
A General Catalytic Enantioselective Transfer Hydrogenation Reaction of β,β-Disubstituted Nitroalkenes Promoted by a Simple Organocatalyst


Abstract
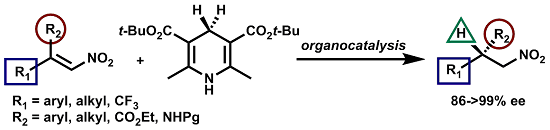
:
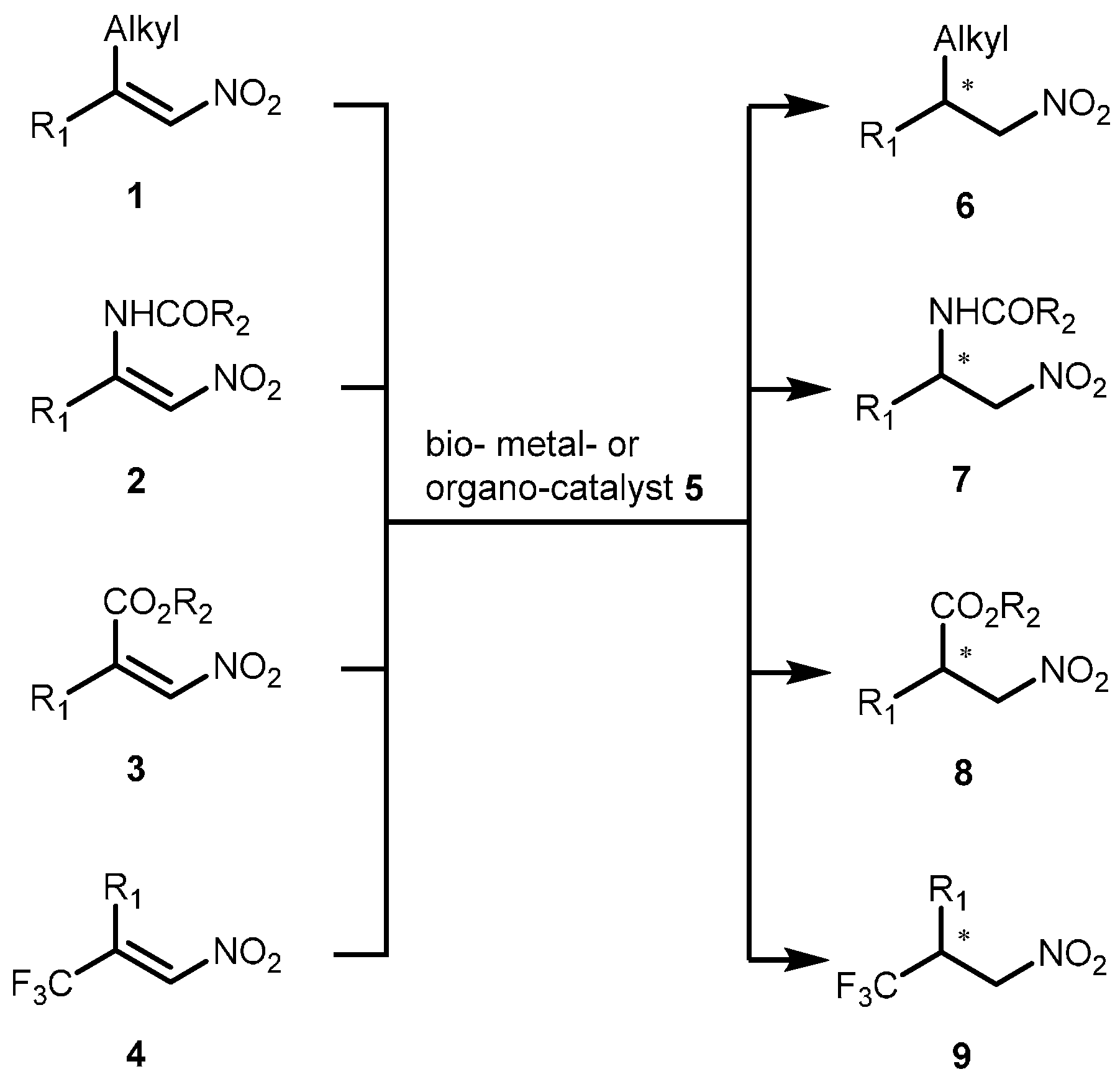
1. Introduction
2. Results and Discussion
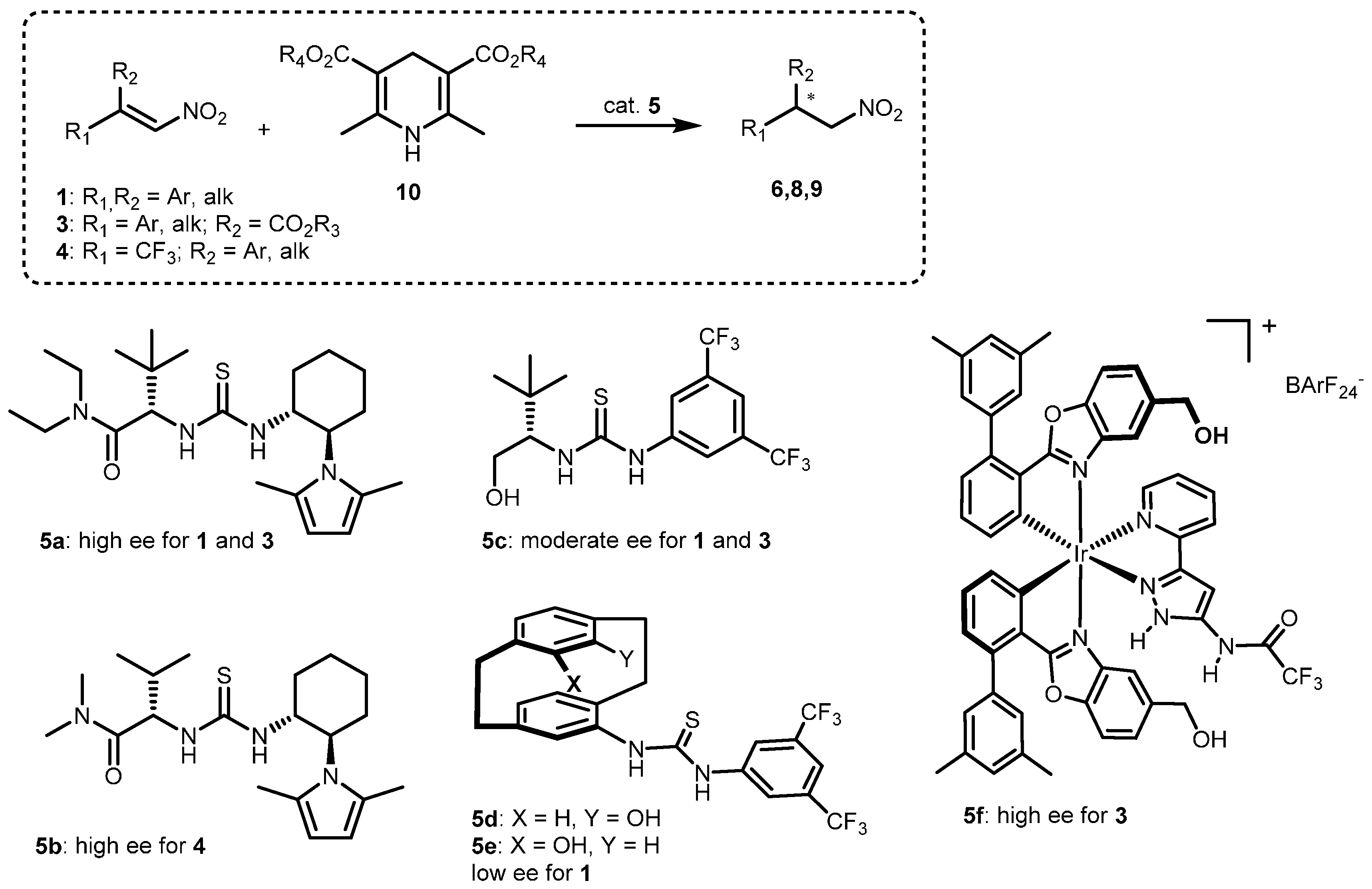
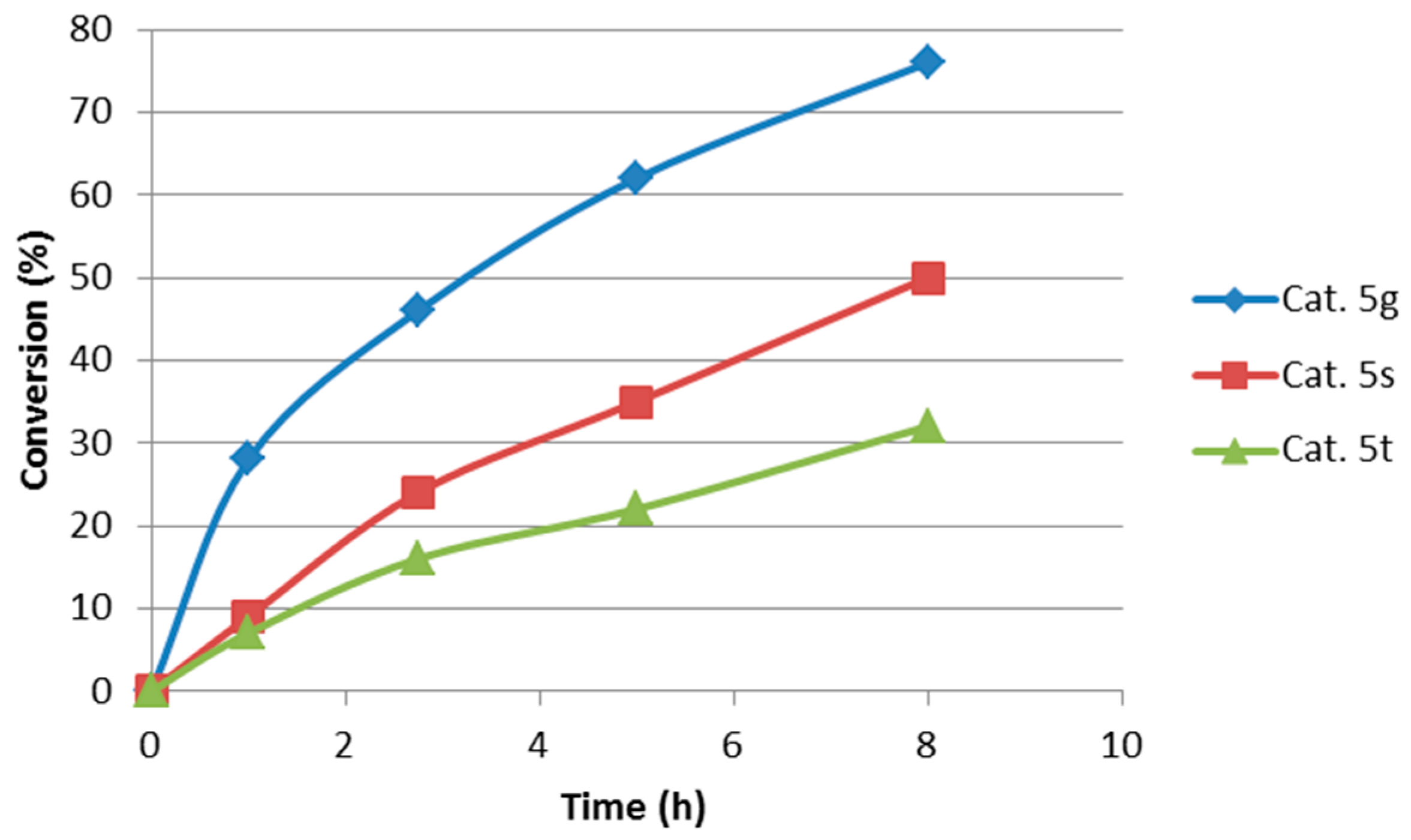
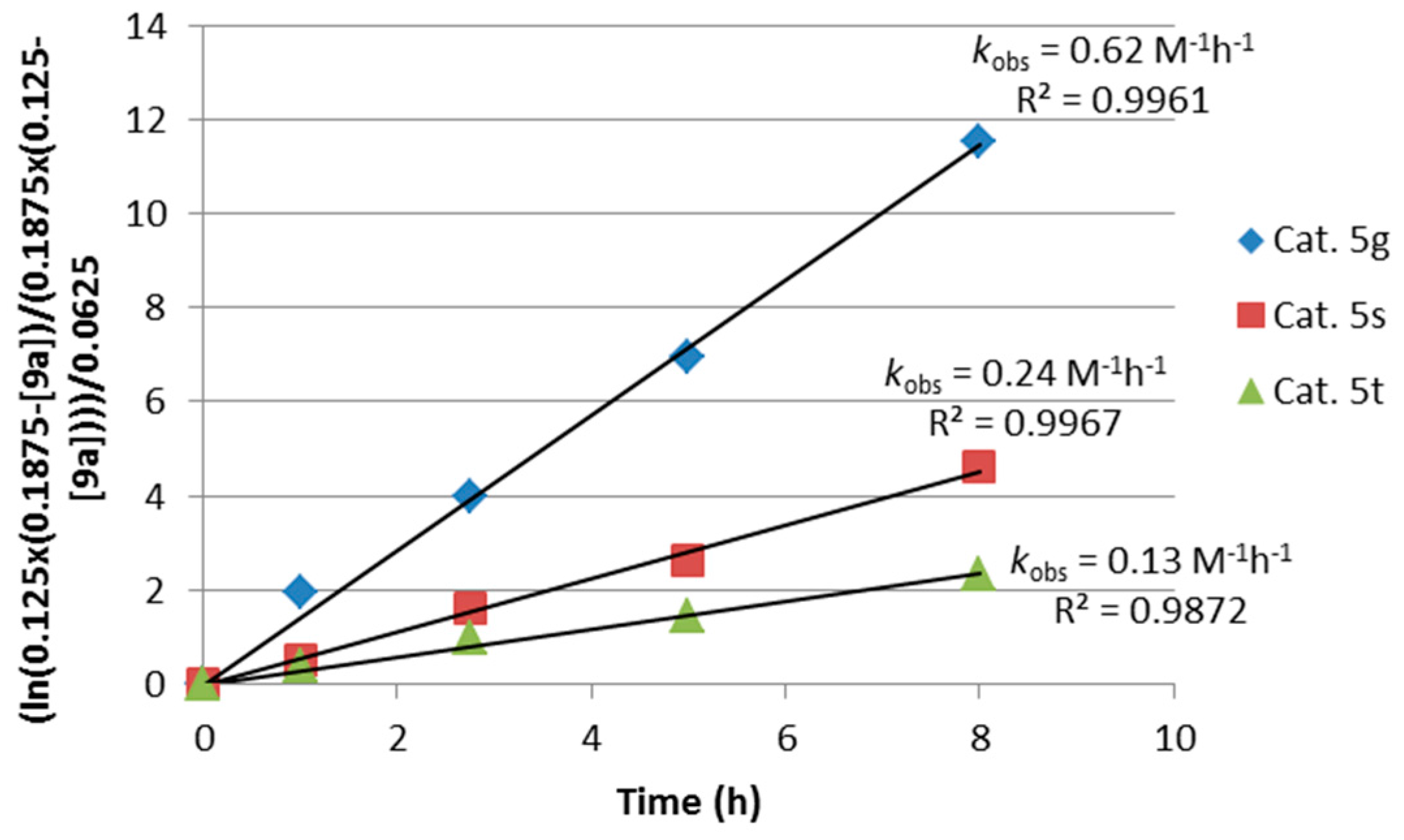
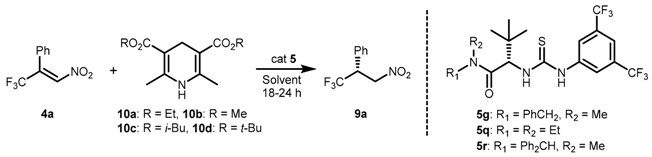
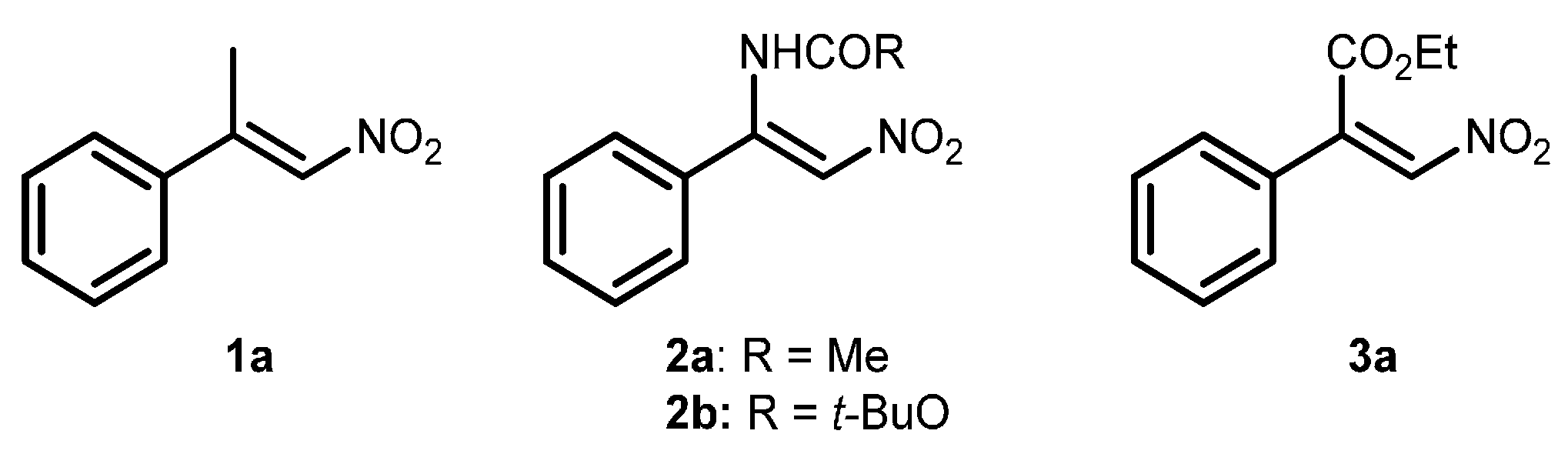
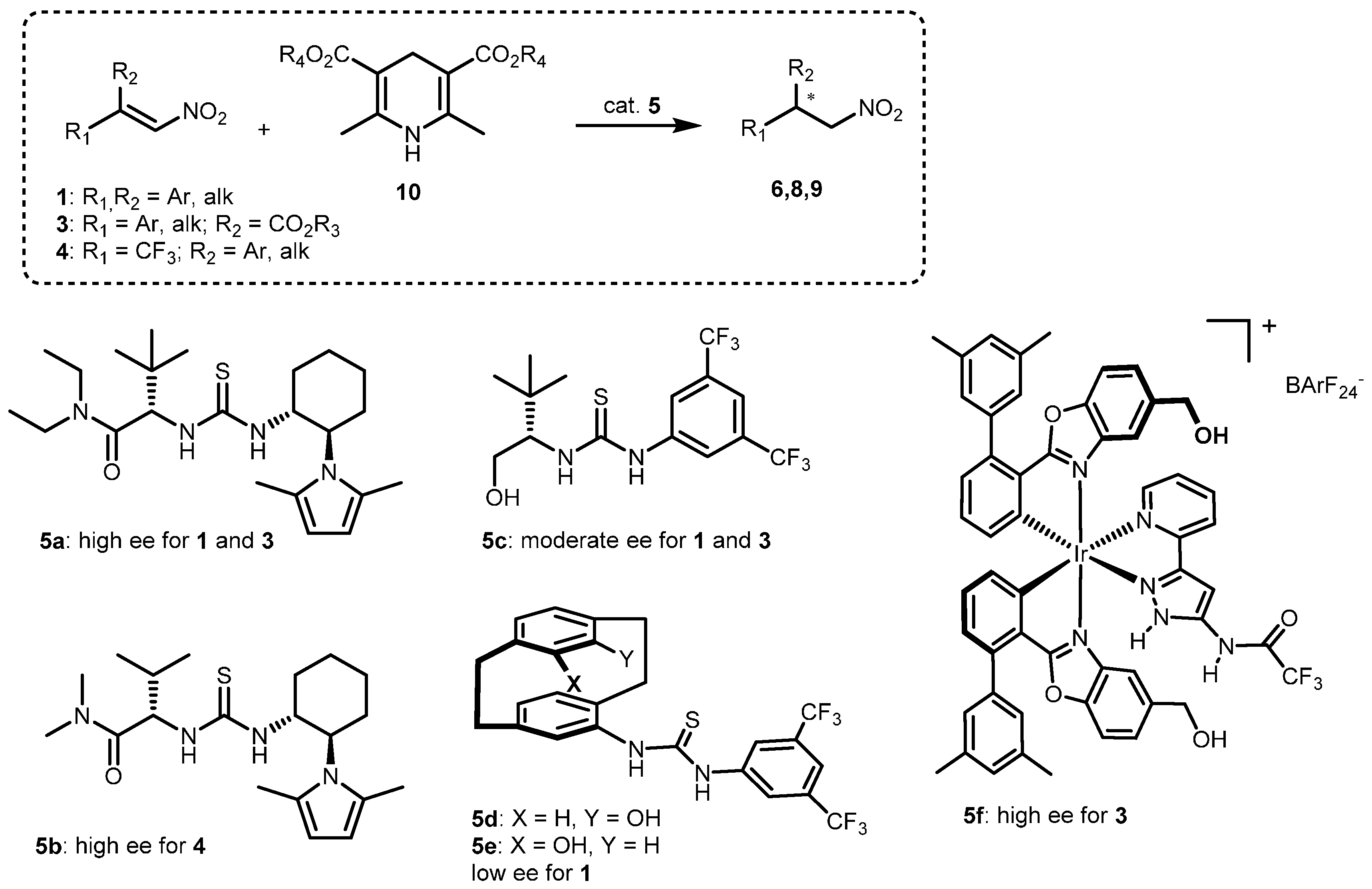
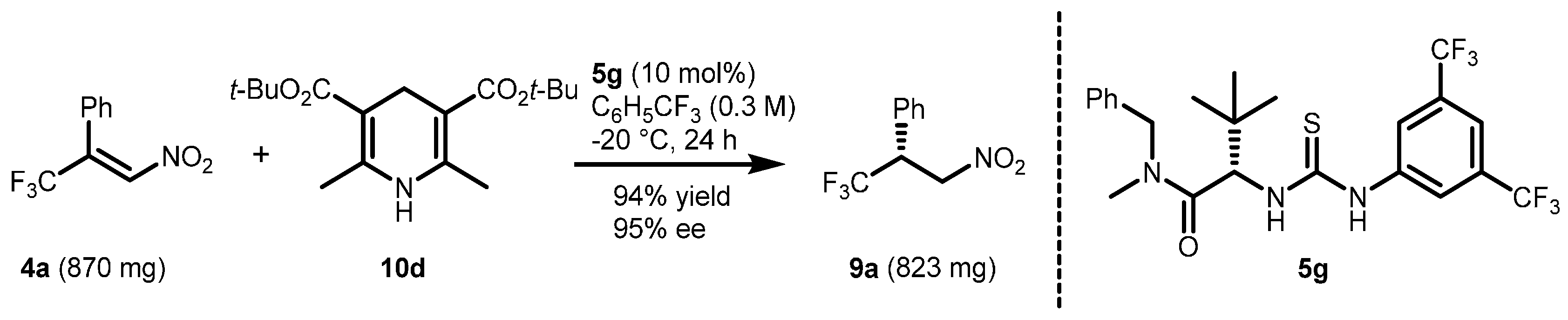
2.1. Optimization and Development of the Transfer Hydrogenation Reaction with β-Trifluoromethylnitroalkenes 4
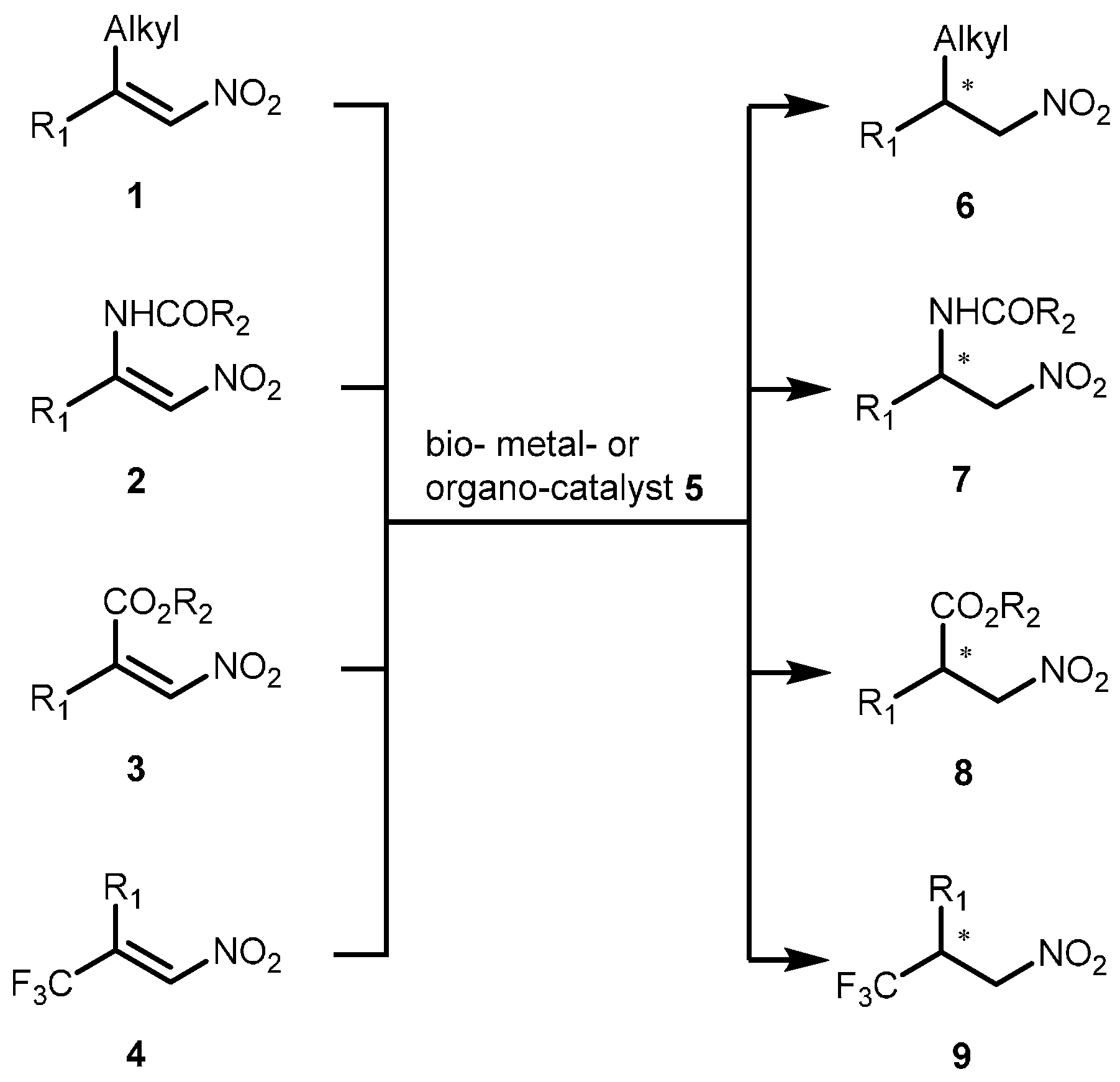
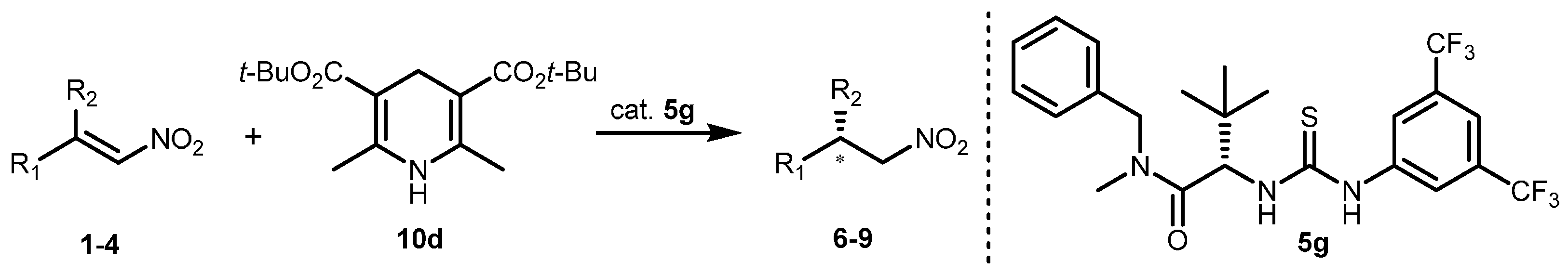
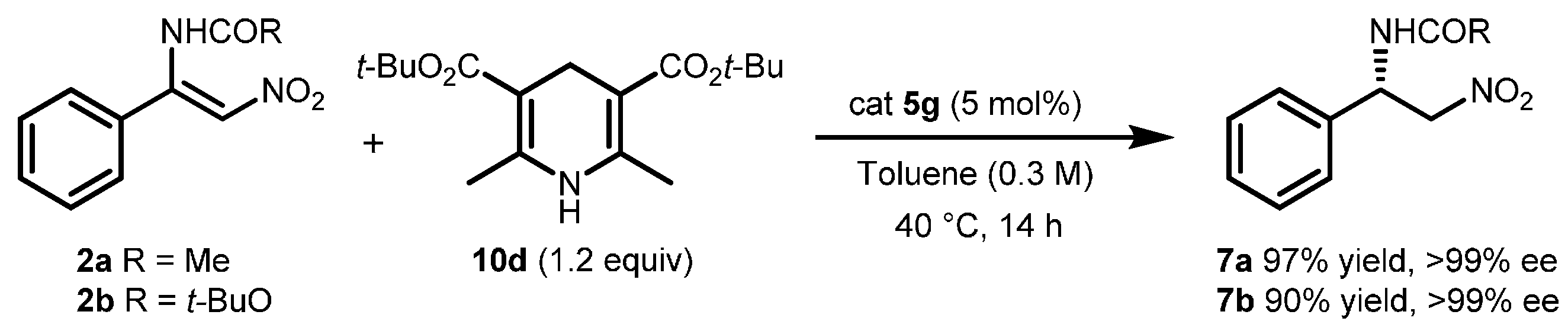
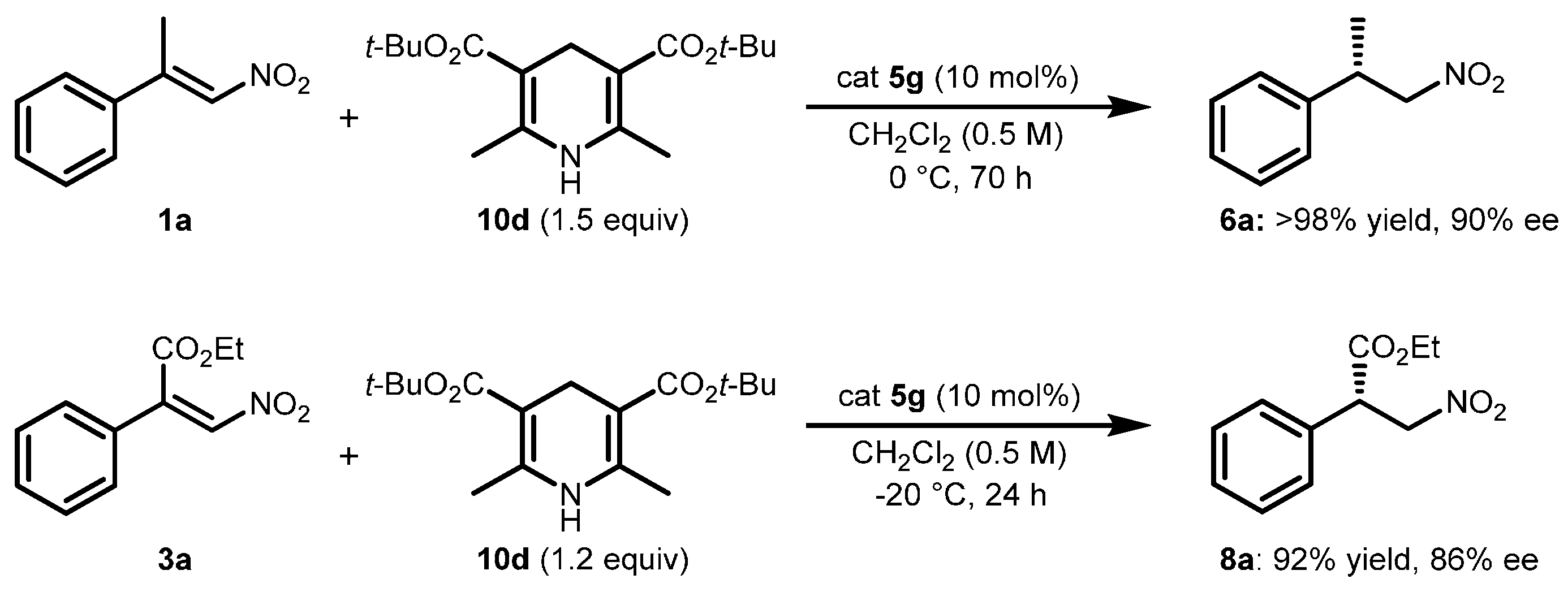
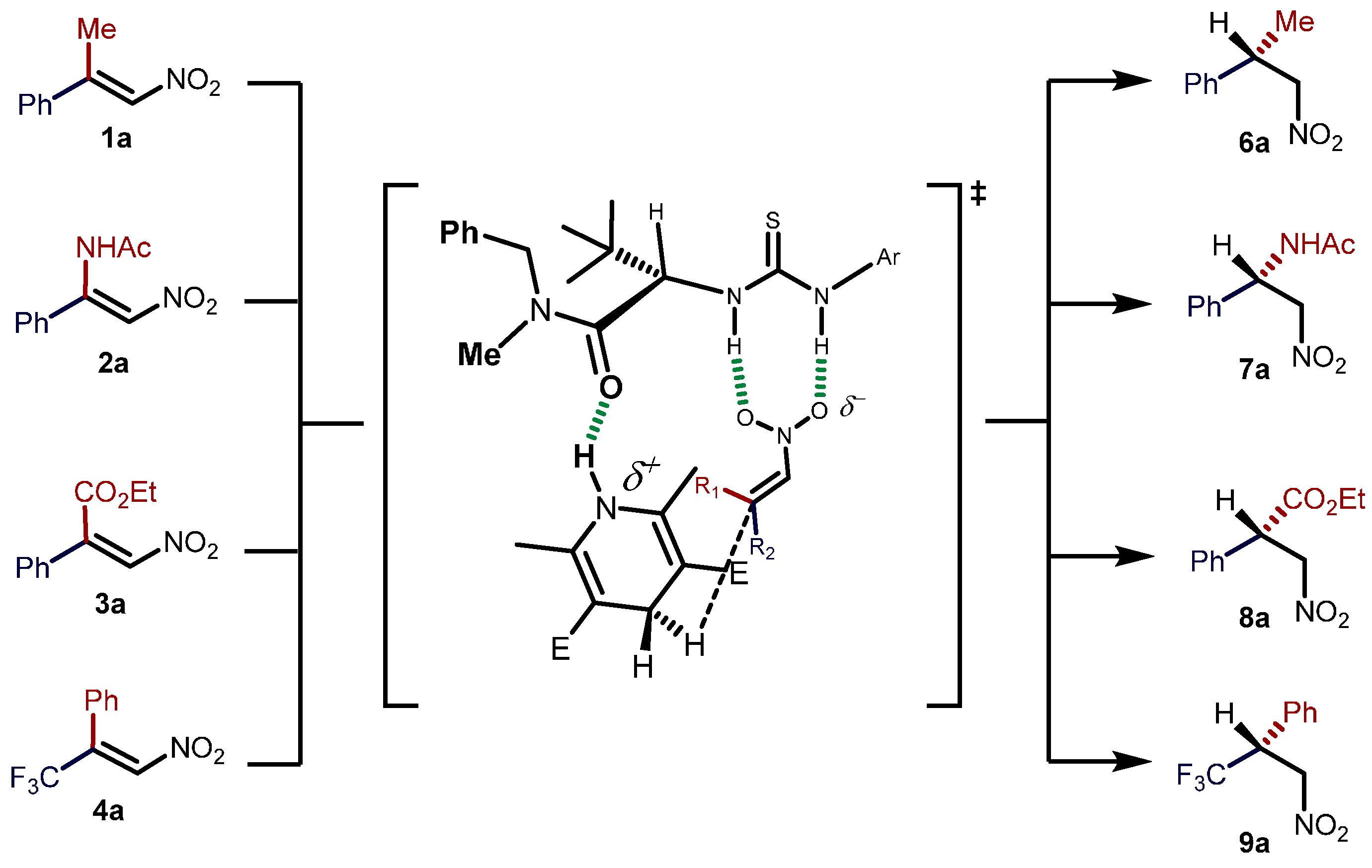
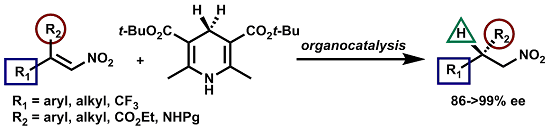
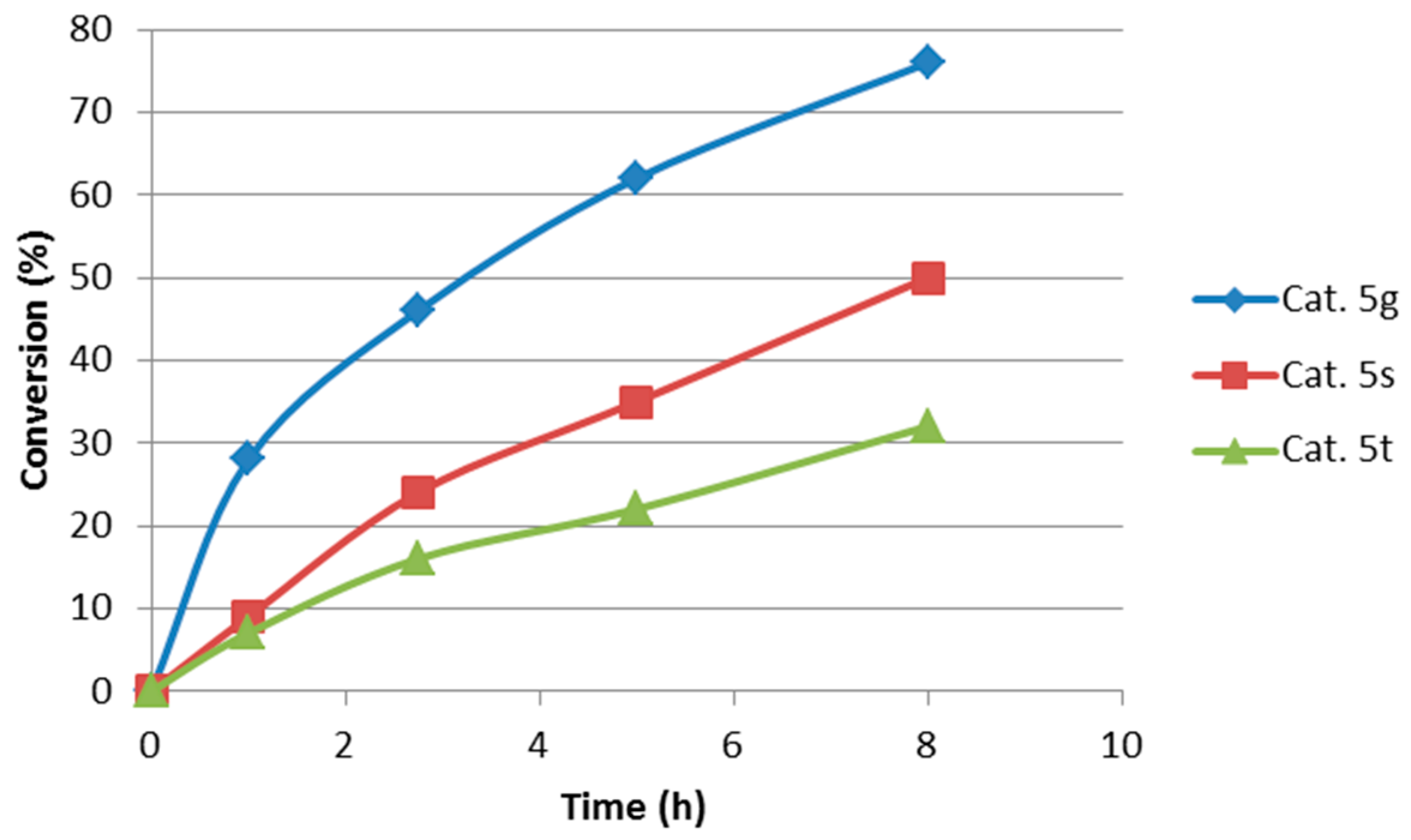
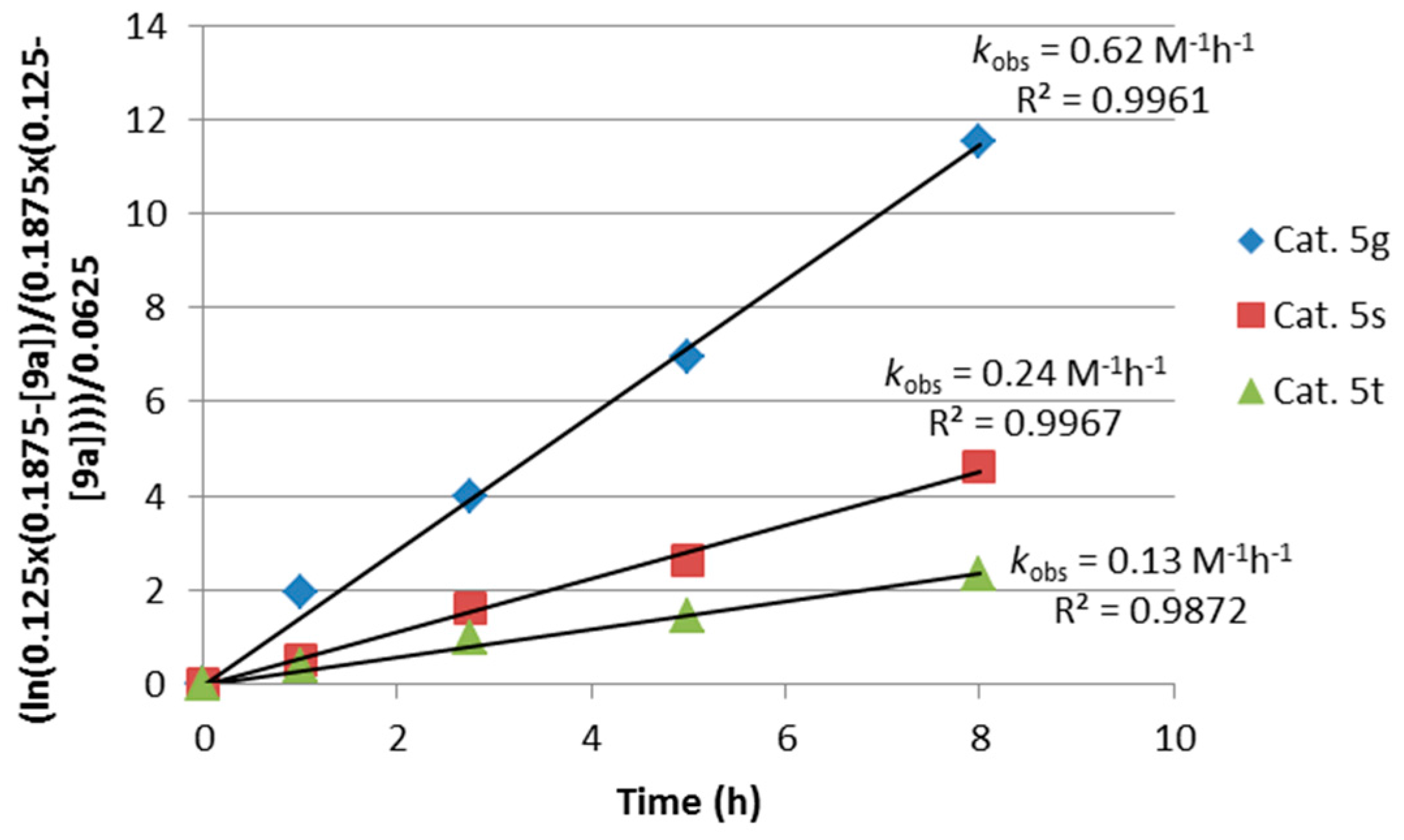
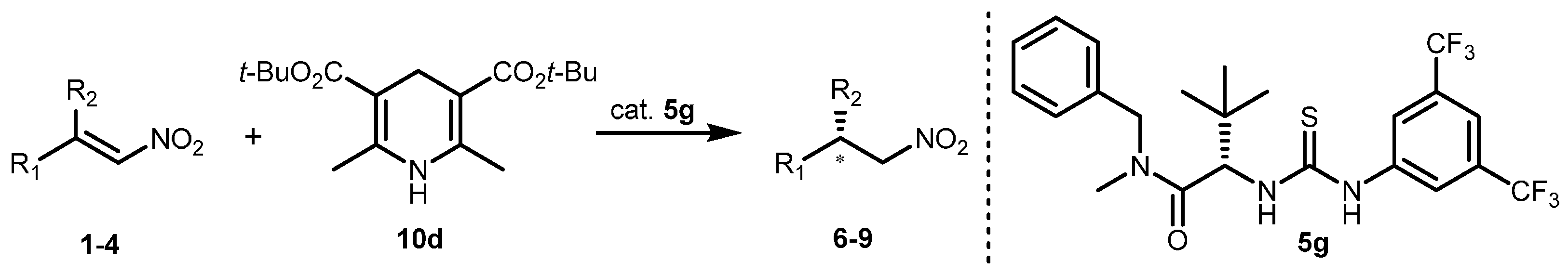
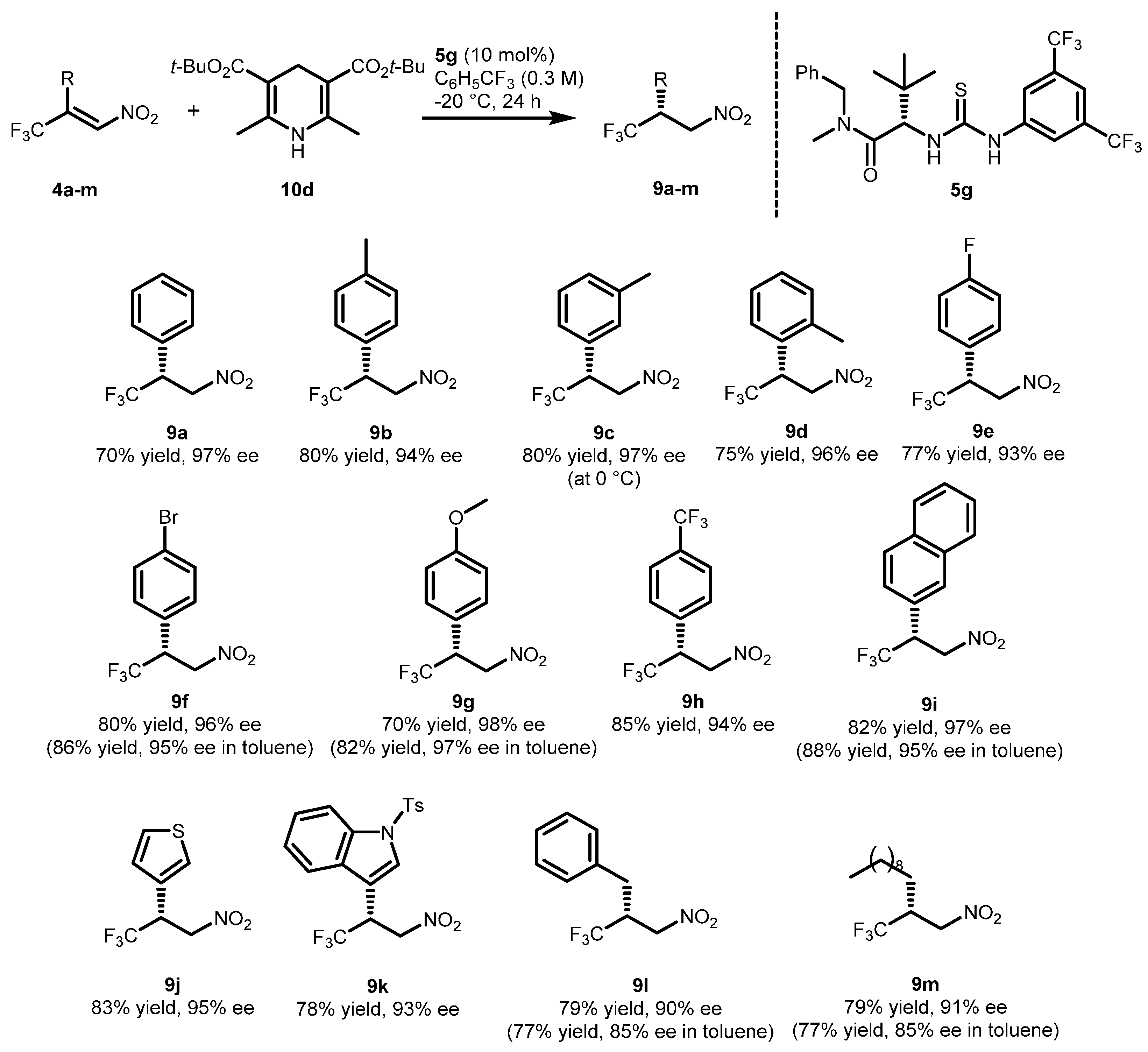
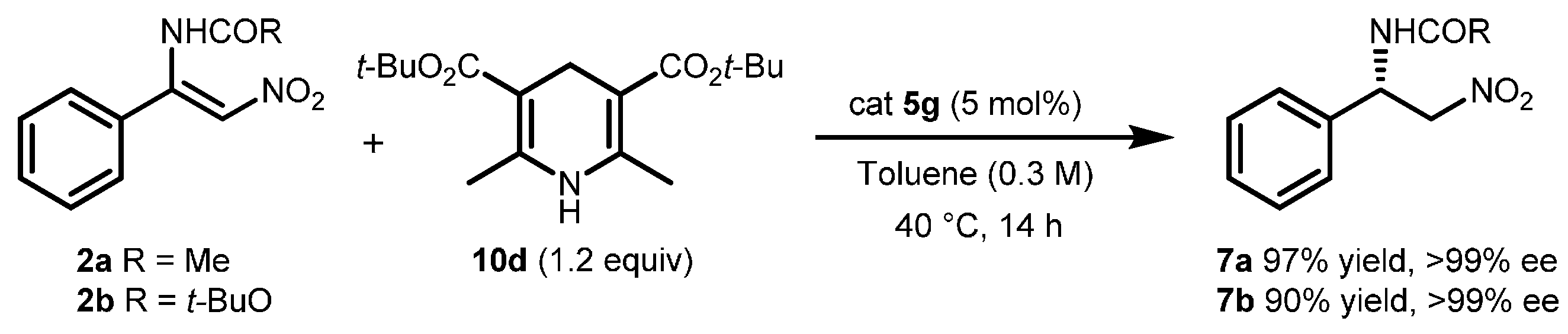
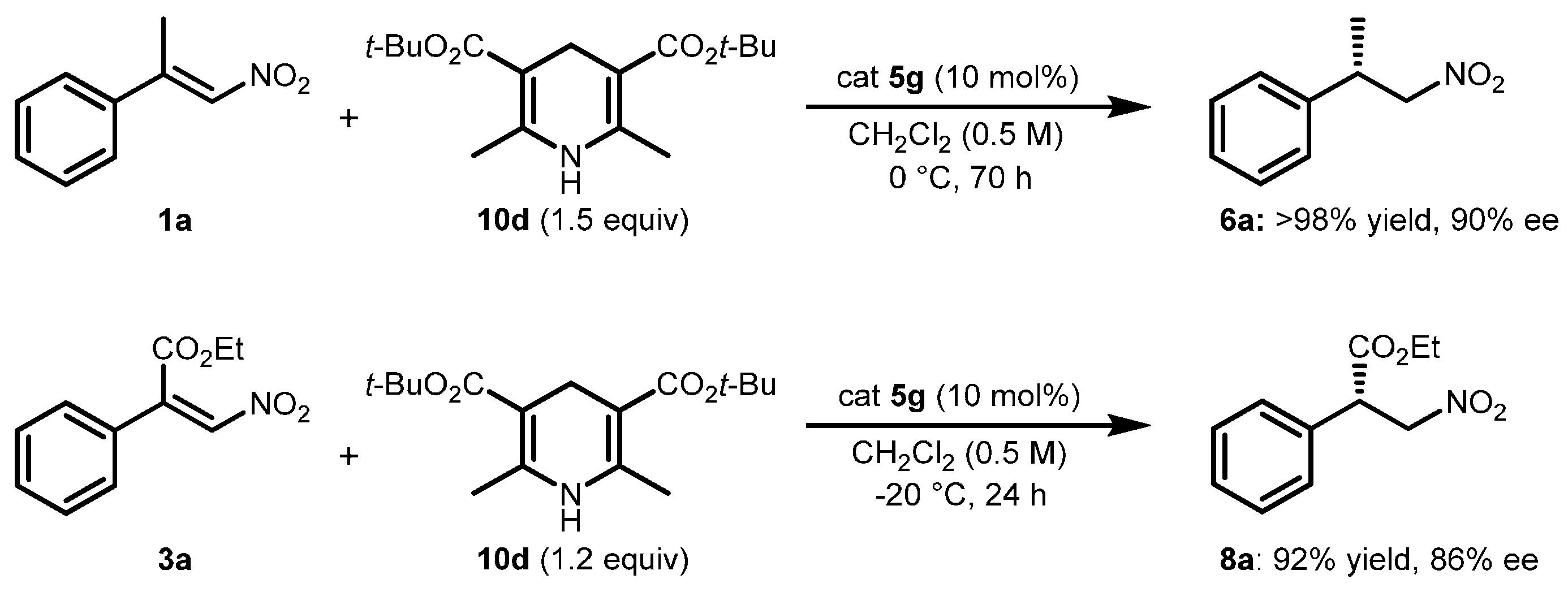
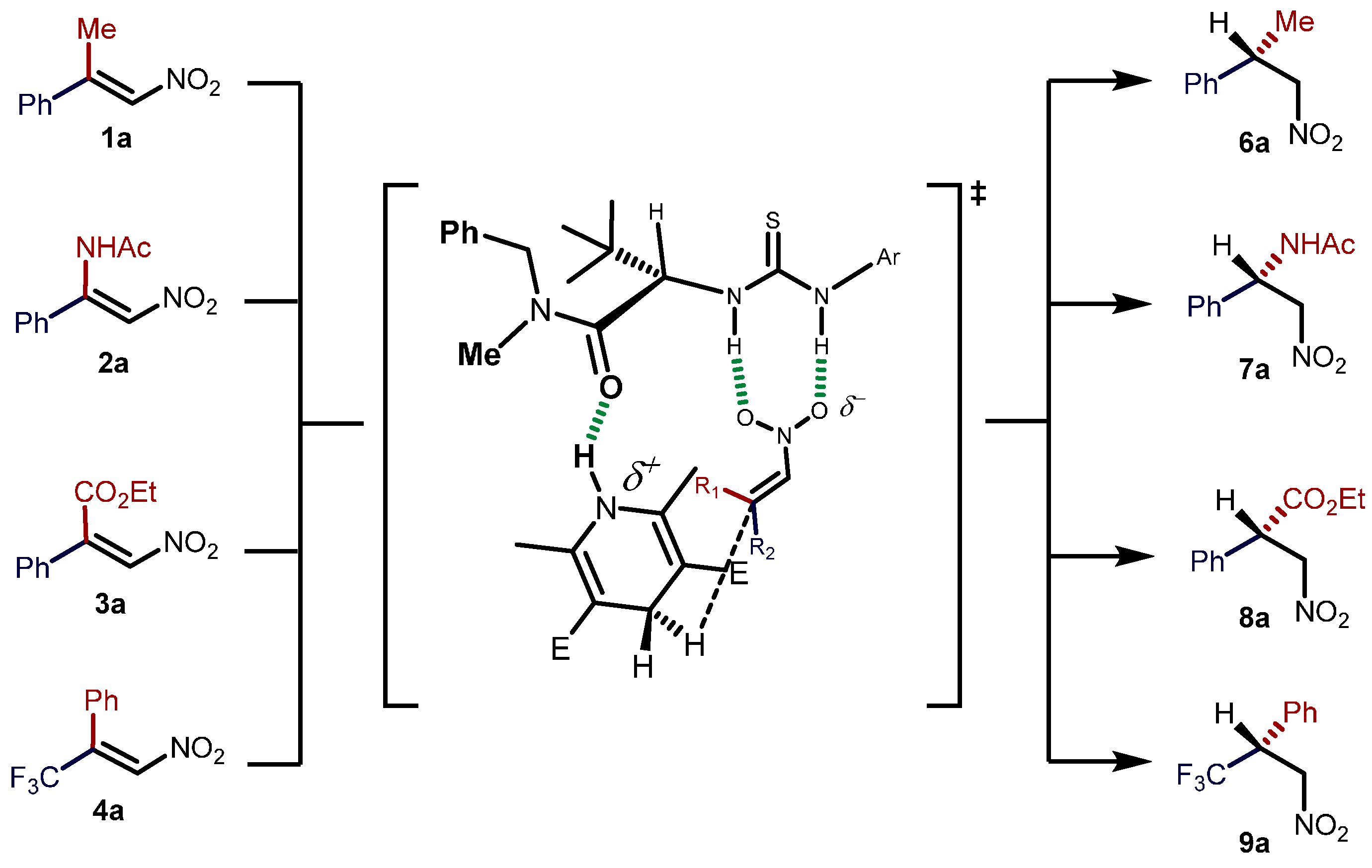
2.2. Extension of the Transfer Hydrogenation Reaction Catalyzed by 5g to Other Nitroalkenes 1–3
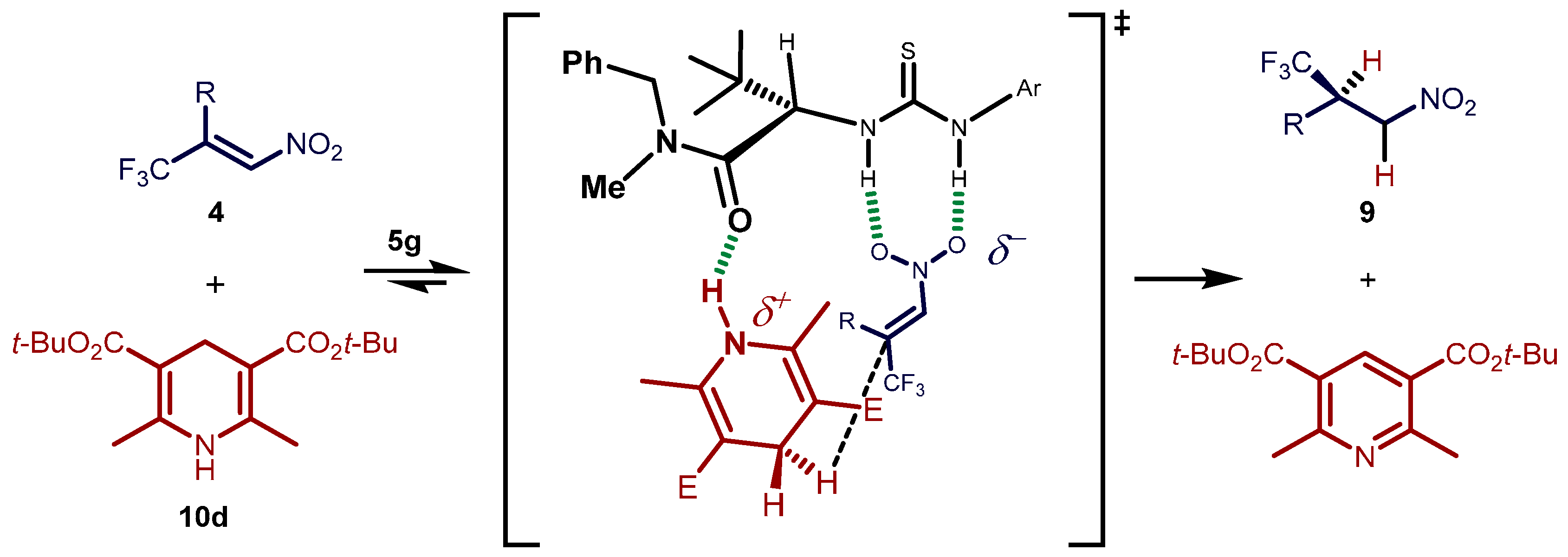
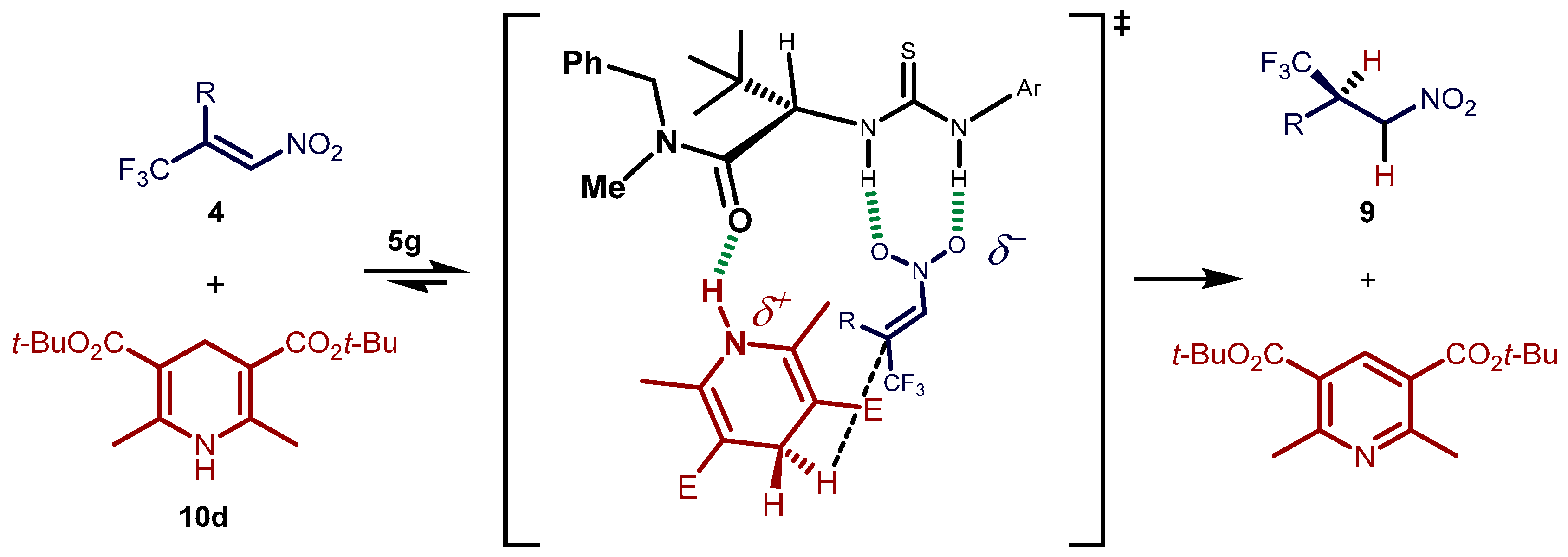
2.3. Proposed Reaction Model for the Transfer Hydrogenation Reactions Catalyzed by 5g with Nitroalkenes
3. Materials and Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ono, N. The Nitro Group in Organic Synthesis; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Ballini, R.; Gabrielli, S.; Palmieri, A.; Petrini, M. Nitroalkanes as key compounds for the synthesis of amino derivatives. Curr. Org. Chem. 2011, 15, 1482–1506. