
Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes

 ,
,  , ,
, , 
Abstract
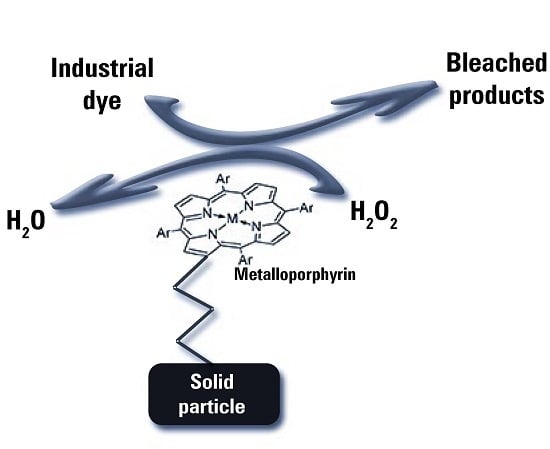
:
1. Introduction
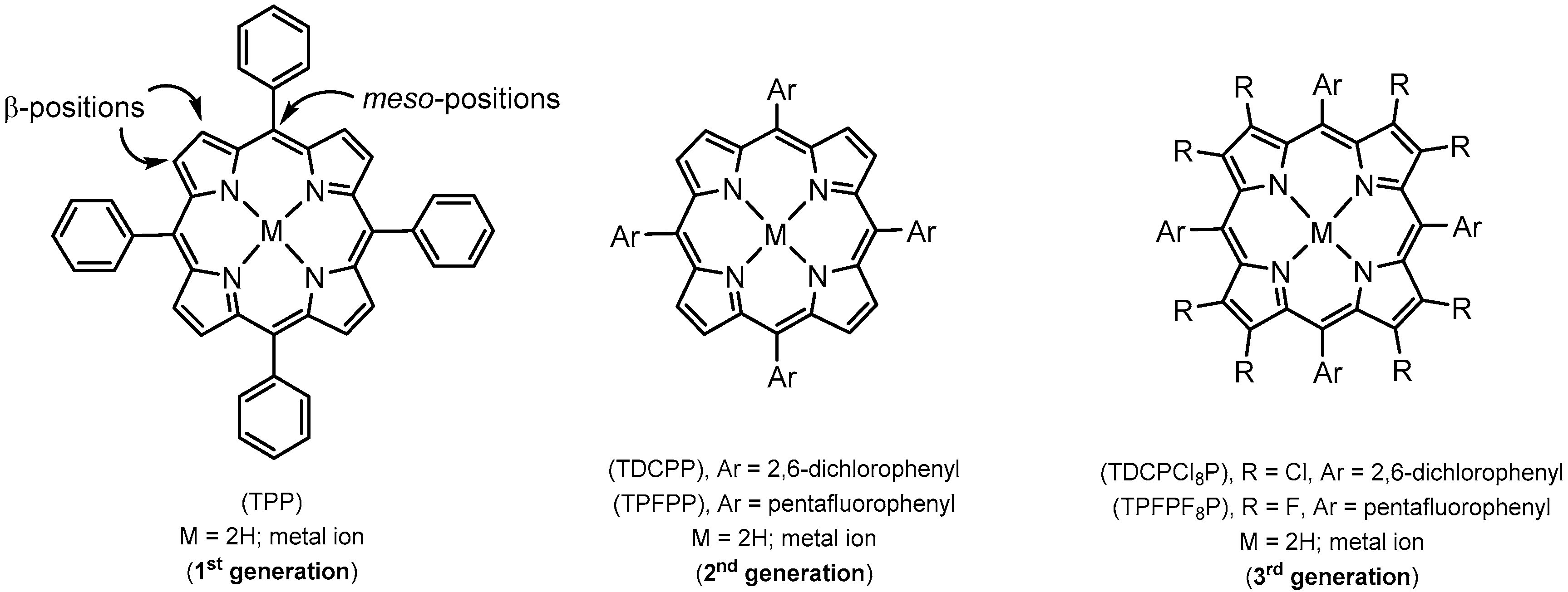
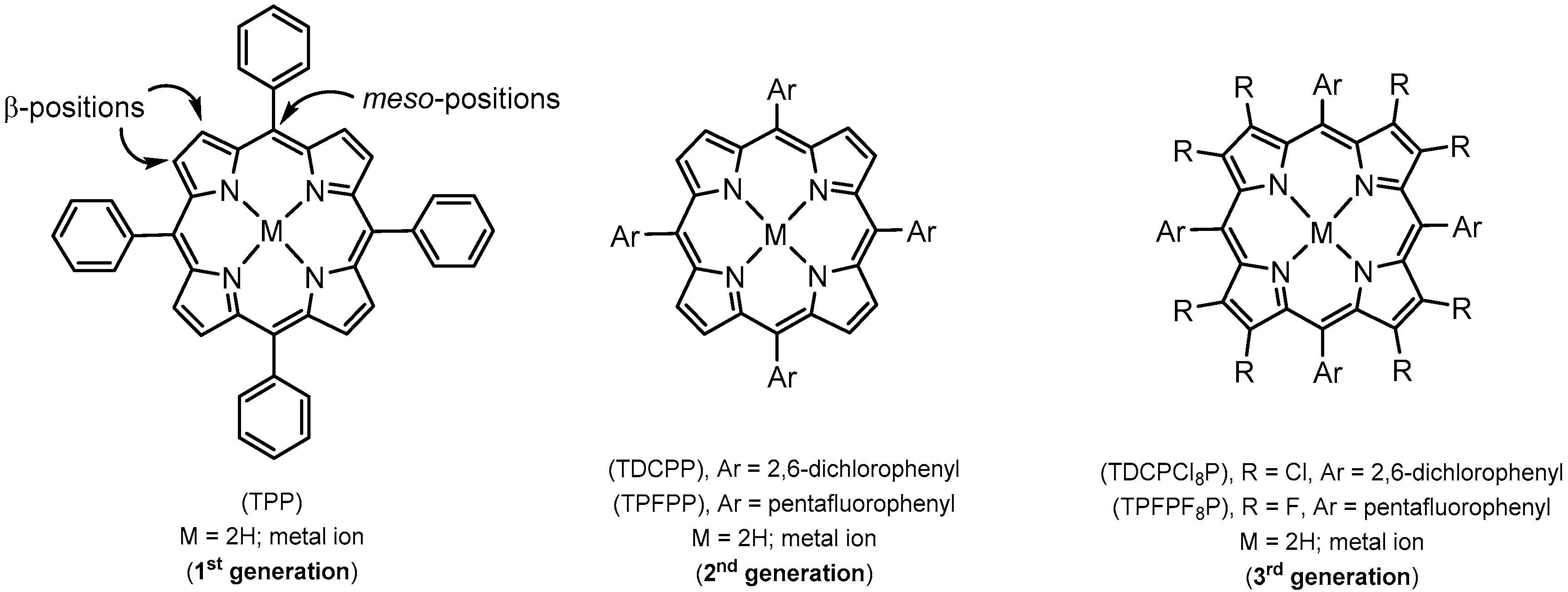
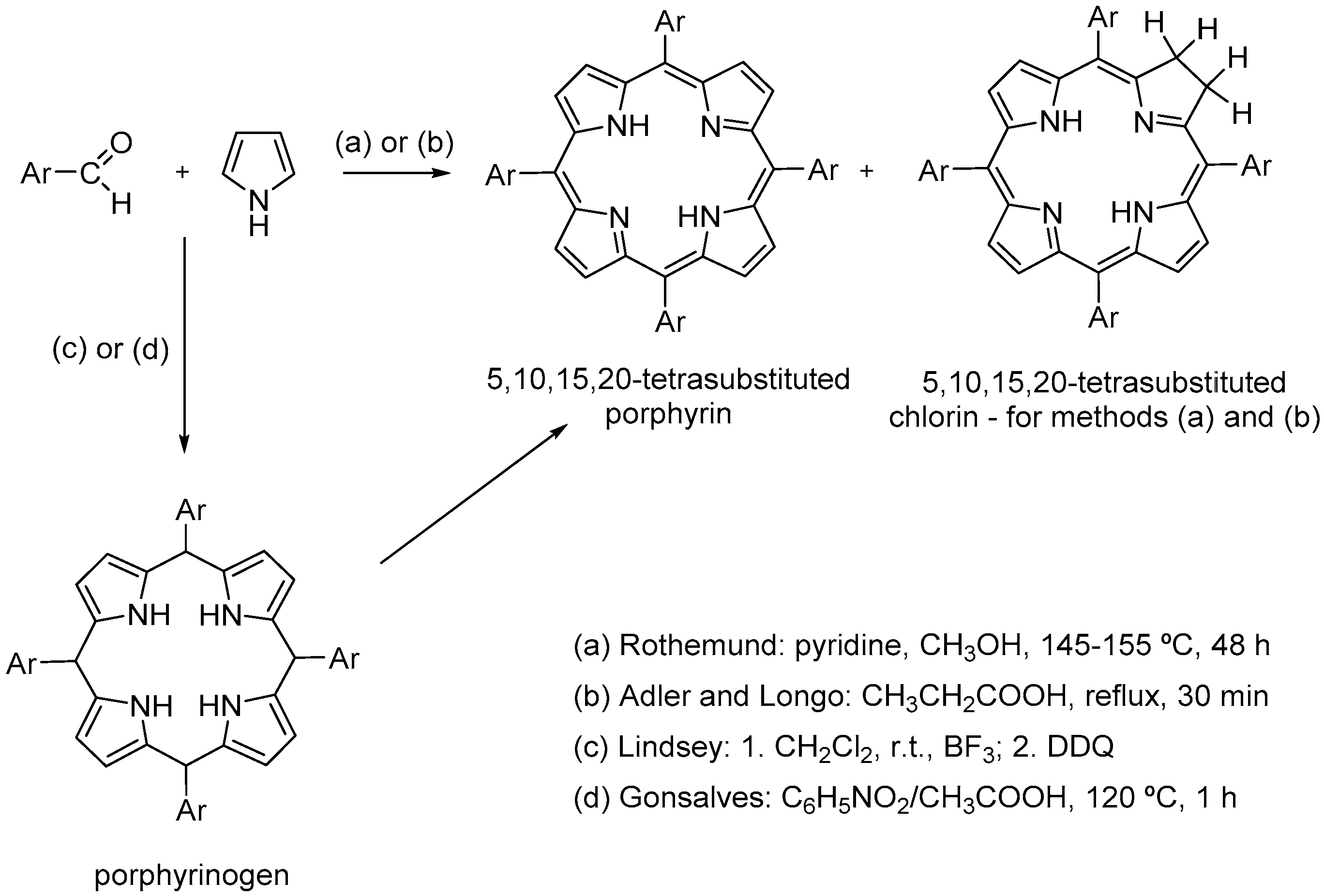
2. Synthetic Metalloporphyrins
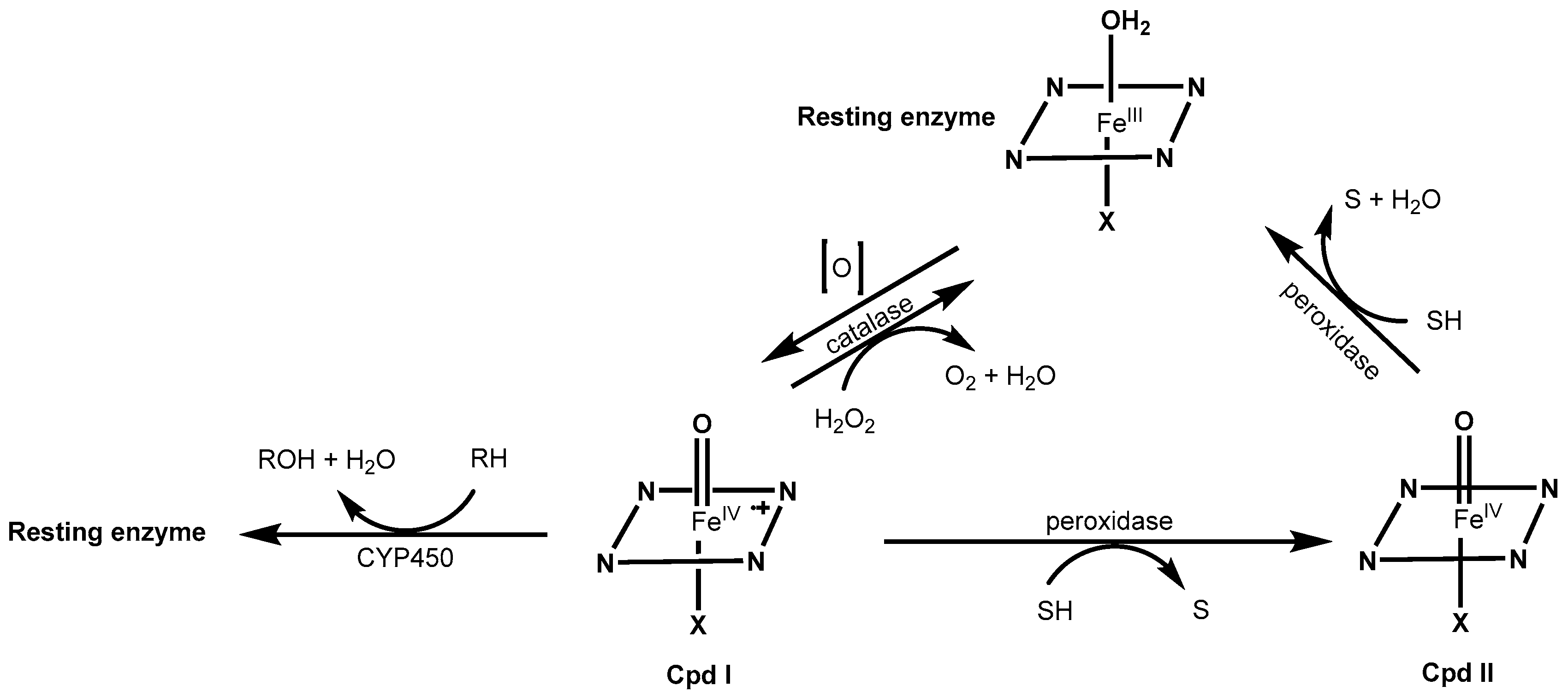
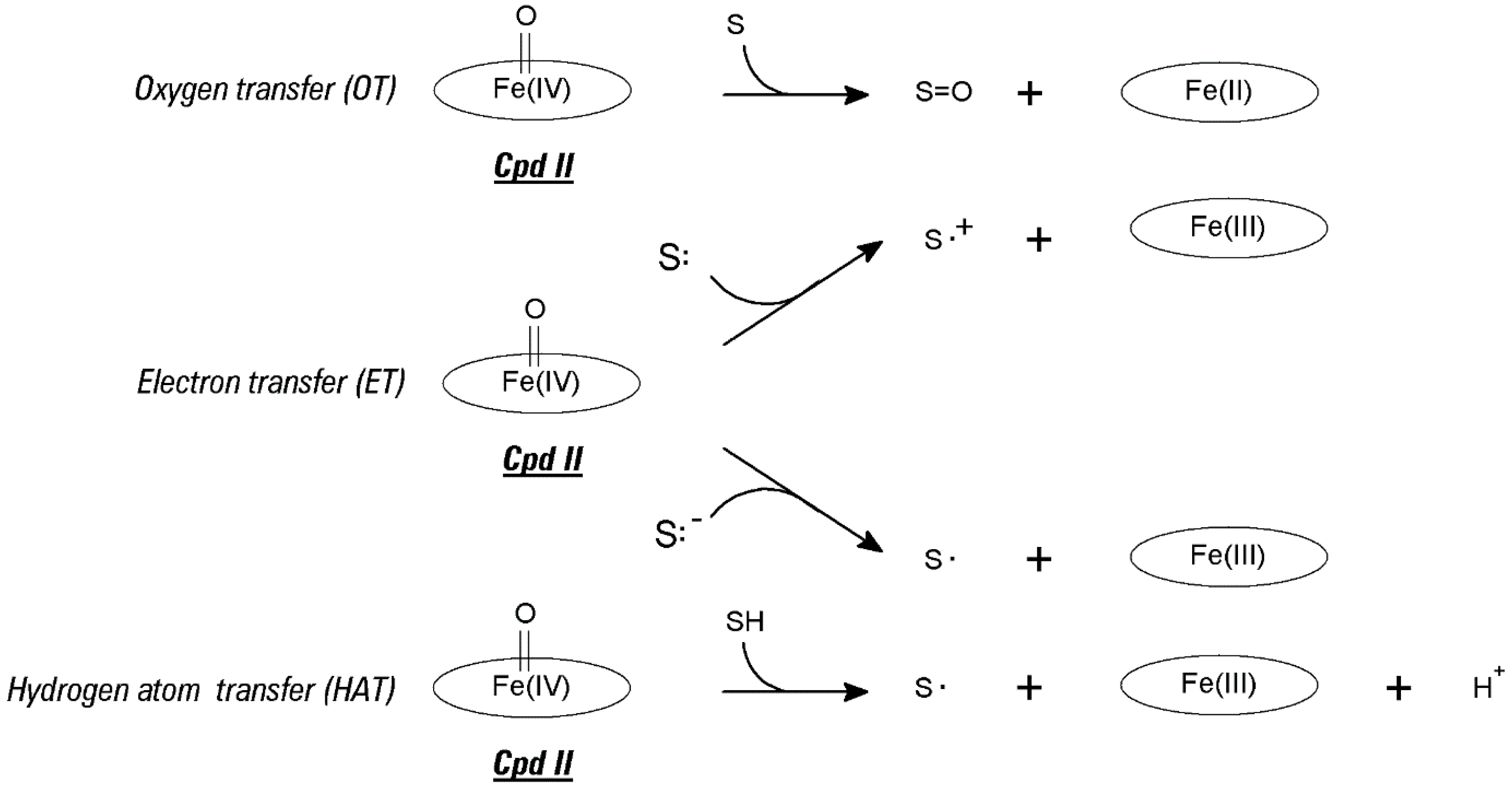
