics of the estrogen receptor. J. Steroid Biochem. 1986, 24, 1–7. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Gustafsson, J.A. The novel estrogen receptor-β subtype: Potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett. 1997, 410, 87–90. [Google Scholar] [CrossRef]
- Filardo, E.J. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 231–238. [Google Scholar] [CrossRef]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.O.; Korach, K.S. Estrogen receptors and endocrine diseases: Lessons from estrogen receptor knockout mice. Curr. Opin. Pharmacol. 2001, 1, 613–619. [Google Scholar] [CrossRef]
- Gilligan, D.M.; Badar, D.M.; Panza, J.A.; Quyyumi, A.A.; Cannon, R.O. Acute vascular effects of estrogen in postmenopausal women. Circulation 1994, 90, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Guetta, V.; Quyyumi, A.A.; Prasad, A.; Panza, J.A.; Waclawiw, M.; Cannon, R.O. The role of nitric oxide in coronary vascular effects of estrogen in postmenopausal women. Circulation 1997, 96, 2795–2801. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.S.; Haynes, M.P.; Sinha, D.; Clerisme, E.; Bender, J.R. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 5930–5935. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Fornari, L.; Mannella, P.; Varone, G.; Caruso, A.; Liao, J.K.; Genazzani, A.R. Novel non-transcriptional mechanisms for estrogen receptor signaling in the cardiovascular system. Interaction of estrogen receptor α with phosphatidylinositol 3-OH kinase. Steroids 2002, 67, 935–939. [Google Scholar] [CrossRef]
- Haynes, M.P.; Sinha, D.; Russell, K.S.; Collinge, M.; Fulton, D.; Morales-Ruiz, M.; Sessa, W.C.; Bender, J.R. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ. Res. 2000, 87, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.; Lu, Y.; Angle, M.; Magder, S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids 2004, 69, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Bender, J.R. Membrane-initiated actions of estrogen on the endothelium. Mol. Cell. Endocrinol. 2009, 308, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma membrane localization and function of the estrogen receptor αvariant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Moriarty, K.; Bender, J.R. Vascular cell signaling by membrane estrogen receptors. Steroids 2008, 73, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of BA for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2004, 24, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.H.; Mullauer, F.B.; de Roo, G.M.; Medema, J.P. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett. 2007, 251, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Kasperczyk, H.; La Ferla-Brühl, K.; Westhoff, M.A.; Behrend, L.; Zwacka, R.M.; Debatin, K.M.; Fulda, S. Betulinic acid as new activator of NF-κB: Molecular mechanisms and implications for cancer therapy. Oncogene 2005, 24, 6945–6956. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.E.; Blum, R.; Wilton, J.; Doto, J.; Galbraith, H.; Burgess, G.L.; Smith, P.C.; Ballow, C. Safety and pharmacokinetics of bevirimat (PA-457), a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Antimicrob. Agents Chemother. 2007, 51, 3063–3066. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp-Fenske, K.; Bollinger, L.; Xu, H.; Yao, Y.; Horke, S.; Förstermann, U.; Li, H. Reciprocal regulation of endothelial nitric-oxide synthase and NADPH oxidase by betulinic acid in human endothelial cells. J. Pharmacol. Exp. Ther. 2007, 322, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.W.; Choi, C.Y.; Hwang, Y.P.; Kim, H.G.; Kim, S.J.; Chung, Y.C.; Lee, K.J.; Jeong, T.C.; Jeong, H.G. Betulinic Acid Increases eNOS Phosphorylation and NO Synthesis via the Calcium-Signaling Pathway. J. Agric. Food Chem. 2016, 64, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Xi, L. eNOS phosphorylation: A pivotal molecular switch in vasodilation and cardioprotection? J. Mol. Cell. Cardiol. 2007, 42, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Greif, D.M.; Kou, R.; Michel, T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: Evidence for crosstalk between phosphorylation sites. Biochemistry 2002, 41, 15845–15853. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Wessler, S.; Follmann, E.; Michaelis, W.; Düsterhöft, T.; Baumann, G.; Stangl, K.; Stangl, V. A constituent of green tea, epigallocatechin-3-gallate, activates endothelial nitric oxide synthase by a phosphatidylinositol-3-OH-kinase-, cAMP-dependent protein kinase-, and Akt-dependent pathway and leads to endothelial-dependent vasorelaxation. J. Biol. Chem. 2004, 279, 6190–6915. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [PubMed]