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. The nitro to carbonyl conversion (Nef reaction): New perspectives for a classical transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Ohta, H.; Kobayashi, N.; Ozaki, K. Asymmetric reduction of nitro olefins by fermenting bakers’ yeast. J. Org. Chem. 1989, 54, 1802–1804. [Google Scholar] [CrossRef]
- Hall, M.; Stueckler, C.; Kroutil, W.; Macheroux, P.; Faber, K. Asymmetric bioreduction of activated alkenes using cloned 12-oxophytodienoate reductase isoenzymes OPR-1 and OPR-3 from lycopersicon esculentum (tomato): A striking change of stereoselectivity. Angew. Chem. Int. Ed. 2007, 46, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Toogood, H.S.; Scrutton, N.S. New developments in ‘ene’-reductase catalyzed biological hydrogenations. Curr. Opin. Chem. Biol. 2014, 19, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Fryszkowska, A.; Fisher, K.; Gardiner, J.M.; Stephens, G.M. Highly enantioselective reduction of β,β-disubstituted aromatic nitroalkenes catalyzed by clostridium sporogenes. J. Org. Chem. 2008, 73, 4295–4298. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Santangelo, S. Substrate scope evaluation of the enantioselective reduction of β-alkyl-β-arylnitroalkenes by old yellow enzymes 1–3 for organic synthesis applications. ChemCatChem 2016, 8, 577–583. [Google Scholar] [CrossRef]
- Czekelius, C.; Carreira, E.M. Catalytic enantioselective conjugate reduction of β,β-disubstituted nitroalkenes. Angew. Chem. Int. Ed. 2003, 42, 4793–4795. [Google Scholar] [CrossRef] [PubMed]
- Czekelius, C.; Carreira, E.M. Enantioselective reduction of nitroalkene mixtures by in situ equilibration. Org. Process Res. Dev. 2007, 11, 633–636. [Google Scholar] [CrossRef]