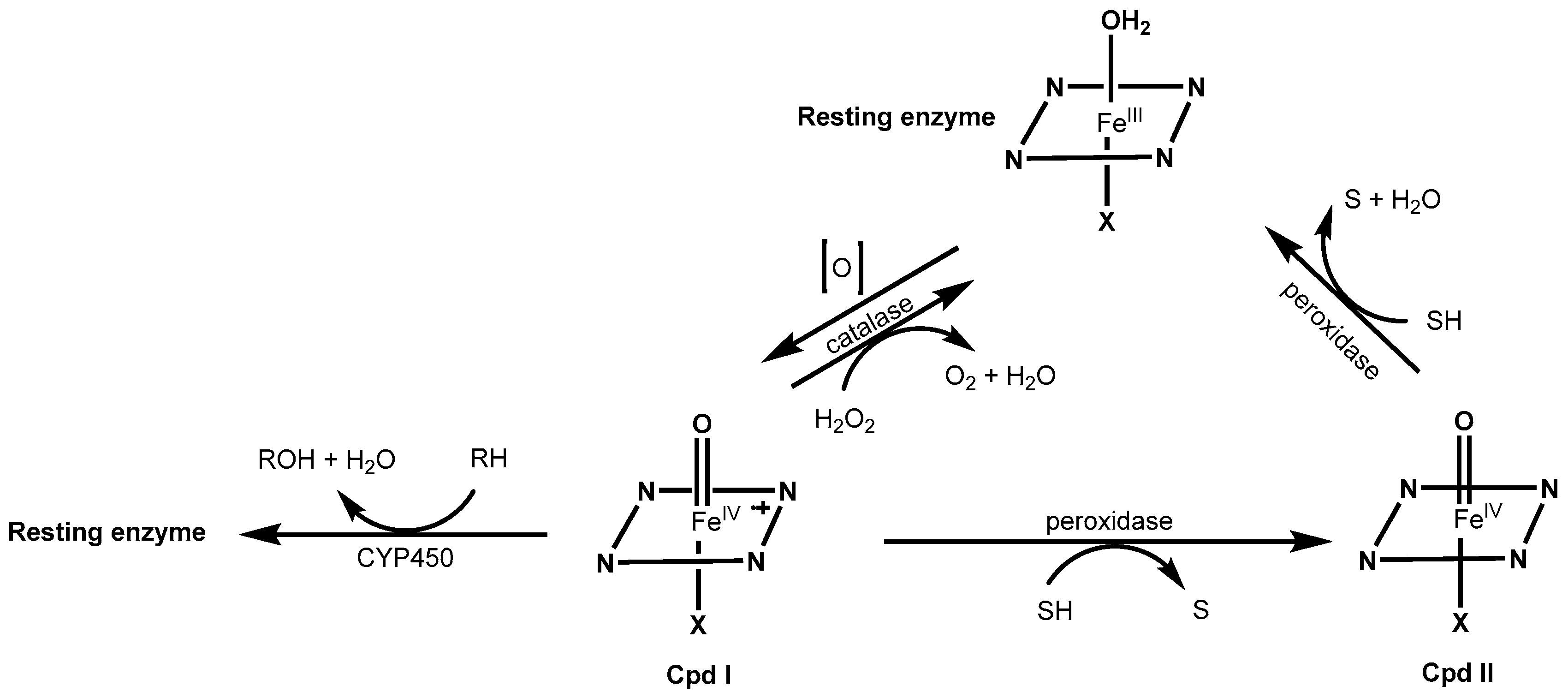
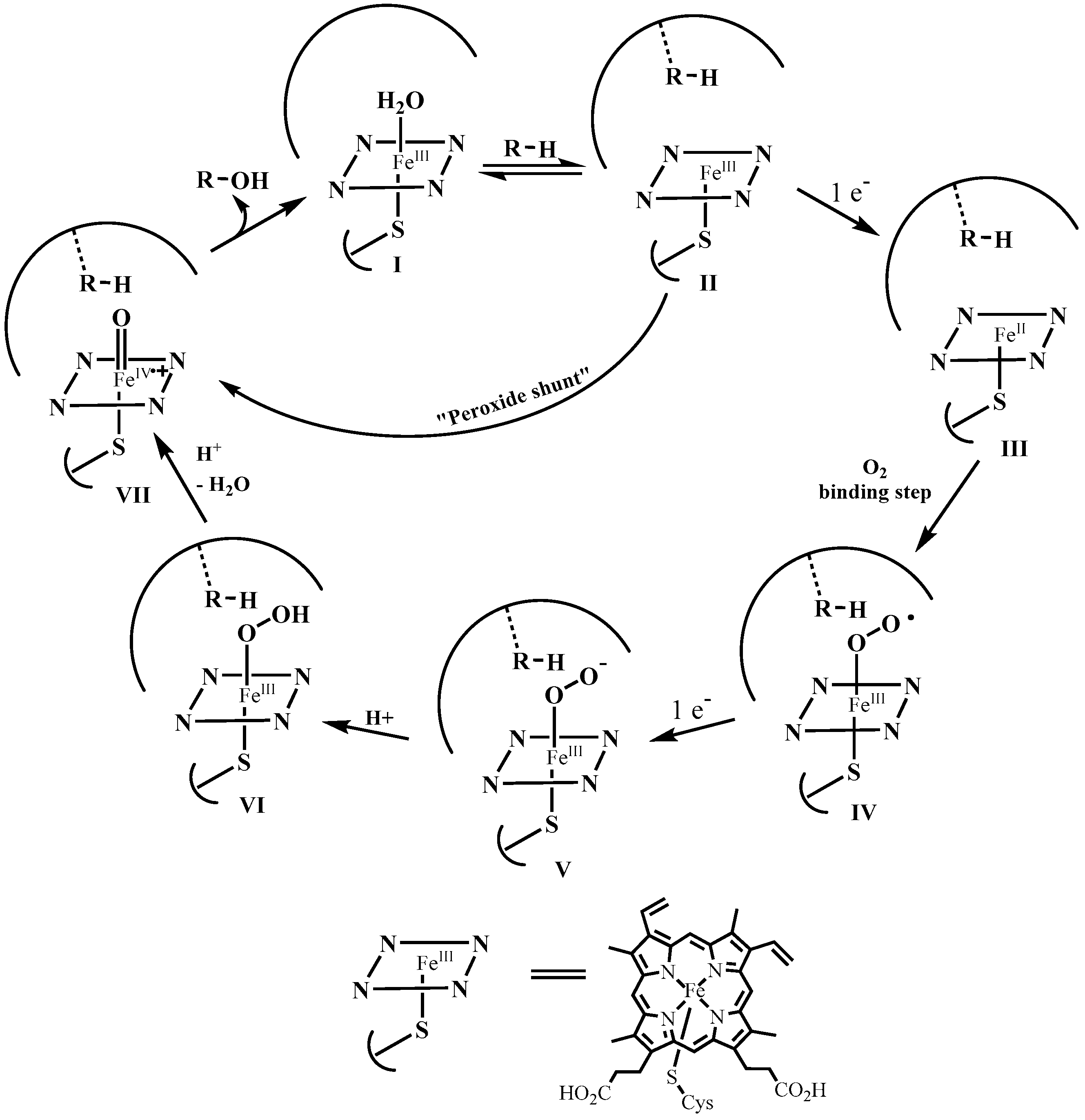
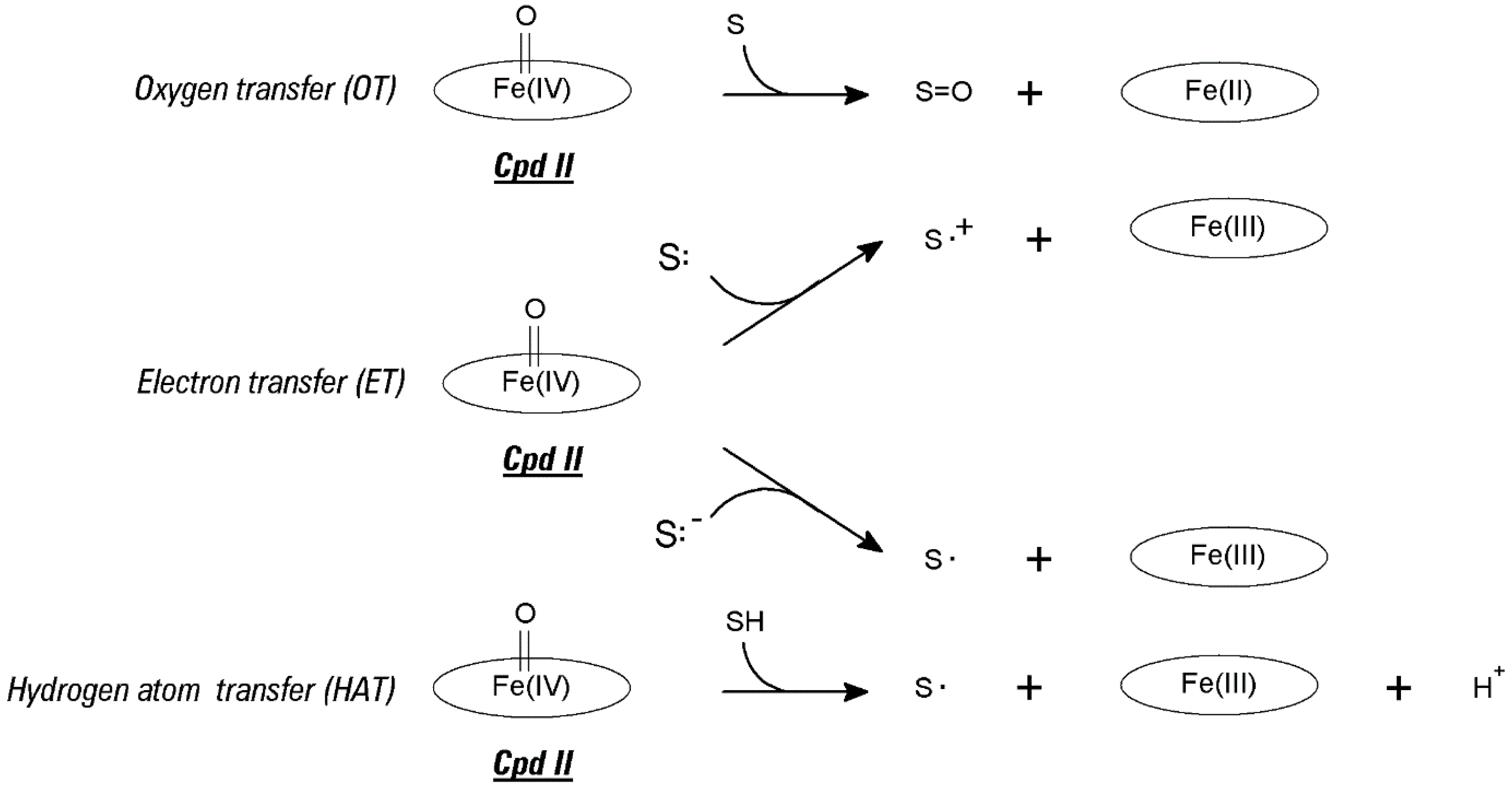
2.1. Redox Metalloporphyrins: An Overview
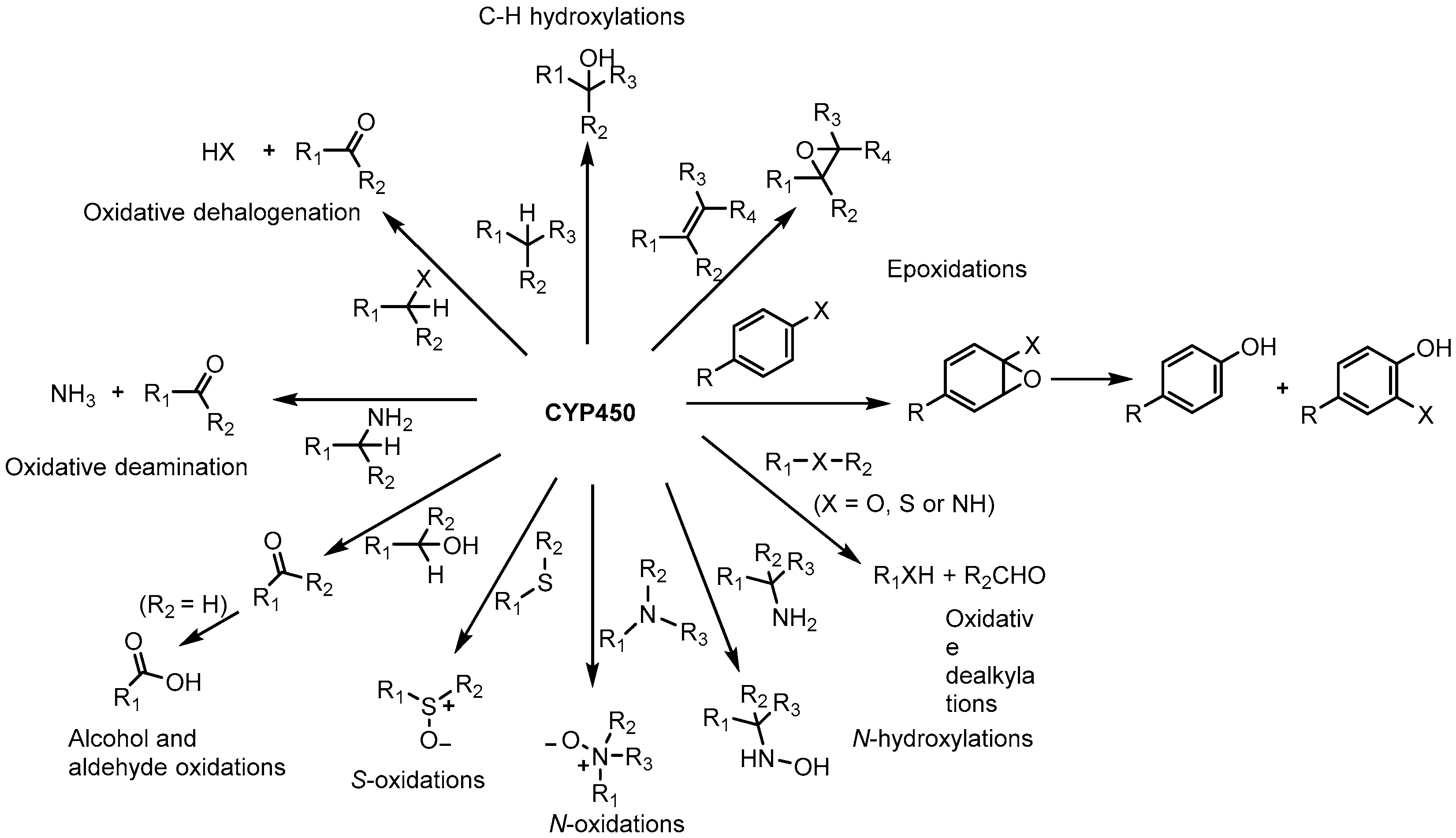
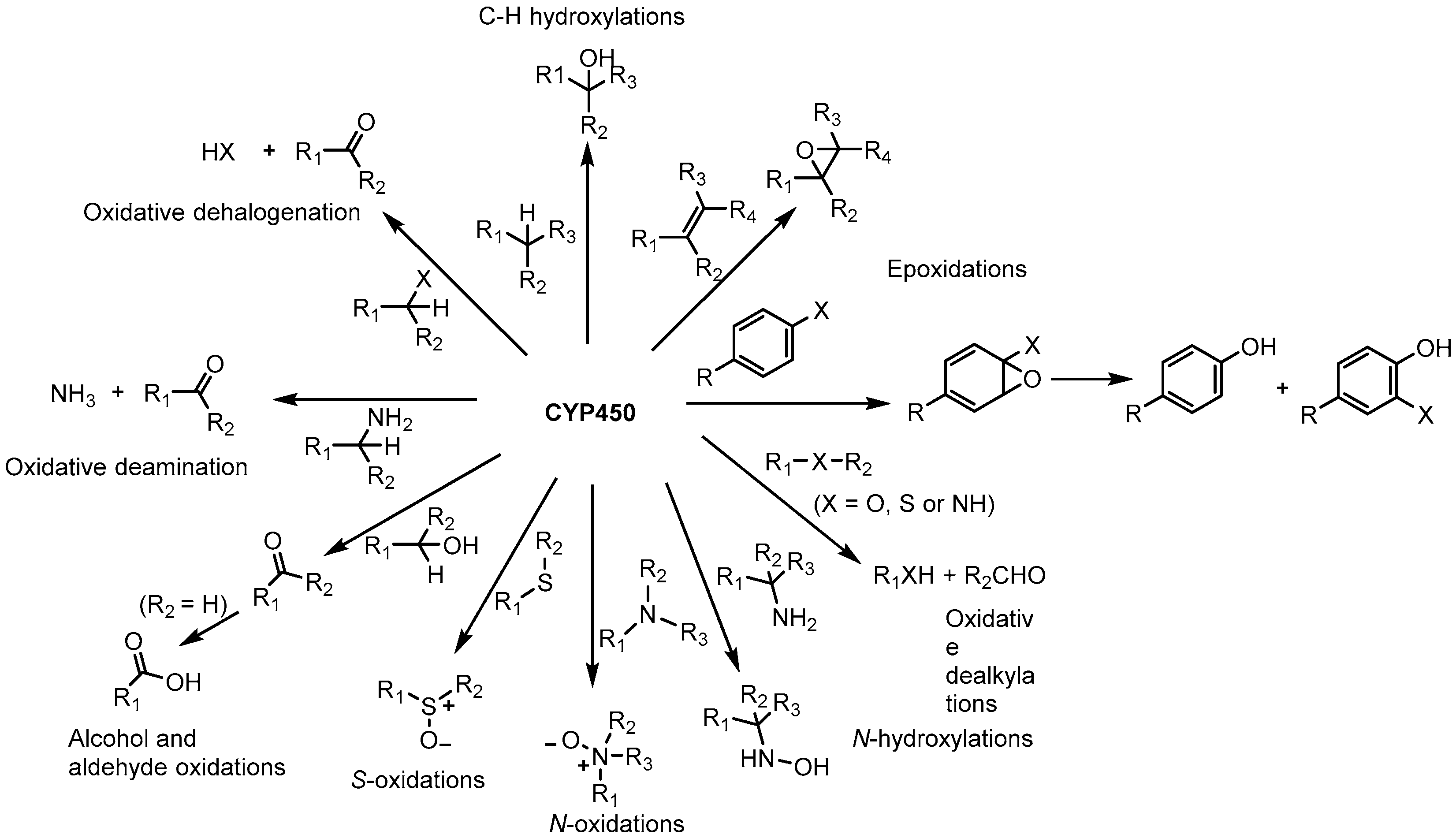
2.2. Substrate Specificity
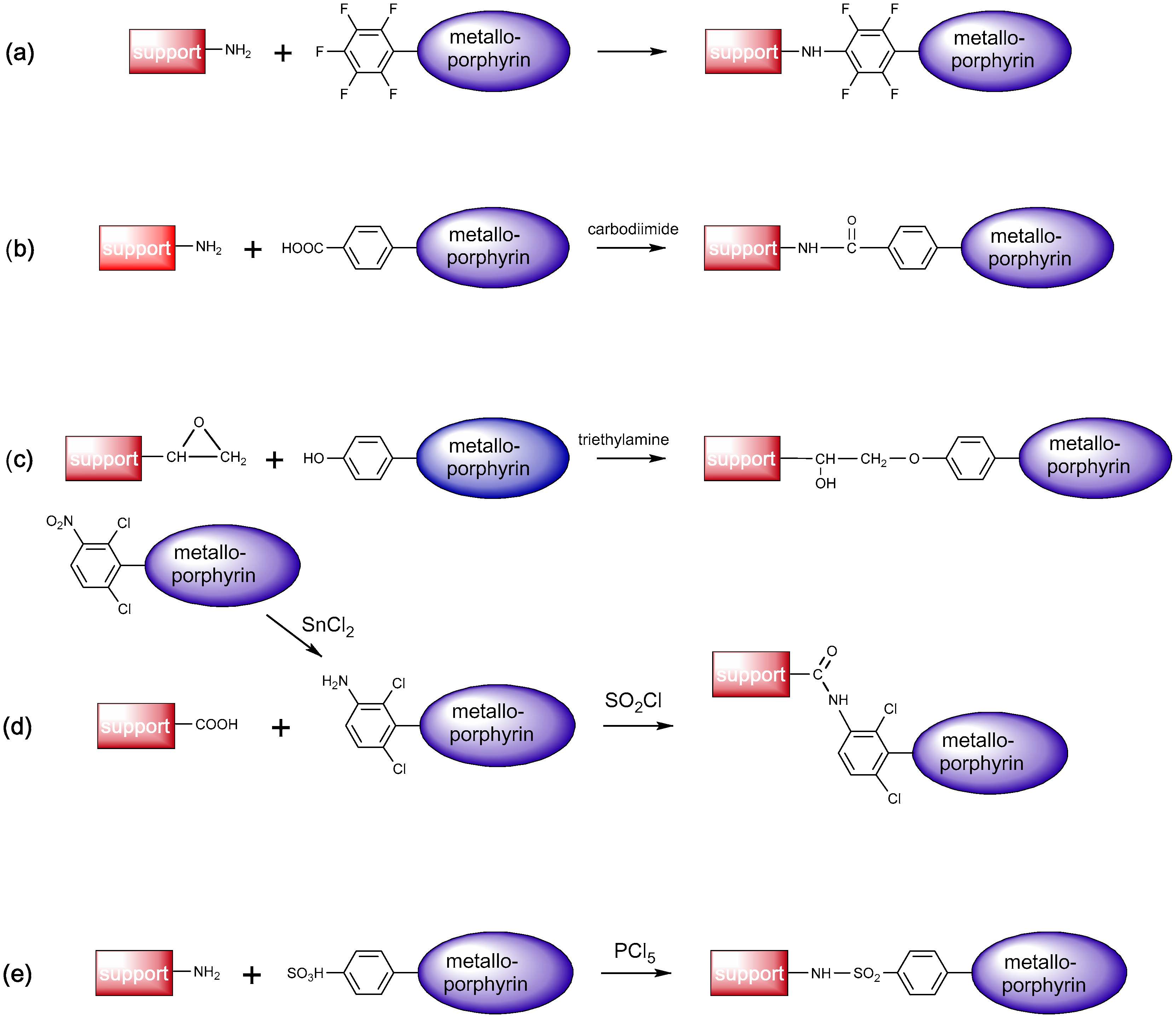
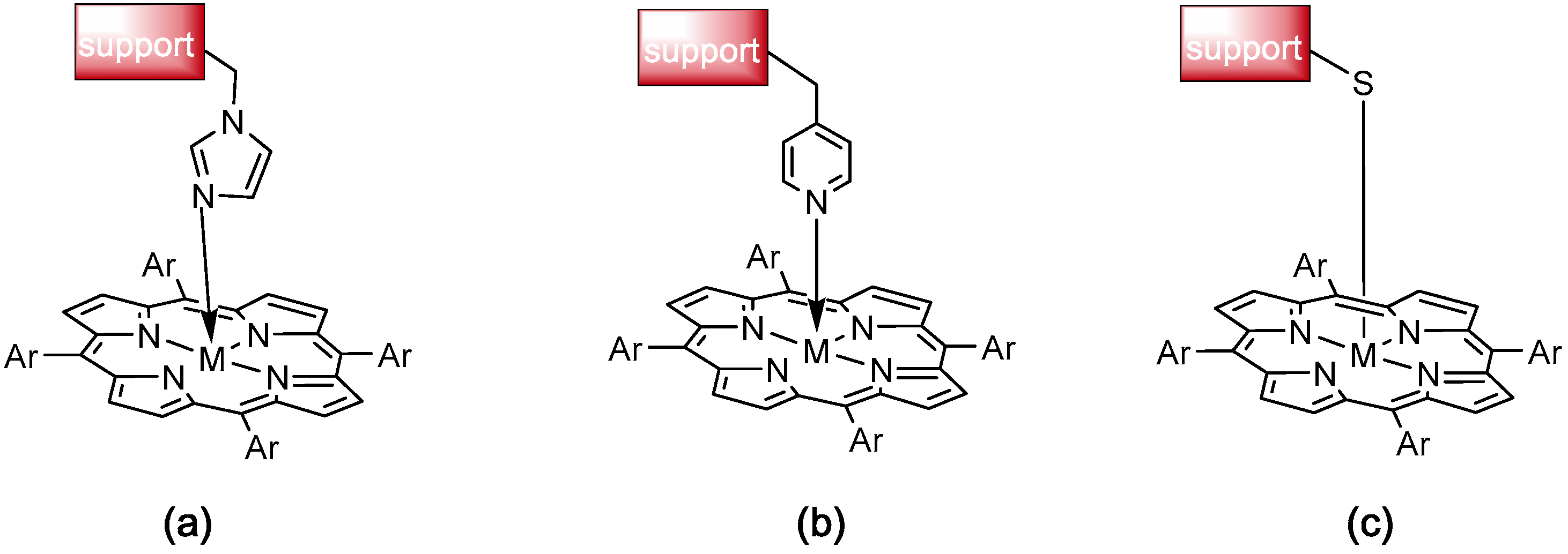
2.3. Immobilization and Emulation of Ligninolytic Enzymes
- Toxicology of metalloporphyrins is still almost unknown. So, a separation from reaction mixture is usually required, further increasing the cost of the process [29];
- Metalloporphyrins are intensely colored, as well as the dye substrates, frustrating the whole purpose of the process when used in free form to bleach dyes.