- Chen, Z.; Yuhanna, I.S.; Galcheva-Gargova, Z.; Karas, R.H.; Mendelsohn, M.E.; Shaul, P.W. Estrogen receptor α mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Investig. 1999, 103, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E. A new estrogen receptor antagonist—An overview of available data. Breast Cancer Res. Treat. 2002, 75 (Suppl. S1), S19–S21. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M.; Wickramasinghe, N.S.; Ivanova, M.M.; Dougherty, S.M. Resveratrol stimulates nitric oxide production by increasing estrogen receptor α-Src-caveolin-1 interaction and phosphorylation in human umbilical vein endothelial cells. FASEB J. 2008, 22, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Hisamoto, K.; Ohmichi, M.; Kurachi, H.; Hayakawa, J.; Kanda, Y.; Nishio, Y.; Adachi, K.; Tasaka, K.; Miyoshi, E.; Fujiwara, N.; et al. Estrogen Induces the Akt-dependent Activation of Endothelial Nitric-oxide Synthase in Vascular Endothelial Cells. J. Biol. Chem. 2001, 276, 3459–3467. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.Y.; Kim, H.G.; Hien, T.T.; Jeong, M.H.; Jeong, T.C.; Jeong, H.G. Puerarin activates endothelial nitric oxide synthase through estrogen receptor-dependent PI3-kinase and calcium-dependent AMP-activated protein kinase. Toxicol. Appl. Pharmacol. 2011, 257, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Xia, N.; Xu, H.; Guo, L.; Wenzel, P.; Daiber, A.; Munzel, T.; Förstermann, U.; Li, H. Betulinic acid protects against cerebral ischemia-reperfusion injury in mice by reducing oxidative and nitrosative stress. Nitric Oxide 2011, 24, 132–138. [Google Scholar] [CrossRef] [PubMed]
- De Melo, C.L.; Queiroz, M.G.; Arruda Filho, A.C.; Rodrigues, A.M.; de Sousa, D.F.; Almeida, J.G.; Pessoa, O.D.; Silveira, E.R.; Menezes, D.B.; Melo, T.S.; et al. Betulinic acid, a natural pentacyclic triterpenoid, prevents abdominal fat accumulation in mice fed a high-fat diet. J. Agric. Food Chem. 2009, 57, 8776–8781. [Google Scholar] [CrossRef] [PubMed]
- Godugu, C.; Patel, A.R.; Doddapaneni, R.; Somagoni, J.; Singh, M. Approaches to improve the oral bioavailability and effects of novel anticancer drugs berberine and betulinic acid. PLoS ONE 2014, 9, e89919. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Gustafson, J.A. Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Edgell, C.J.; McDonald, C.C.; Graham, J.B. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. USA 1983, 80, 3734–3737. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Oehrlein, S.A.; Wallerath, T.; Ihrig-Biedert, I.; Wohlfart, P.; Ulshöfer, T.; Jessen, T.; Herget, T.; Förstermann, U.; Kleinert, H. Activation of protein kinase C α and/or epsilon enhances transcription of the human endothelial nitric oxide synthase gene. Mol. Pharmacol. 1998, 53, 630–637. [Google Scholar] [PubMed]
- Li, H.; Förstermann, U. Structure-activity relationship of staurosporine analogs in regulating expression of endothelial nitric-oxide synthase gene. Mol. Pharmacol. 2000, 57, 427–435. [Google Scholar] [PubMed]
- Ishii, K.; Sheng, H.; Warner, T.D.; Förstermann, U.; Murad, F. A simple and sensitive bioassay method for detection of EDRF with RFL-6 rat lung fibroblasts. Am. J. Physiol. 1991, 261, H598–H603. [Google Scholar] [PubMed]
- Steiner, A.L.; Parker, C.W.; Kipnis, D.M. Radioimmunoassay for cyclic nucleotides. I. Preparation of antibodies and iodinated cyclic nucleotides. J. Biol. Chem. 1972, 247, 1106–1113. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compound (betulinic acid) are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohmann, N.; Xia, N.; Steinkamp-Fenske, K.; Förstermann, U.; Li, H. Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation. Molecules 2016, 21, 973. https://doi.org/10.3390/molecules21080973
Hohmann N, Xia N, Steinkamp-Fenske K, Förstermann U, Li H. Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation. Molecules. 2016; 21(8):973. https://doi.org/10.3390/molecules21080973
Chicago/Turabian StyleHohmann, Nicolas, Ning Xia, Katja Steinkamp-Fenske, Ulrich Förstermann, and Huige Li. 2016. "Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation" Molecules 21, no. 8: 973. https://doi.org/10.3390/molecules21080973
APA StyleHohmann, N., Xia, N., Steinkamp-Fenske, K., Förstermann, U., & Li, H. (2016). Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation. Molecules, 21(8), 973. https://doi.org/10.3390/molecules21080973