- Soltani, O.; Ariger, M.A.; Carreira, E.M. Transfer hydrogenation in water: Enantioselective, catalytic reduction of (E)-β,β-disubstituted nitroalkenes. Org. Lett. 2009, 11, 4196–4198. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, K.; Zhang, J.; Wu, W.; Zhang, X. Rh-catalyzed highly enantioselective hydrogenation of nitroalkenes under basic conditions. Chem. Eur. J. 2013, 19, 10840–10844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, S.; Huang, K.; Wang, R.; Zhang, X. A novel chiral bisphosphine-thiourea ligand for asymmetric hydrogenation of β,β-disubstituted nitroalkenes. Org. Lett. 2013, 15, 4014–4017. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-B.; Cheng, L.; Li, Y.-P.; Fu, Y.; Zhu, S.-F.; Zhou, Q.-L. Enantioselective iridium-catalyzed hydrogenation of β,β-disubstituted nitroalkenes. Chem. Commun. 2016, 52, 4812–4815. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xiang, J.; Cun, L.; Wang, Y.; Zhu, J.; Liao, J.; Deng, J. Chemoselective and enantioselective transfer hydrogenation of β,β-disubstituted nitroalkenes catalyzed by a water-insoluble chiral diamine-rhodium complex in water. Tetrahedron Asymmetry 2010, 21, 1900–1905. [Google Scholar] [CrossRef]
- Li, S.; Huang, K.; Cao, B.; Zhang, J.; Wu, W.; Zhang, X. Highly enantioselective hydrogenation of β,β-disubstituted nitroalkenes. Angew. Chem. Int. Ed. 2012, 51, 8573–8576. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, M.; Zhao, Q.; Wu, W.; Hu, X.; Dong, X.-Q.; Zhang, X. Synthesis of chiral β-amino nitroalkanes via rhodium-catalyzed asymmetric hydrogenation. Org. Lett. 2016, 18, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Liu, M.; Kong, D.; Zi, G.; Hou, G. Highly efficient iridium-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins. Chem. Commun. 