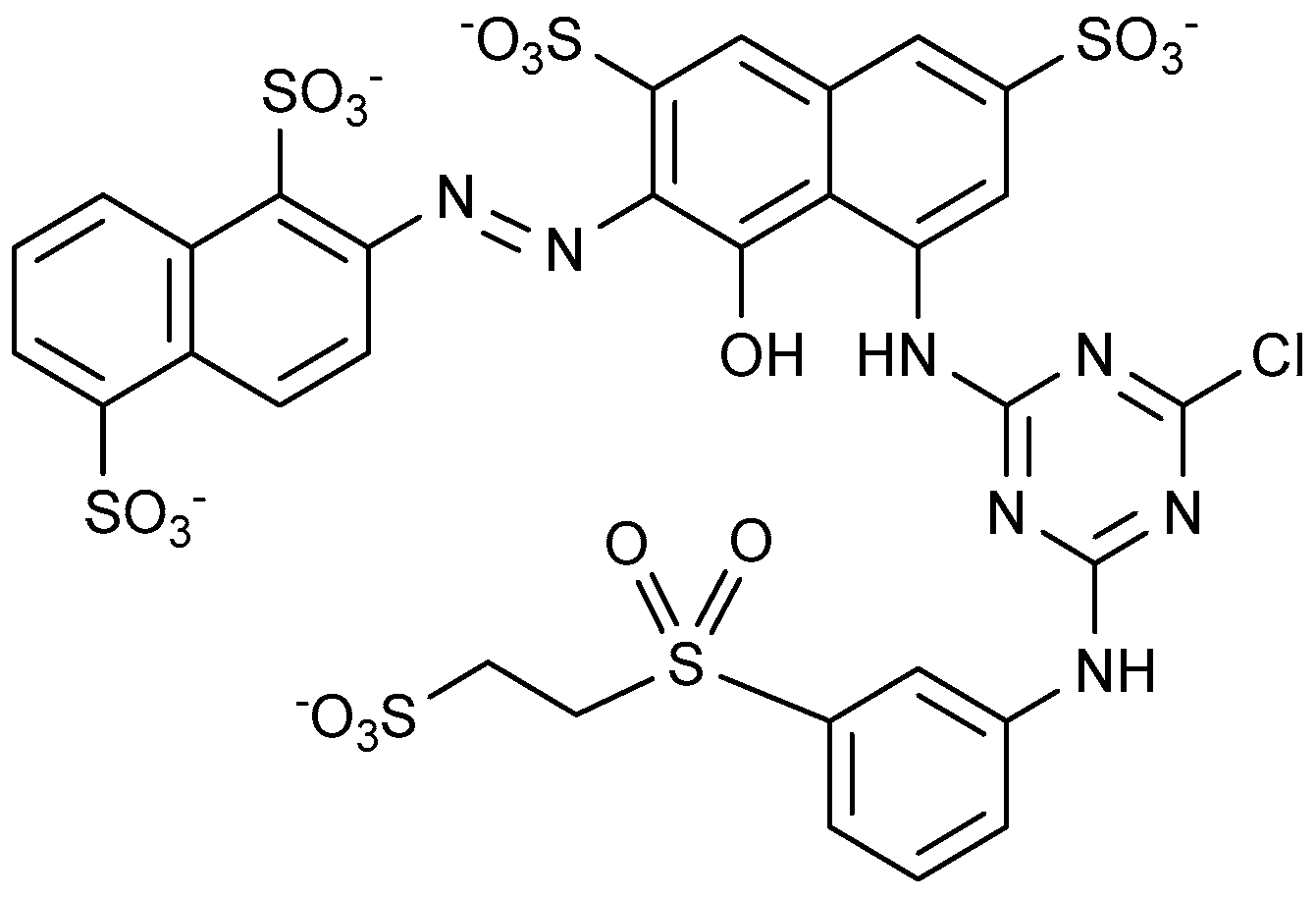
3. Textile Dyes
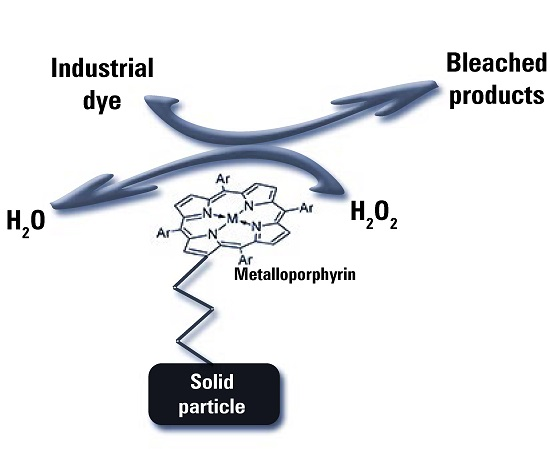
4. Application of Metalloporphyrins in the Decolorization of Textile Dyes
4.1. Biomimetic Bleaching of Industrial Dyes
- It is reasonably stable and safe for production, transport, storage, delivery, and usage.
- Its relatively low price has allowed a more and more wide use in many technological applications.
- Last but not least, side products arising from its action are only water and molecular oxygen, causing no environmental and toxicological concern.
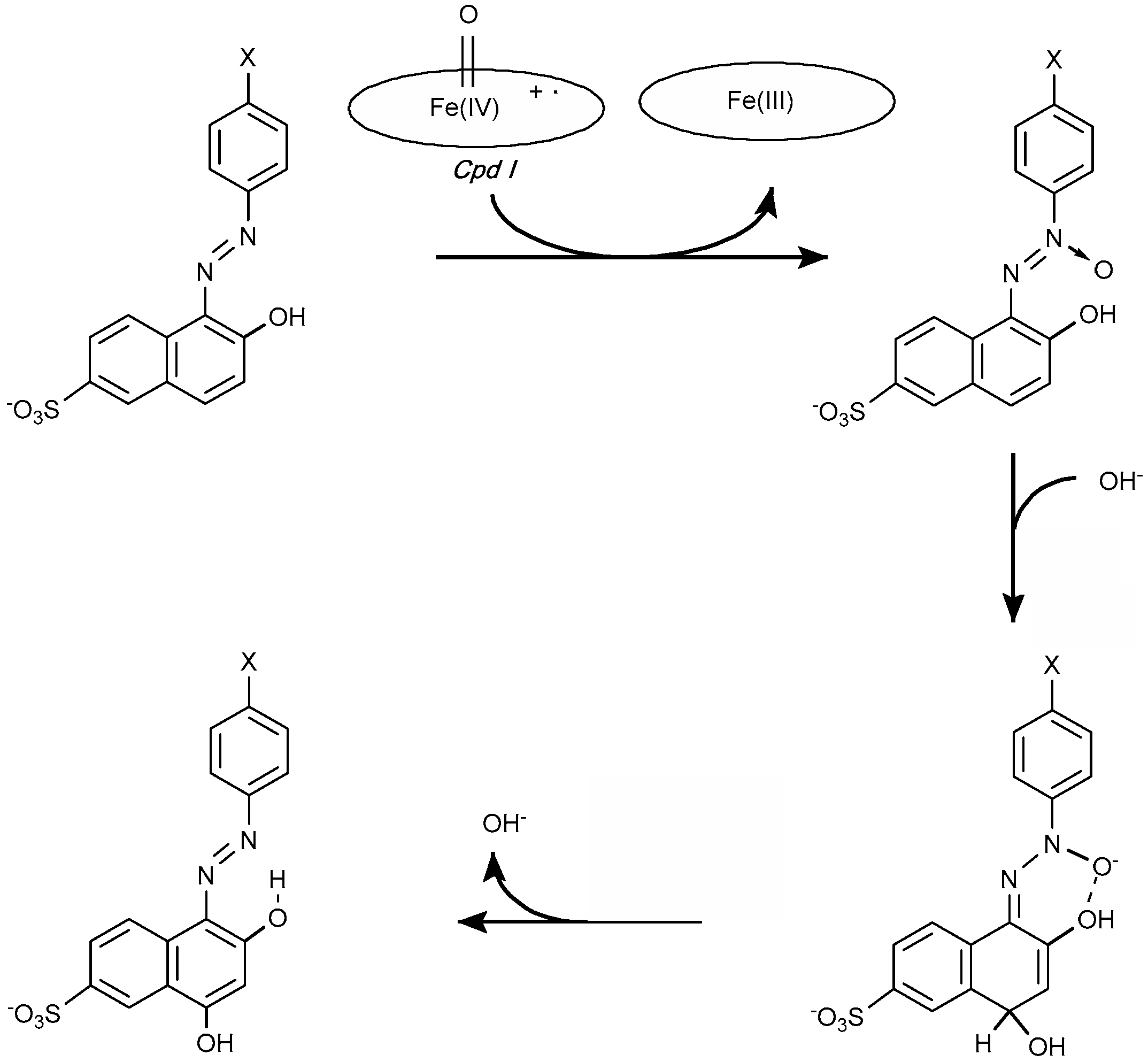
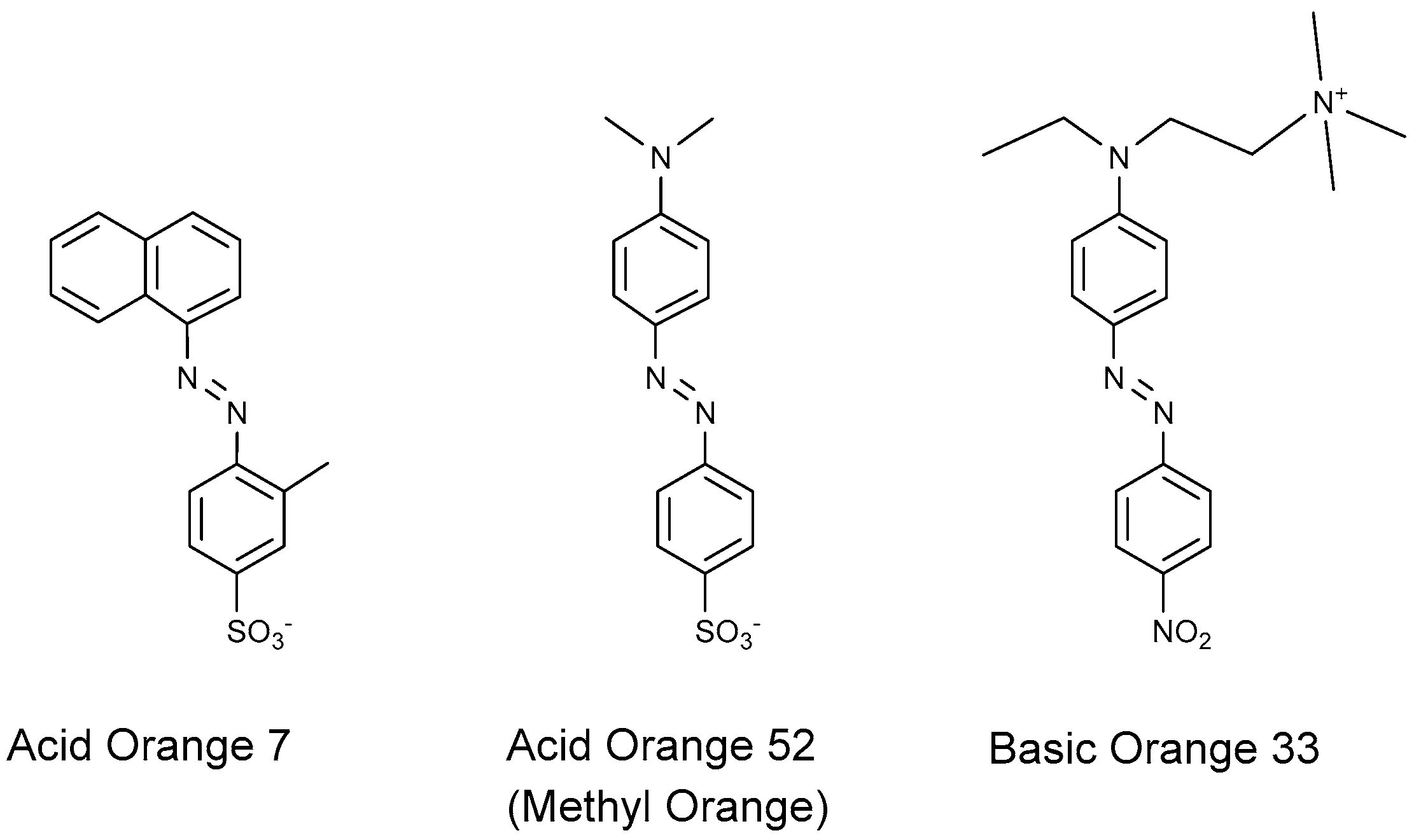
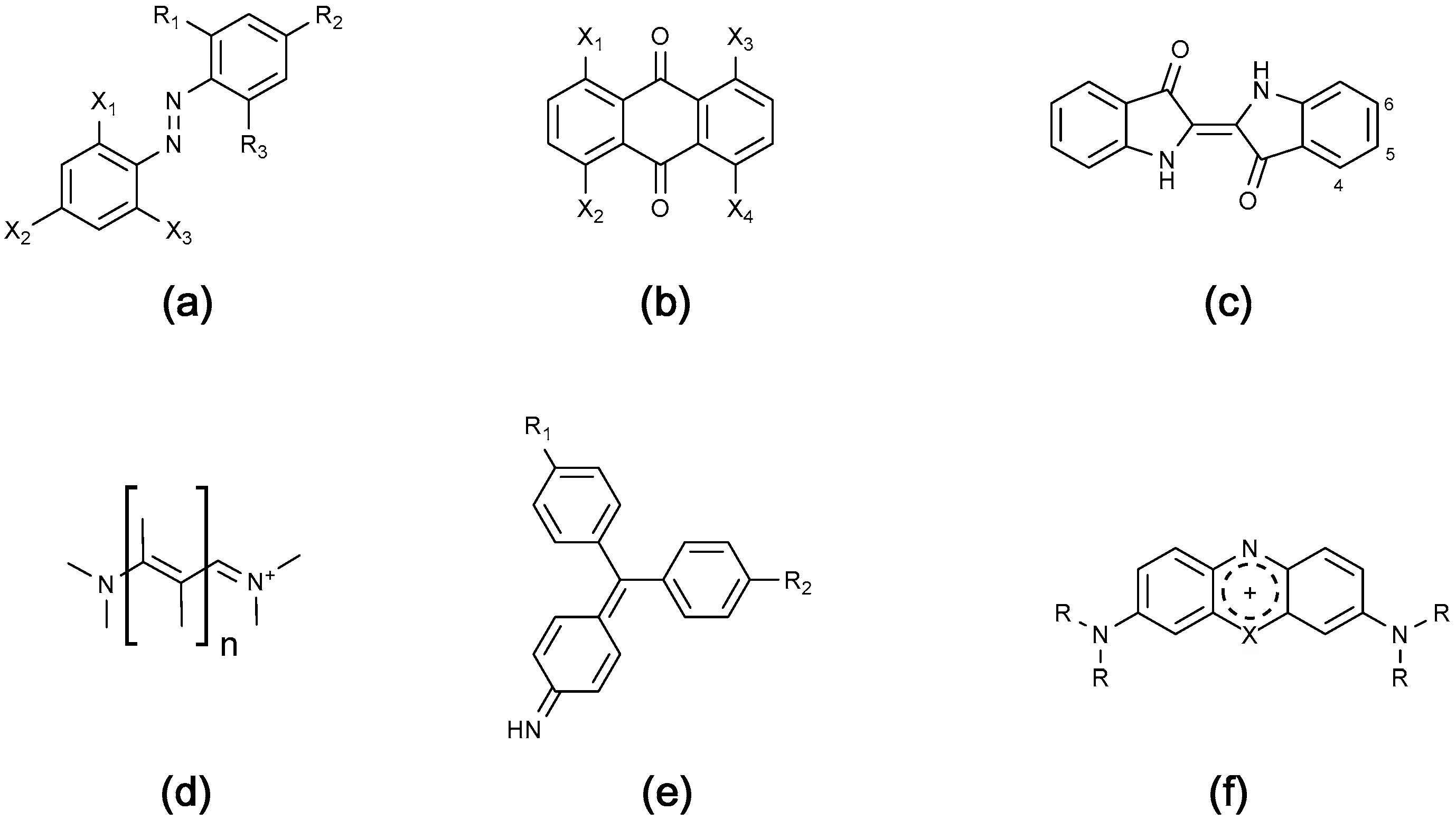
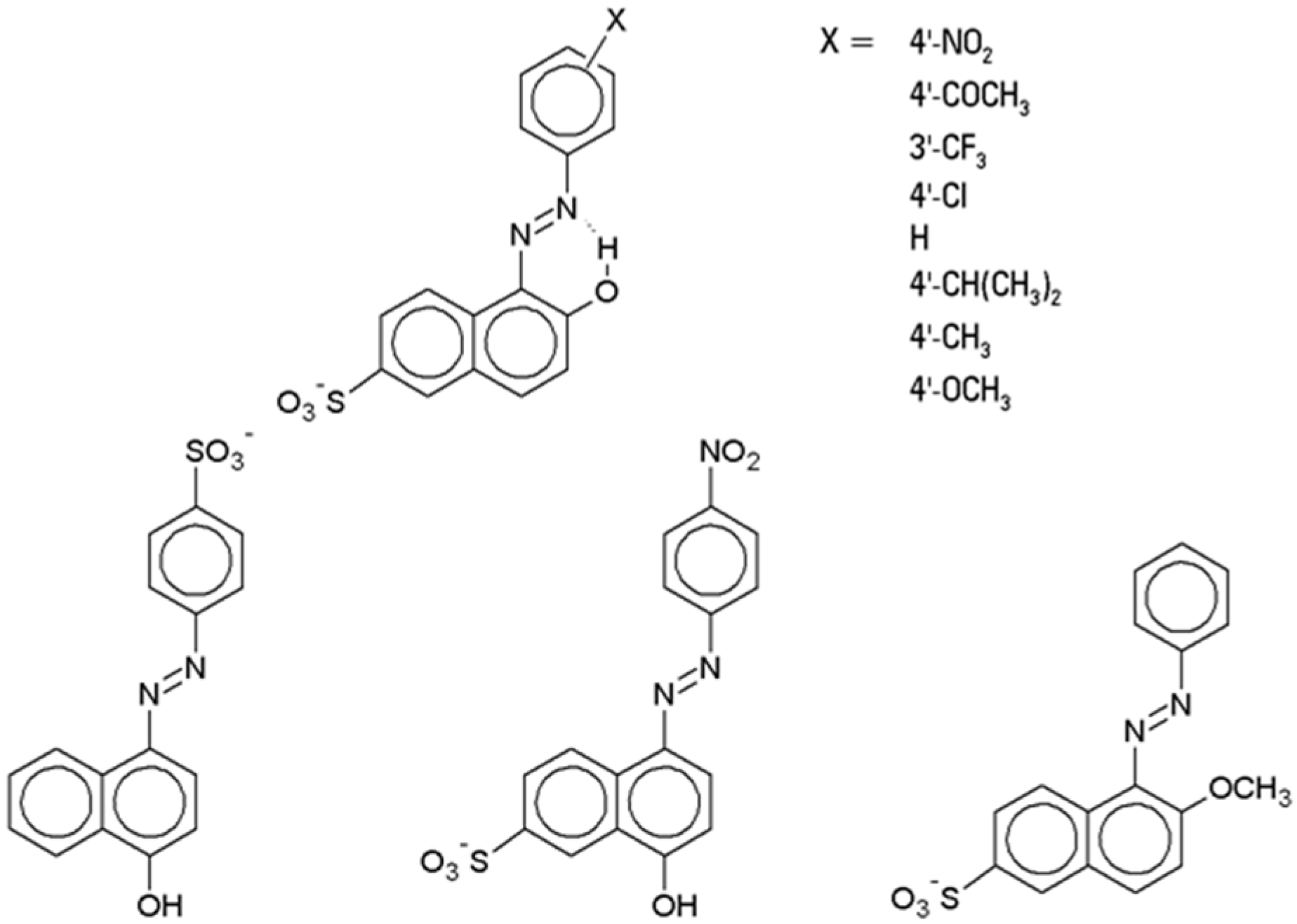
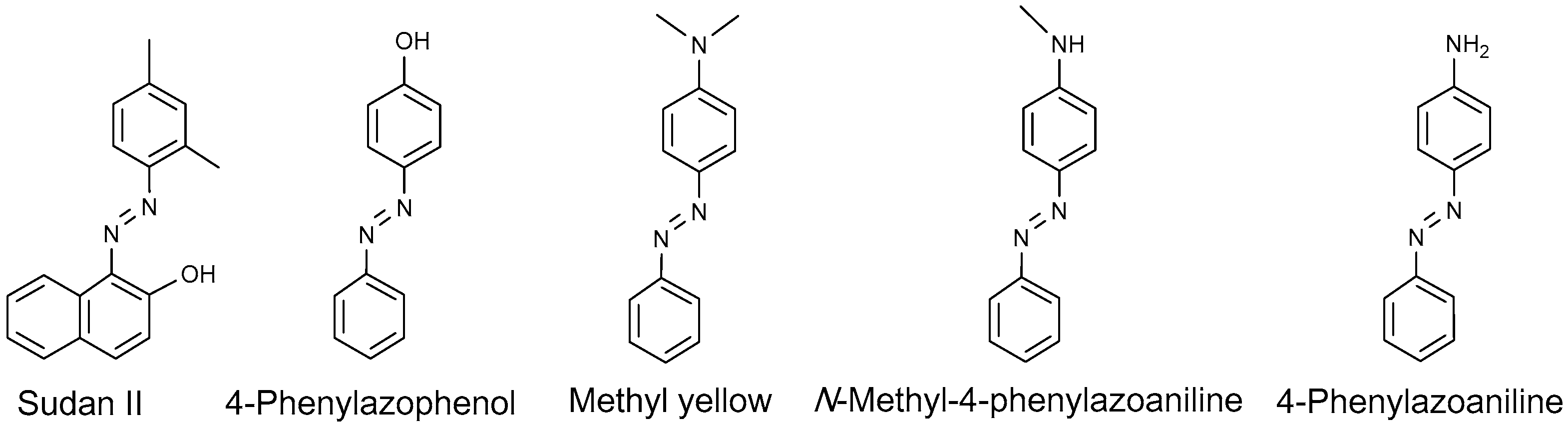
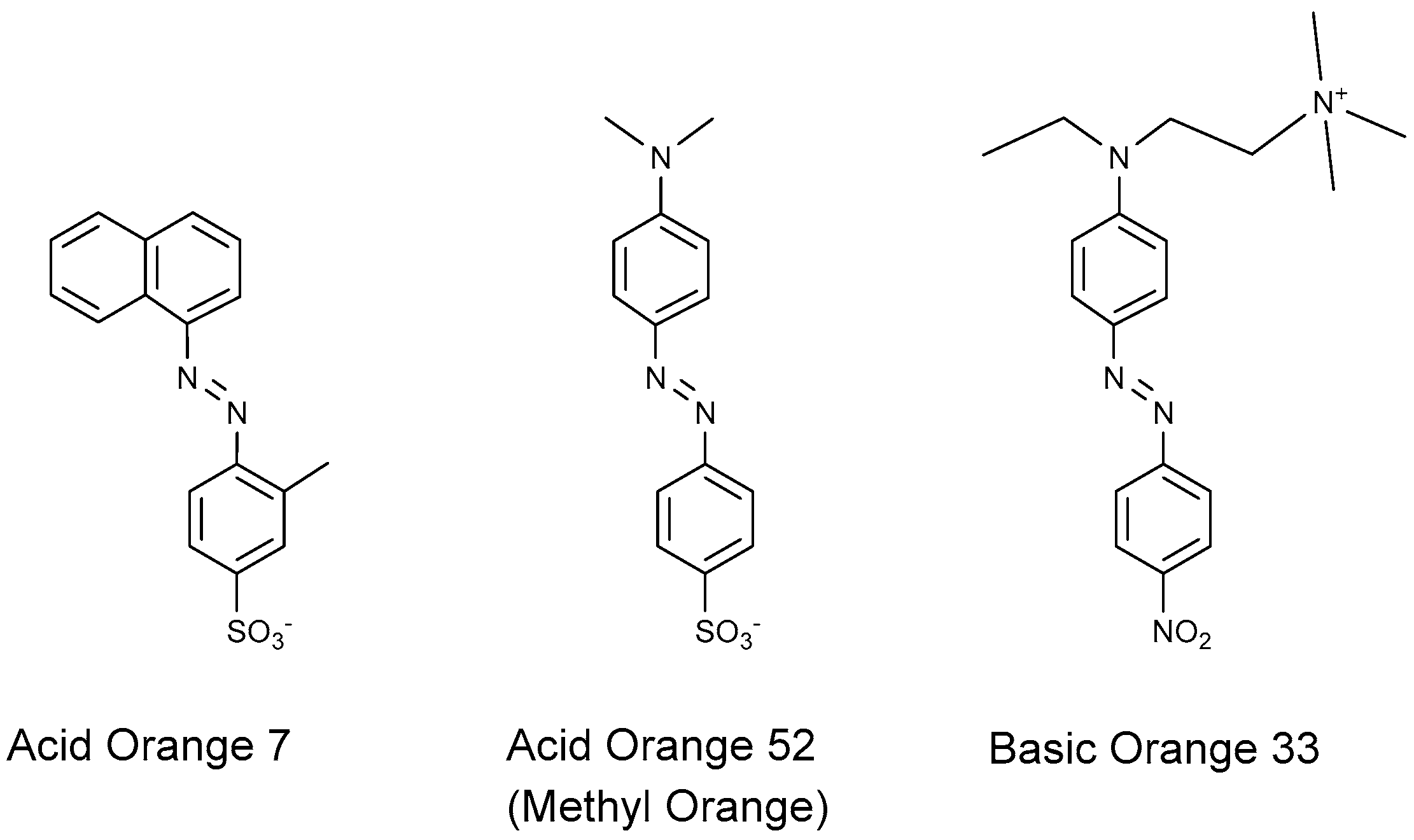
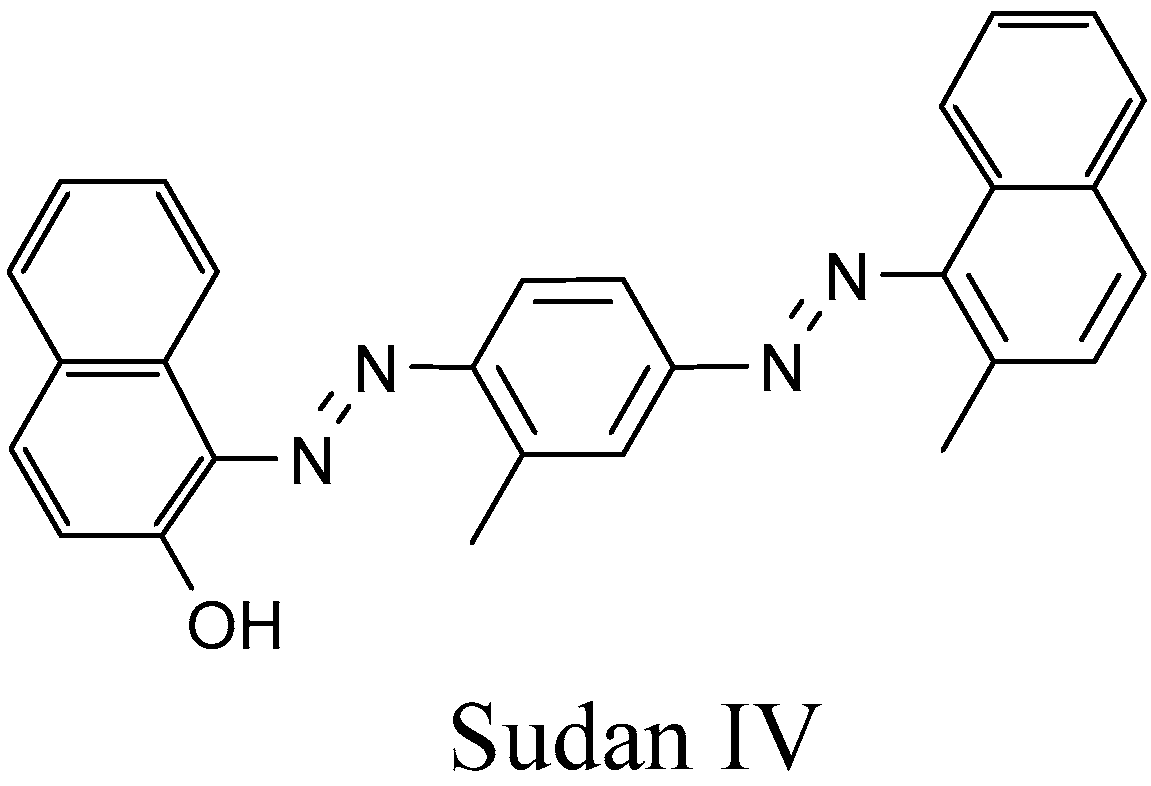
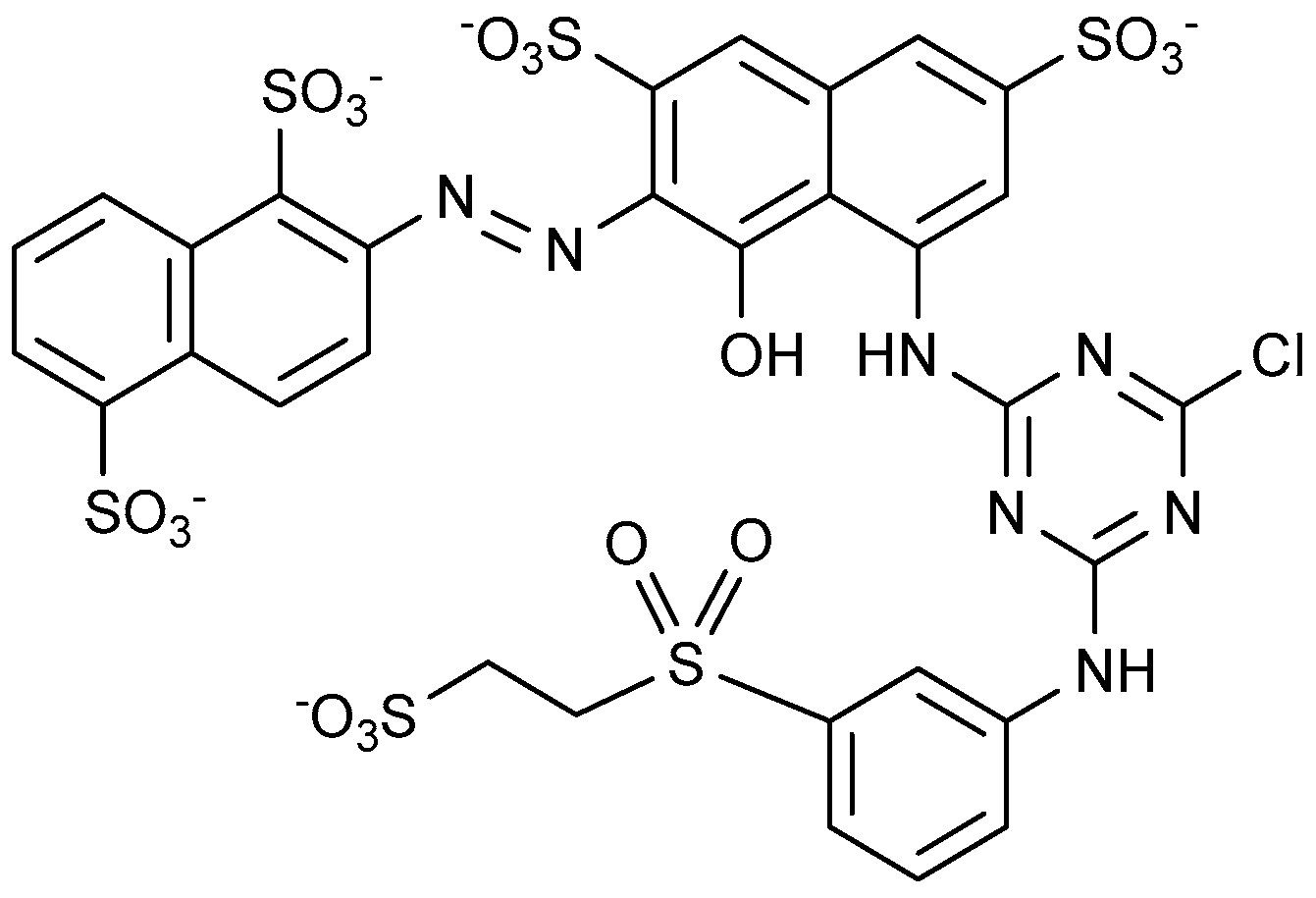
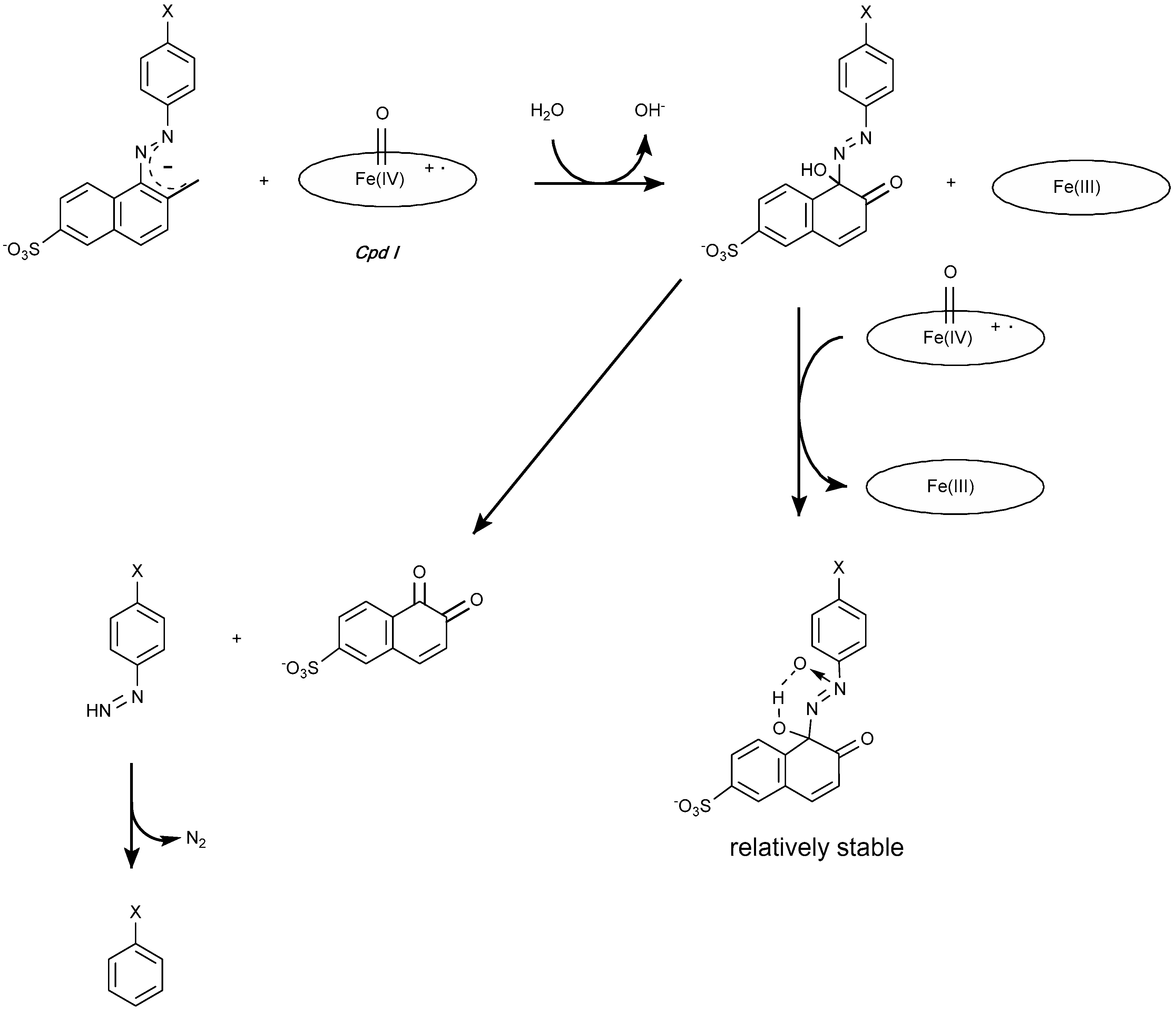
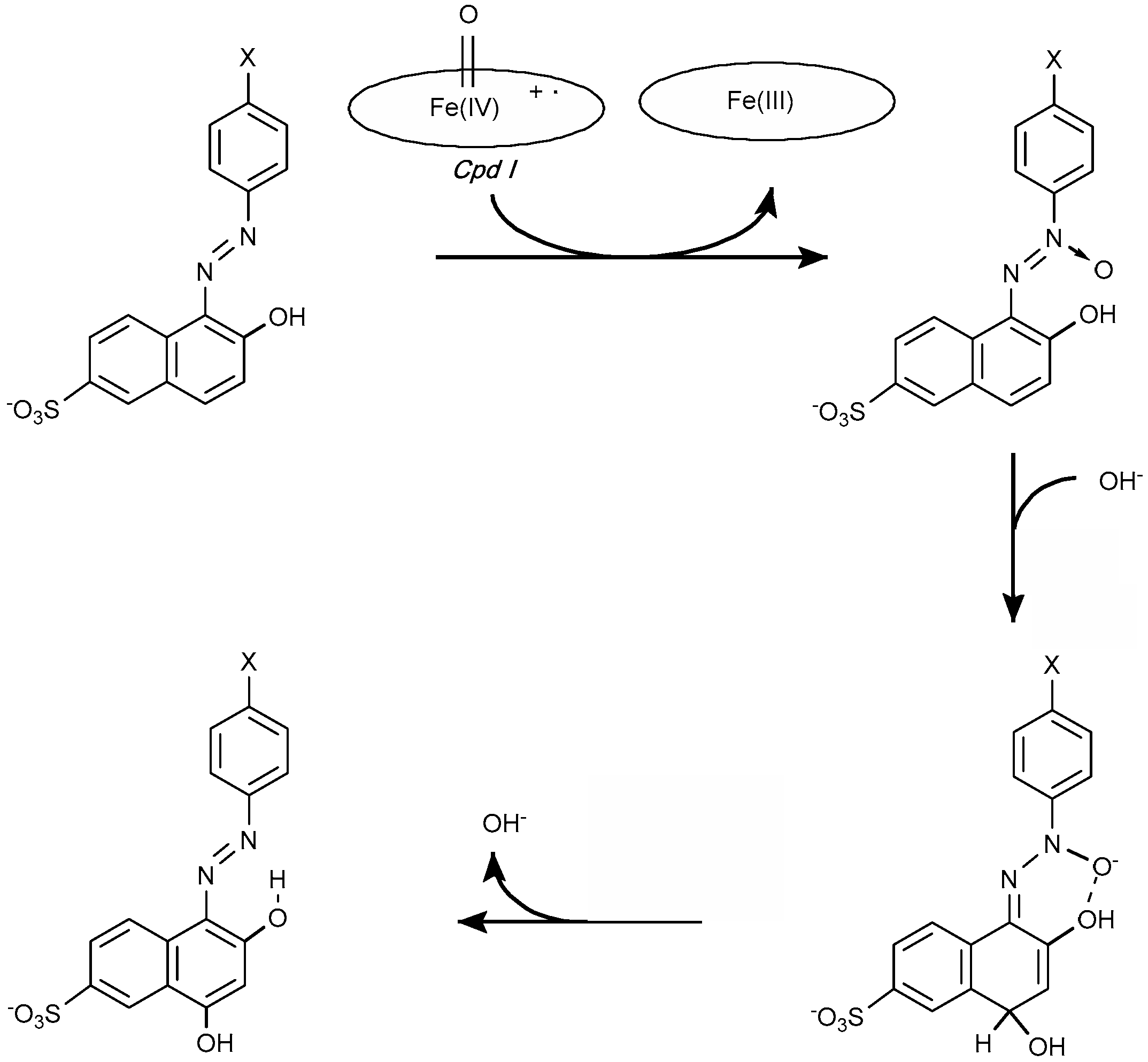
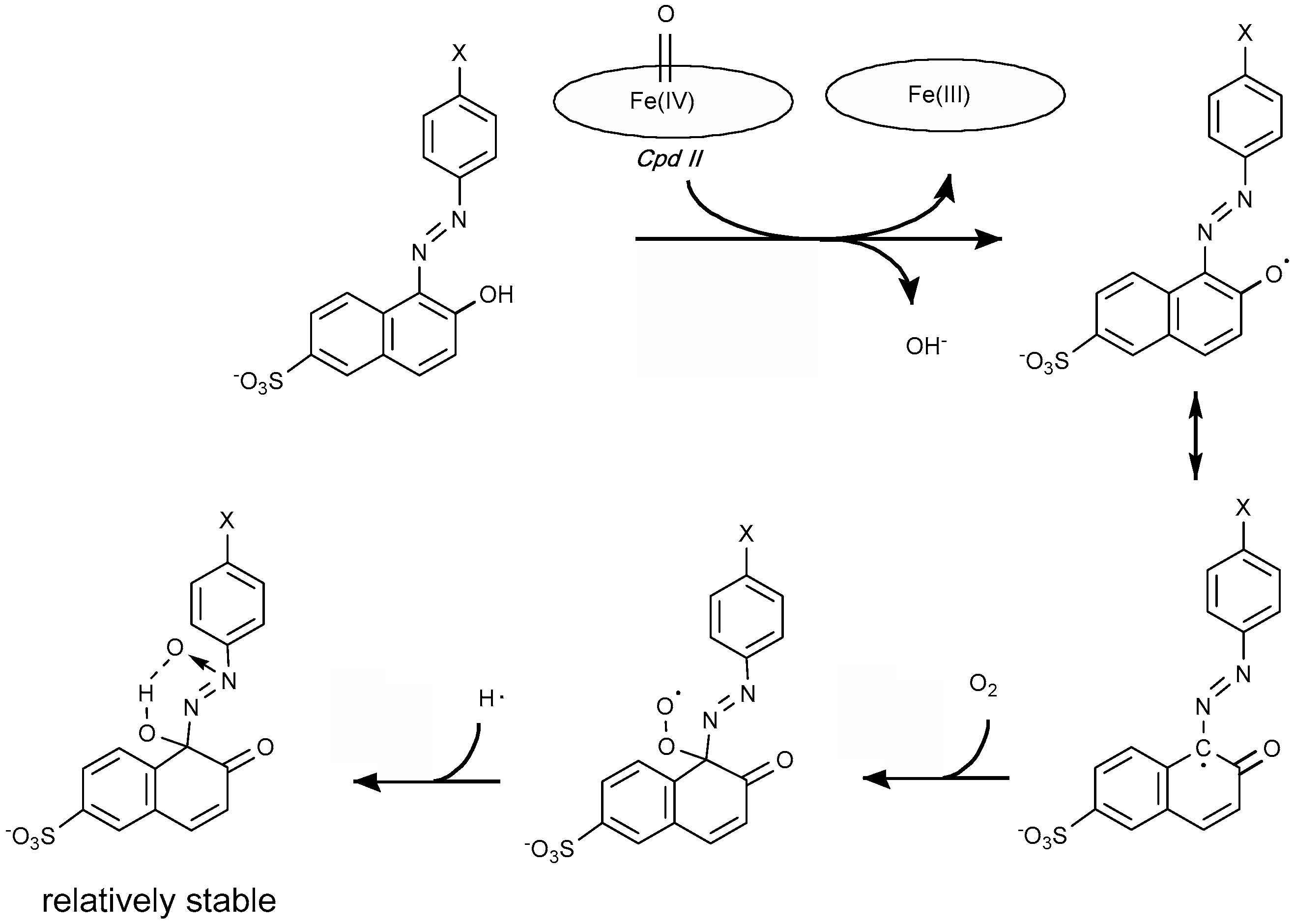
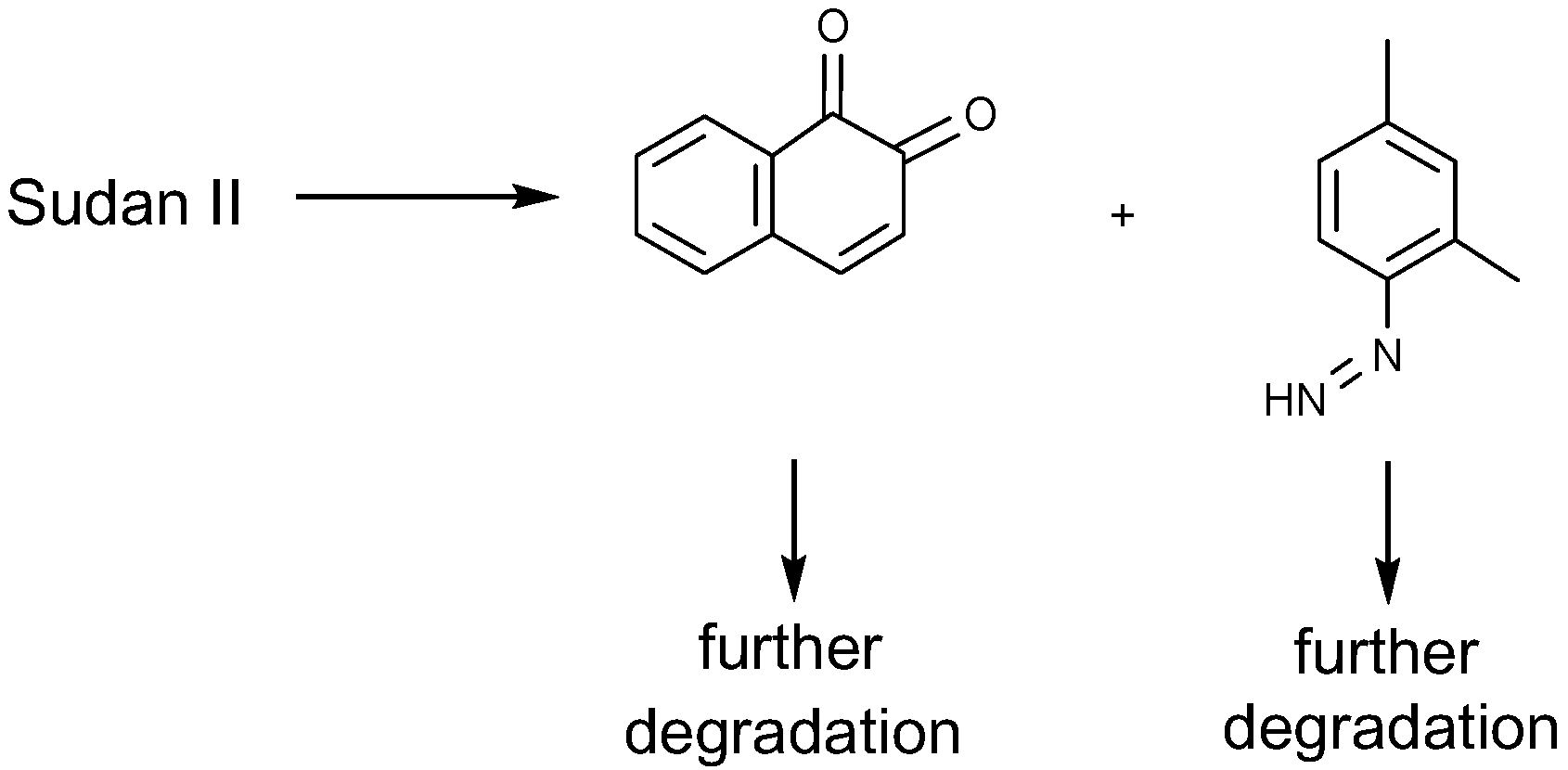
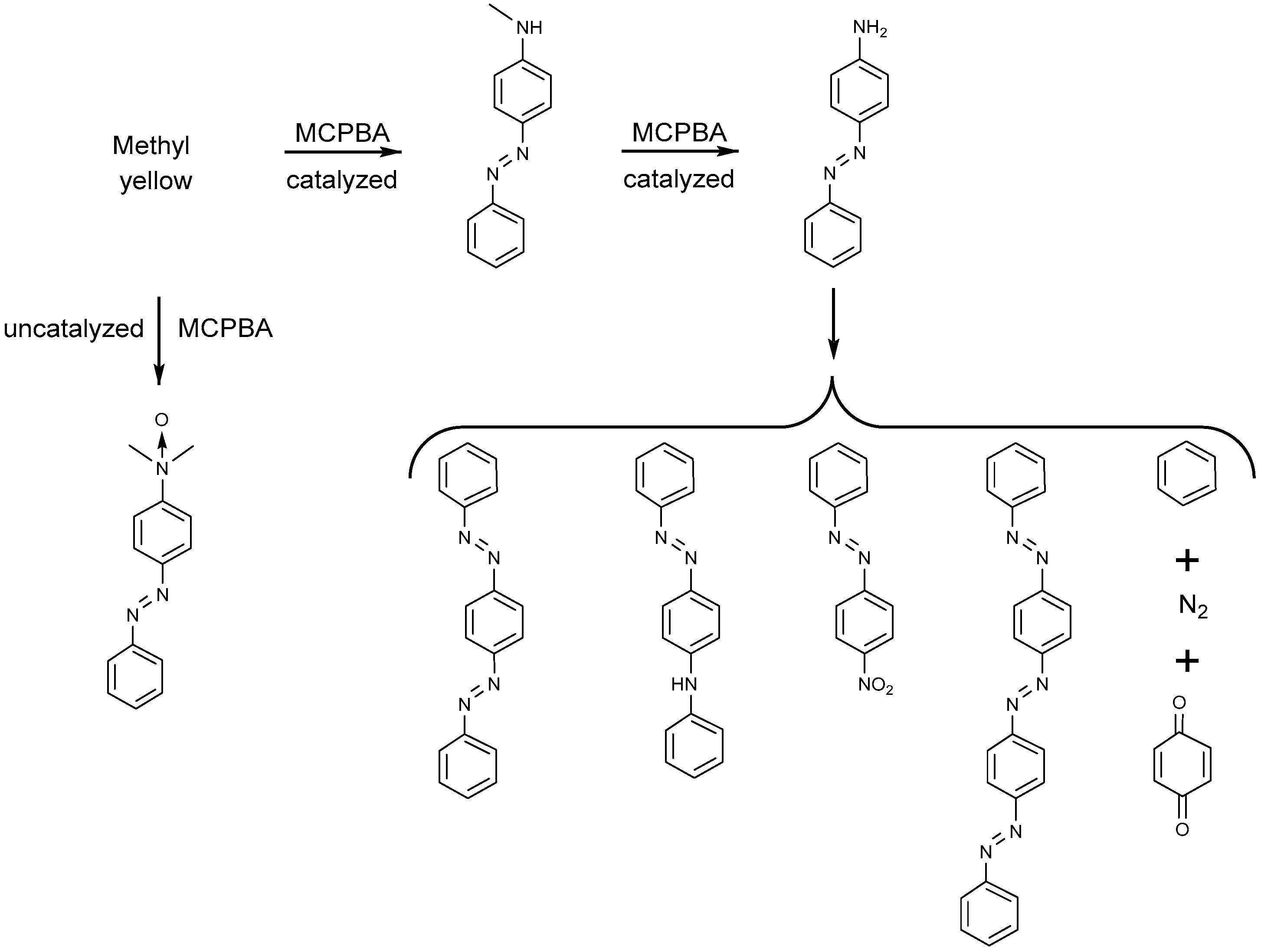
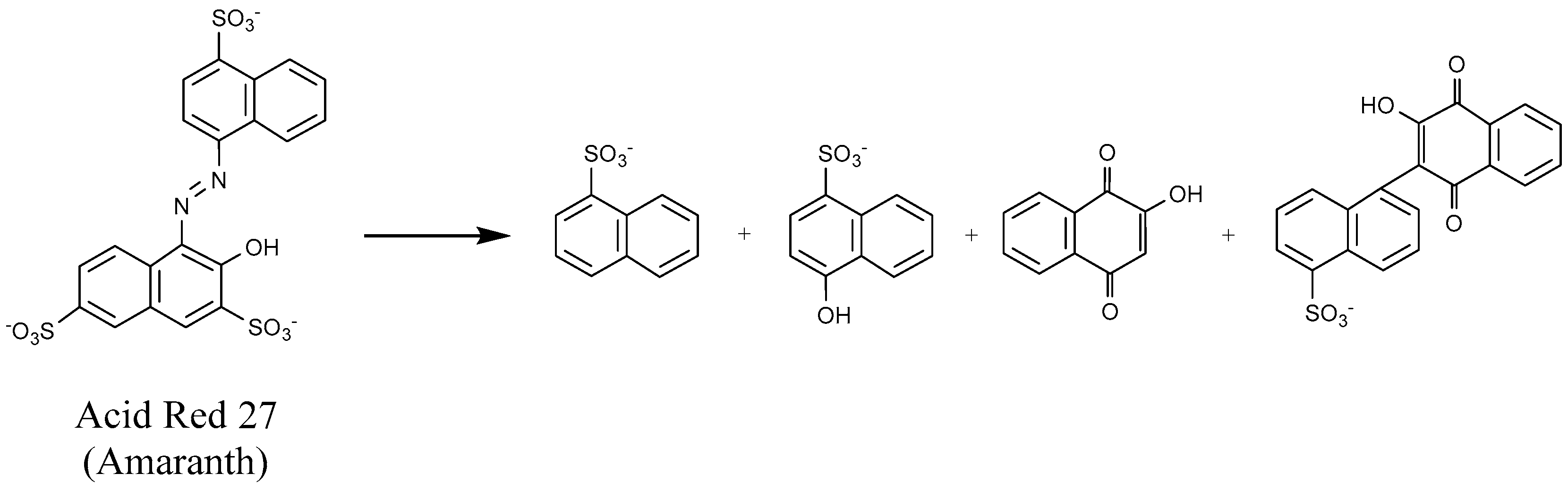
4.1.1. Azo Dyes
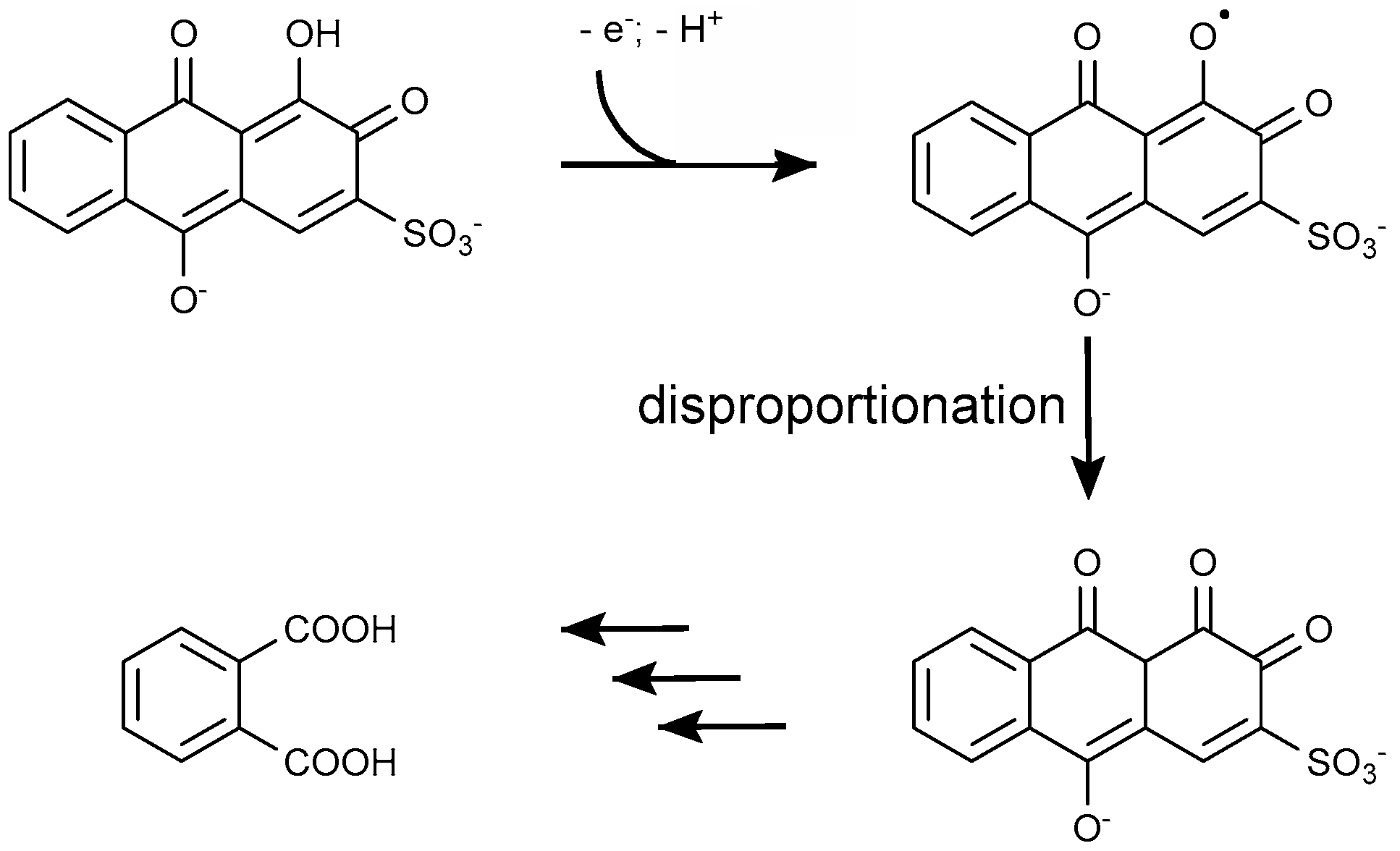
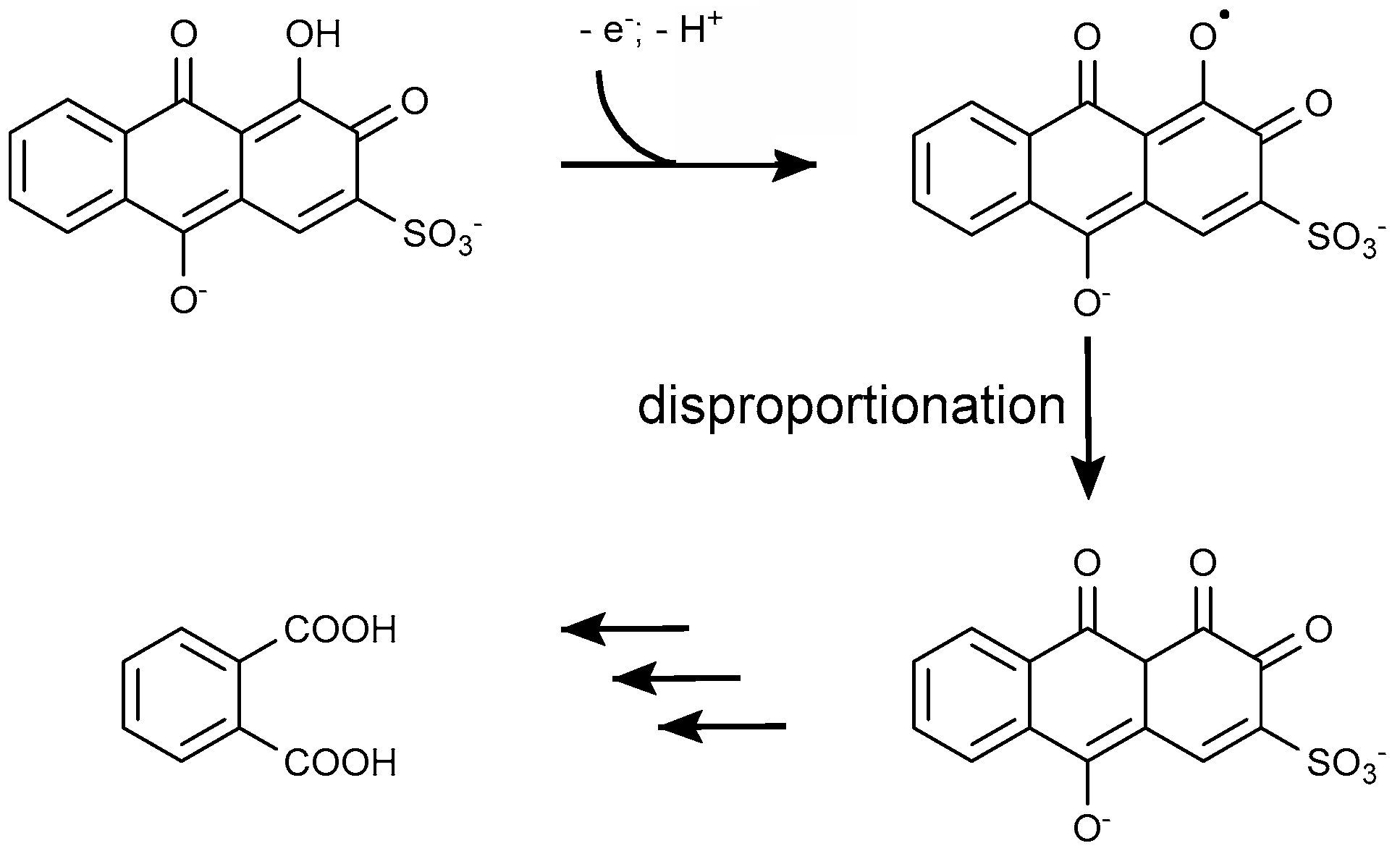
4.1.2. Anthraquinone Dyes
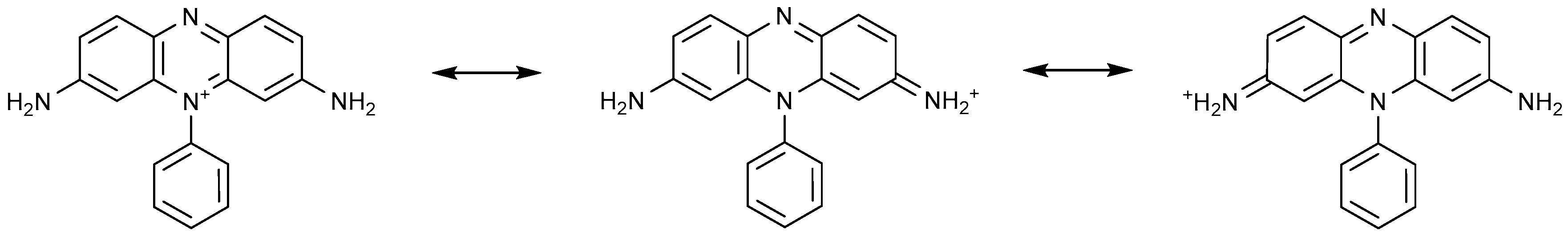
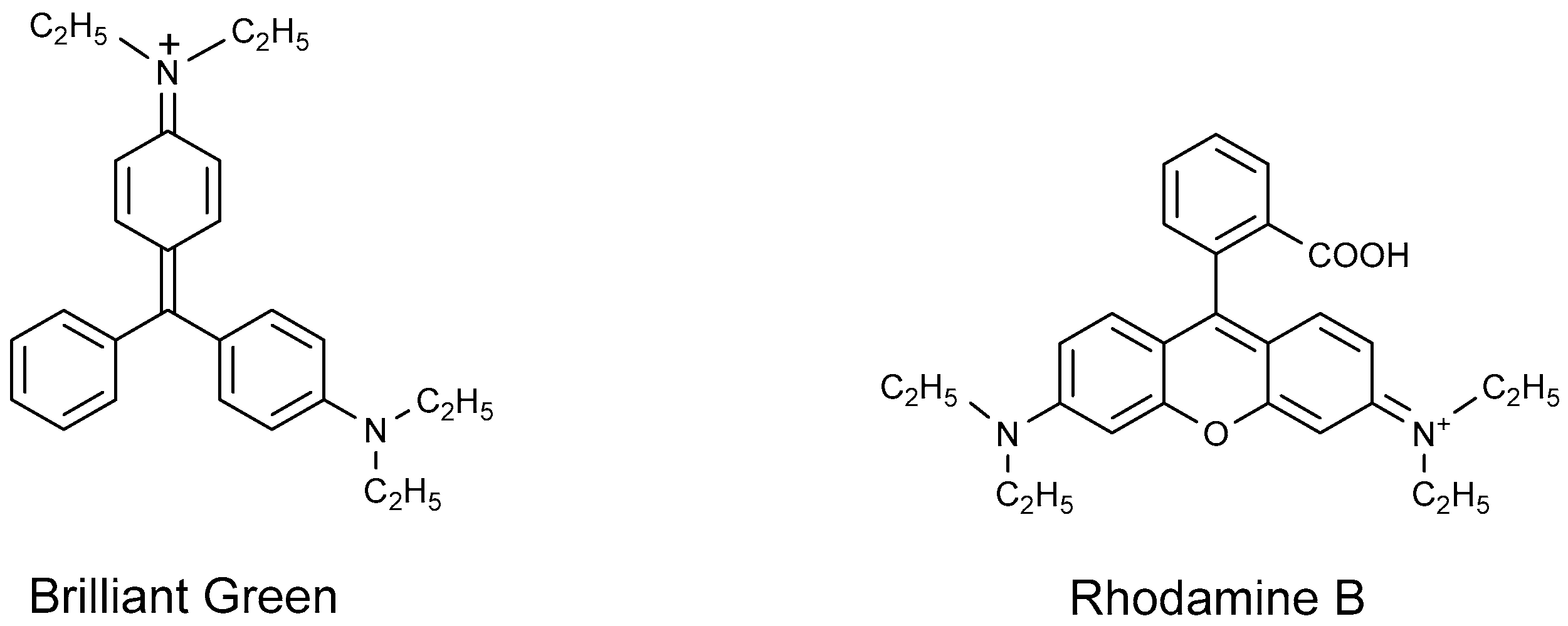
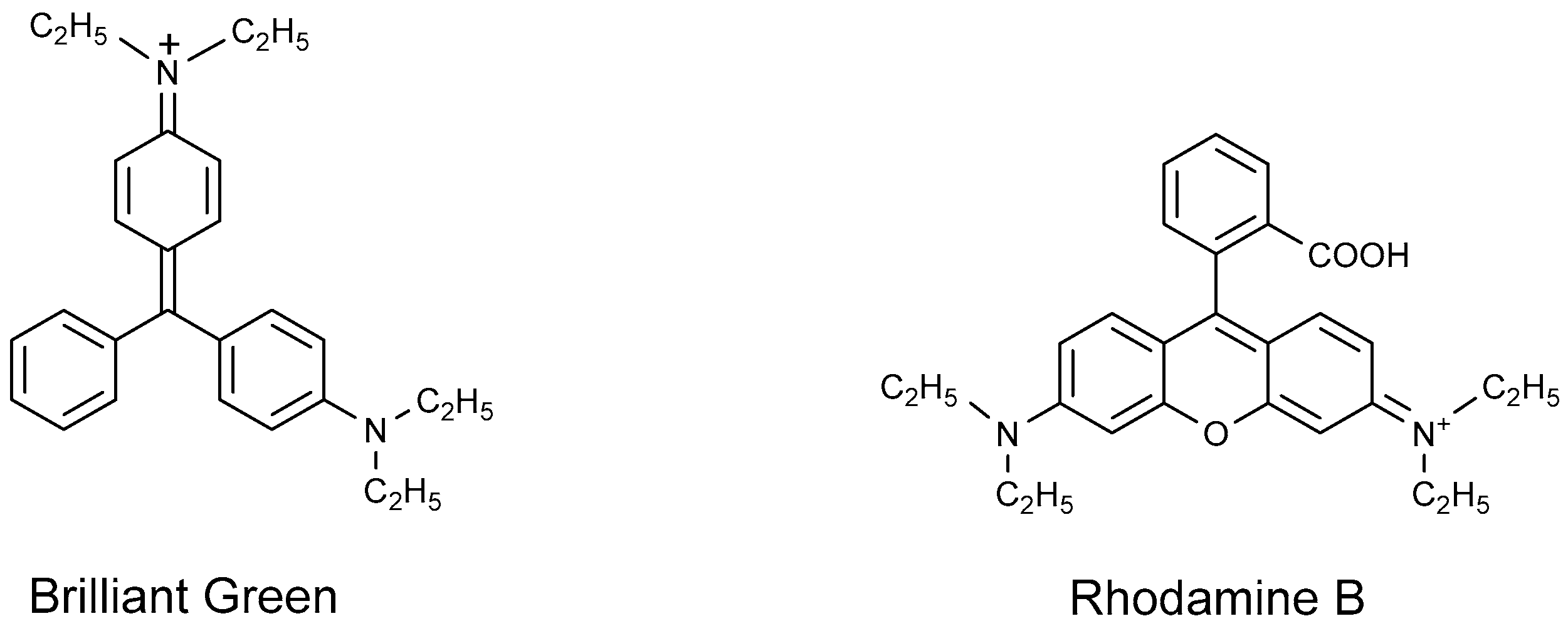