2014, 50, 12870–12872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Dong, D.; Zhu, B.; Geng, H.; Wang, Y.; Zhang, X. Rhodium-catalyzed enantioselective hydrogenation of β-acylamino nitroolefins: A new approach to chiral β-amino nitroalkanes. Org. Lett. 2013, 15, 5524–5527. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiao, T.; Li, D.; Zhang, X. First iridium-catalyzed highly enantioselective hydrogenation of β-nitroacrylates. Org. Lett. 2015, 17, 3782–3785. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-W.; Yan, Y.; Wang, Y.-Q.; Wang, C.; Sun, J. Highly enantioselective reduction of β-amino nitroolefins with a simple N-sulfinyl urea as bifunctional catalyst. Chem. Eur. J. 2012, 18, 9204–9207. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, S.G.; Walji, A.M.; MacMillan, D.W.C. Enantioselective organocatalytic transfer hydrogenation reactions using Hantzsch esters. Acc. Chem. Res. 2007, 40, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Benaglia, M.; Massolo, E.; Raimondi, L. Organocatalytic strategies for enantioselective metal-free reductions. Catal. Sci. Technol. 2014, 4, 2708–2723. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P. Organocatalytic transfer hydrogenation and hydrosilylation reactions. Top. Curr. Chem. 2016, 374, 29. [Google Scholar] [CrossRef]
- Rueping, M.; Dufour, J.; Schoepke, S.J. Advances in catalytic metal-free reductions: From bio-inspired concepts to applications in the organocatalytic synthesis of pharmaceuticals and natural products. Green Chem. 2011, 13, 1084–1105. [Google Scholar] [CrossRef]
- Bernardi, L.; Fochi, M.; Comes Franchini, M.; Ricci, A. Bioinspired organocatalytic asymmetric reactions. Org. Biomol. Chem. 2012, 10, 2911–2922. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, H.; Takemoto, Y. Discovery and application of asymmetric reaction by multi-functional thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.J.A.; Ozores, L.; List, B. Organocatalytic asymmetric transfer hydrogenation of nitroolefins. J. Am. Chem. Soc. 2007, 129, 8976–8977. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.J.A.; Cheng, X.; List, B. Organocatalytic asymmetric transfer hydrogenation of β-nitroacrylates: Accessing β2-amino acids. J. Am. Chem. Soc. 2008, 130, 13862–13863. [Google Scholar] [CrossRef] [PubMed]
- Massolo, E.; Benaglia, M.; Orlandi, M.; Rossi, S.; Celentano, G. Enantioselective organocatalytic reduction of β-trifluoromethyl nitroalkenes: An efficient strategy for the synthesis of chiral β-trifluoromethyl amines. Chem. Eur. J. 2015, 21, 3589–3595. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.F.; Lauber, M.B.; Muhr, V.; Kratzer, D.; Paradies, J. Readily available hydrogen bond catalysts for the asymmetric transfer hydrogenation of nitroolefins. Org. Biomol. Chem. 2011, 9, 4323–4327. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.F.; Falk, F.C.; Fröhlich, R.; Paradies, J. Planar-chiral thioureas as hydrogen-bond catalysts. Eur. J. Org. Chem. 2010, 2265–2269. [Google Scholar] [CrossRef]
- Chen, L.-A.; Xu, W.; Huang, B.; Ma, J.; Wang, L.; Xi, J.; Harms, K.; Gong, L.; Meggers, E. Asymmetric catalysis with an inert chiral-at-metal iridium complex. J. Am. Chem. Soc. 2013, 135, 10598–10601. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.C.; Koovits, P.J. An enantioselective tandem reduction/nitro-Mannich reaction of nitroalkenes using a simple thiourea organocatalyst. Chem. Sci. 2013, 4, 2897–2901. [Google Scholar] [CrossRef]