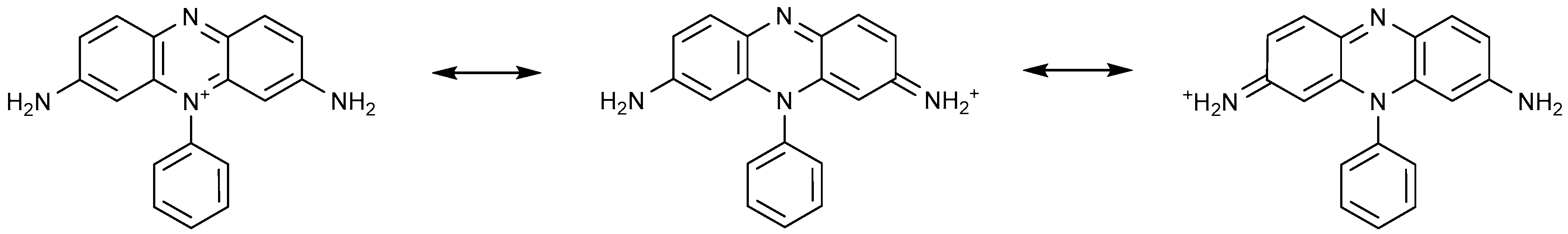
4.1.3. Cationic Dyes
4.1.4. Applications of Metalloporphyrins as Generic Decolorizing Catalysts
4.2. Effect of Redox Mediators
4.3. Comparison with Enzymatic Decolorization
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vajnhandl, S.; Valh, J.V. The status of water reuse in European textile sector. J. Environ. Manag. 2014, 141, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Maguire, R.J. Occurrence and persistence of dyes in a Canadian river. Water Sci. Technol. 1992, 26, 265–270. [Google Scholar]
- Pierce, J. Colour in textile effluents—The origins of the problem. J. Soc. Dyers Colour. 1994, 110, 131–133. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Decontamination of textile wastewater via TiO2/activated carbon composite materials. Adv. Colloid Interface Sci. 2010, 159, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Sarayu, K.; Sandhya, S. Current technologies for biological treatment of textile wastewater—A review. Appl. Biochem. Biotechnol. 2012, 167, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Muhd Julkapli, N.; Bagheri, S.; Bee Abd Hamid, S. Recent advances in heterogeneous photocatalytic decolorization of synthetic dyes. Sci. World J. 2014, 2014, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Karthik, V.; Saravanan, K.; Bharathi, P.; Dharanya, V.; Meiaraj, C. An overview of treatments for the removal of textile dyes. J. Chem. Pharm. Sci. 2014, 7, 301–307. [Google Scholar]
- Tsuda, S.; Murakami, M.; Matsusaka, N.; Kano, K.; Taniguchi, K.; Sasaki, Y.F. DNA Damage induced by red food dyes orally administered to pregnant and male mice. Toxicol. Sci. 2001, 61, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Luan, Z.; Wei, N.; Li, J.; Liu, C. The color removal of dye wastewater by magnesium chloride/red mud (MRM) from aqueous solution. J. Hazard. Mater. 2009, 170, 690–698. [Google Scholar] [CrossRef] [PubMed]
- European Union. European Directive Relating to Restrictions on the Marketing and Use of Certain Dangerous Substances and Preparation (Azocolourants). Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32002L0061 (accessed on 17 July 2016).
- European Union. European Water Framework Directive—Integrated River Basin Management for Europe. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52012DC0670 (accessed on 17 July 2016).
- Dasgupta, J.; Sikder, J.; Chakraborty, S.; Curcio, S.; Drioli, E. Remediation of textile effluents by membrane based treatment techniques: A state of the art review. J. Environ. Manage. 2015, 147, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Karthik, V.; Saravanan, K.; Thomas, T.; Devi, M. Review on microbial decolourisation of textile dyes. J. Chem. Pharm. Sci. 2014, 7, 293–300. [Google Scholar]
- Chiavola, A. Textiles. Water Environ. Res. 2012, 84, 1511–1532. [Google Scholar] [CrossRef]
- Türgay, O.; Ersöz, G.; Atalay, S.; Forss, J.; Welander, U. The treatment of azo dyes found in textile industry wastewater by anaerobic biological method and chemical oxidation. Sep. Purif. Technol. 2011, 79, 26–33. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sánchez-Pérez, J.A. Combination of Advanced Oxidation Processes and biological treatments for wastewater decontamination—A review. Sci. Total Environ. 2011, 409, 4141–4166. [Google Scholar] [CrossRef] [PubMed]
- Ong, Y.K.; Li, F.Y.; Sun, S.P.; Zhao, B.W.; Liang, C.Z.; Chung, T.S. Nanofiltration hollow fiber membranes for textile wastewater treatment: Lab-scale and pilot-scale studies. Chem. Eng. Sci. 2014, 114, 51–57. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, X.; Gao, B.; Zhao, S.; Wang, Y.; Yue, Q.; Li, Q. Flocculation kinetics and floc characteristics of dye wastewater by polyferric chloride-poly-epichlorohydrin-dimethylamine composite flocculant. Sep. Purif. Technol. 2013, 118, 583–590. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Ji, H.-B.; Huang, X.-J. Photocatalytic Degradation of Methyl Orange over Metalloporphyrins Supported on TiO2 Degussa P25. Molecules 2012, 17, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Aleboyeh, A.; Moussa, Y.; Aleboyeh, H. The effect of operational parameters on UV/H2O2 decolourisation of Acid Blue 74. Dyes and Pigments 2005, 66, 129–134. [Google Scholar] [CrossRef]