- Martinelli, E.; Vicini, A.C.; Mancinelli, M.; Mazzanti, A.; Zani, P.; Bernardi, L.; Fochi, M. Catalytic highly enantioselective transfer hydrogenation of β-trifluoromethyl nitroalkenes. An easy and general entry to optically active β-trifluoromethyl amines. Chem. Commun. 2015, 51, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, A.; Bernardi, L.; Fochi, M. Organocatalytic enantioselective transfer hydrogenation of β-amino nitroolefins. Adv. Synth. Catal. 2016, 358, 1561–1565. [Google Scholar] [CrossRef]
- Zuend, S.J.; Jacobsen, E.N. Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: Transition state stabilization via multiple non-covalent interactions. J. Am. Chem. Soc. 2009, 131, 15358–15374. [Google Scholar] [CrossRef] [PubMed]
- Zuend, S.J.; Coughlin, M.P.; Lalonde, M.P.; Jacobsen, E.N. Scaleable catalytic asymmetric Strecker syntheses of unnatural α-amino acids. Nature 2009, 461, 968–971. [Google Scholar] [CrossRef] [PubMed]
- CAS 959979-30-7, Sigma-Aldrich code 693316.
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Sigman, M.S. Systematically probing the effect of catalyst acidity in a hydrogen-bond-catalyzed enantioselective reaction. Angew. Chem. Int. Ed. 2007, 47, 4748–4750. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; Portland Press: London, UK, 1995. [Google Scholar]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef] [PubMed]
- Romanini, S.; Galletti, E.; Caruana, L.; Mazzanti, A.; Himo, F.; Santoro, S.; Fochi, M.; Bernardi, L. Catalytic asymmetric reactions of 4-substituted indoles with nitroethene: A direct entry to Ergot alkaloid structures. Chem. Eur. J. 2015, 21, 17578–17582. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Han, P.; Wu, X.; Tang, M. The mechanism investigation of chiral phosphoric acid-catalyzed Friedel-Crafts reactions—How the chiral phosphoric acid regains the proton. Comp. Theor. Chem. 2014, 1050, 39–45. [Google Scholar] [CrossRef]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [PubMed]
- It should be noted that the Re-Si descriptors for the pro-chiral face of nitroalkenes 1–4 are not relevant to this discussion, since they are related to the CIP priorities of the substituents.
- Leutbecher, H.; Greiner, G.; Amann, R.; Stolz, A.; Beifuss, U.; Conrad, J. Laccase-catalyzed phenol oxidation. Rapid assignment of ring-proton deficient polycyclic benzofuran regioisomers by experimental 1H-13C long-range coupling contants and DFT-predicted product formation. Org. Biomol. Chem. 2011, 9, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W. Porphyria-inducing activity of a series of pyridine and dihydropyridine compounds. Investigation in a cell culture system. J. Med. Chem. 1975, 18, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-R.; Wu, H.; Xiang, B.; Yu, W.-B.; Han, L.; Jia, Y.-X. Highly enantioselective construction of trifluoromethylated all-carbon quaternary stereocenters via nickel-catalyzed Friedel-Crafts alkylation reaction. J. Am. Chem. Soc. 2013, 135, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Foli, G.; Sasso D’Elia, C.; Fochi, M.; Bernardi, L. Reversible modulation of the activity of thiourea catalysts with anions: A simple approach to switchable asymmetric catalysis. RSC Adv. 2016, 6, 66490–66494. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 7b and 9a are available from the authors.
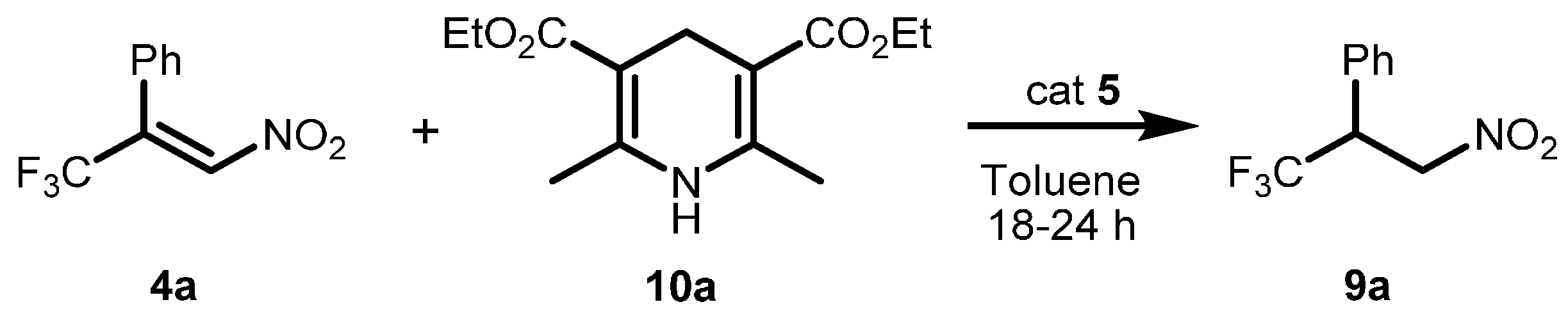
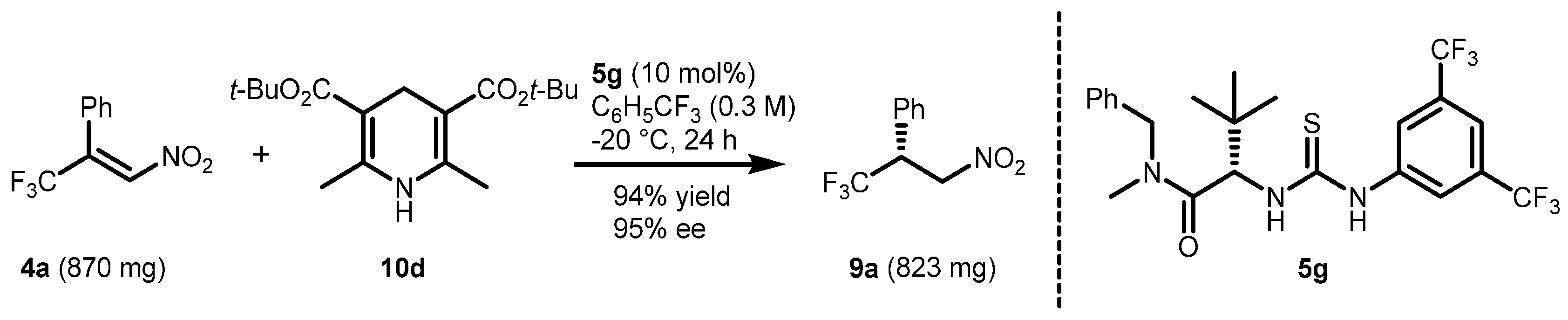
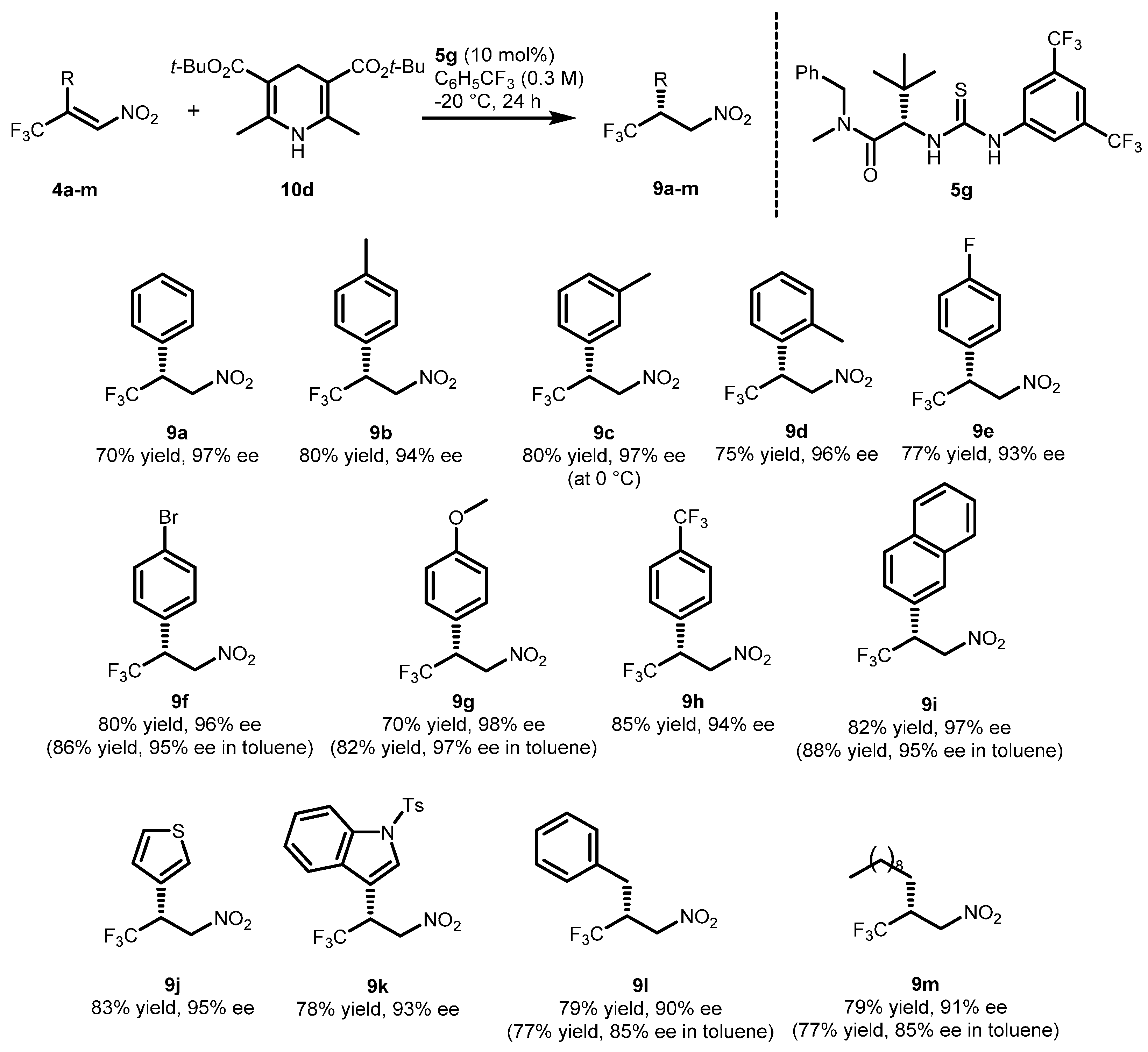
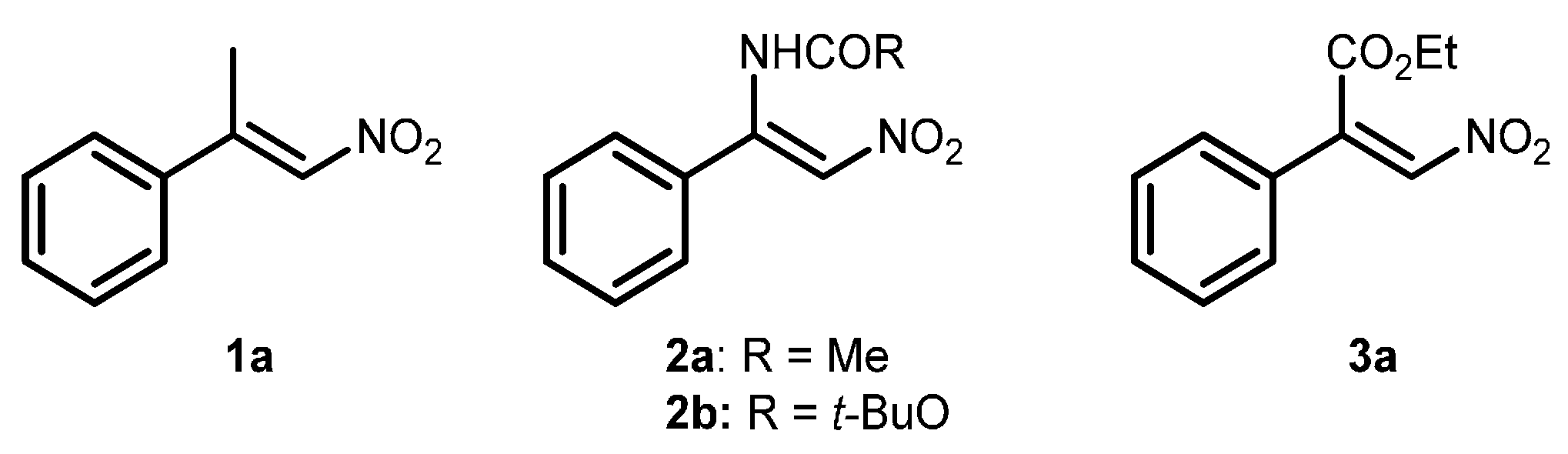
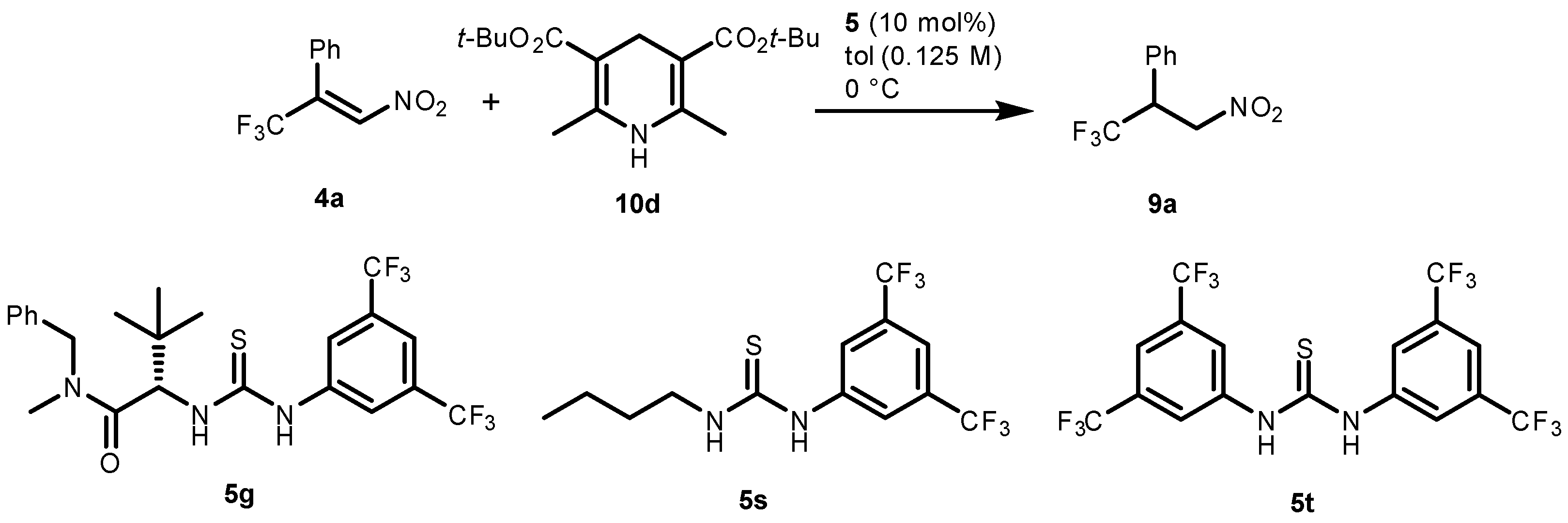

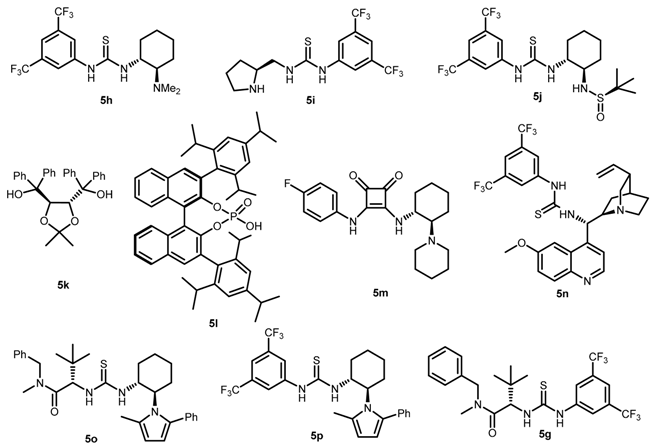
| Entry | Catalyst 5 | Conversion 2 (%) | ee 3 (%) |
|---|---|---|---|
| 1 | 5h | 97 | +12 |
| 2 | 5i | 80 | −9 |
| 3 | 5j | >95 | +20 |
| 4 | 5k | >95 | +5 |
| 5 | 5l | 60 | −14 |
| 6 | 5m | >95 | −17 |
| 7 | 5n | 87 | −24 |
| 8 | 5o | >95 | +48 |
| 9 | 5p | 85 | −15 |
| 10 | 5g | >95 | +77 |
| Entry | 10 | 5 (mol %) | Solvent (M) | T (°C) | Conversion 2 (%) | ee 3 (%) |
|---|---|---|---|---|---|---|
| 1 | 10a | 5g (10) | toluene (0.625) | 40 | >95 | 77 |
| 2 | 10b | 5g (10) | toluene (0.625) | 40 | >95 | 69 |
| 3 | 10c | 5g (10) | toluene (0.625) | 40 | >95 | 70 |
| 4 | 10d | 5g (10) | toluene (0.625) | 40 | >95 | 89 |
| 5 | 10d | 5g (10) | CH2Cl2 (0.13) | 40 | 61 | 66 |
| 6 | 10d | 5g (10) | MTBE (0.13) | 40 | 60 | 39 |
| 7 | 10d | 5g (10) | THF (0.13) | 40 | 60 | 4 |
| 8 | 10d | 5g (10) | toluene (0.625) | −20 | >95 | 94 |
| 9 | 10d | 5q (10) | toluene (0.625) | −20 | >95 | 77 |
| 10 | 10d | 5r (10) | toluene (0.625) | −20 | >95 | 92 |
| 11 | 10d | 5g (10) | PhCF3 (0.625) | −20 | >95 | 95 |
| 12 | 10d | 5g (5) | PhCF3 (0.625) | −20 | >95 | 92 |
| 13 | 10d | 5g (10) | PhCF3 (0.3) | −20 | >95 | 97 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, L.; Fochi, M. A General Catalytic Enantioselective Transfer Hydrogenation Reaction of β,β-Disubstituted Nitroalkenes Promoted by a Simple Organocatalyst. Molecules 2016, 21, 1000. https://doi.org/10.3390/molecules21081000
Bernardi L, Fochi M. A General Catalytic Enantioselective Transfer Hydrogenation Reaction of β,β-Disubstituted Nitroalkenes Promoted by a Simple Organocatalyst. Molecules. 2016; 21(8):1000. https://doi.org/10.3390/molecules21081000
Chicago/Turabian StyleBernardi, Luca, and Mariafrancesca Fochi. 2016. "A General Catalytic Enantioselective Transfer Hydrogenation Reaction of β,β-Disubstituted Nitroalkenes Promoted by a Simple Organocatalyst" Molecules 21, no. 8: 1000. https://doi.org/10.3390/molecules21081000