- Correia, V.M.; Stephenson, T.; Judd, S.J. Characterisation of textile wastewaters—A review. Environ. Technol. 1994, 15, 917–929. [Google Scholar] [CrossRef]
- Merzouk, B.; Yakoubi, M.; Zongo, I.; Leclerc, J.P.; Paternotte, G.; Pontvianne, S.; Lapicque, F. Effect of modification of textile wastewater composition on electrocoagulation efficiency. Desalination 2011, 275, 181–186. [Google Scholar] [CrossRef]
- Nordin, N.; Amir, S.F.M.; Riyanto; Othman, M.R. Textile industries wastewater treatment by electrochemical oxidation technique using metal plate. Int. J. Electrochem. Sci. 2013, 8, 11403–11415. [Google Scholar]
- Bisschops, I.; Spanjers, H. Literature review on textile wastewater characterisation. Environ. Technol. 2003, 24, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Gonsalves, A.M.A.; Pereira, M.M. State of the art in the development of biomimetic oxidation catalysts. J. Mol. Catal. A Chem. 1996, 113, 209–221. [Google Scholar] [CrossRef]
- Zucca, P.; Cocco, G.; Manca, S.; Steri, D.; Sanjust, E. Imidazole versus pyridine as ligands for metalloporphine immobilization in ligninolytic peroxidases-like biomimetic catalysts. J. Mol. Catal. A Chem. 2014, 394, 129–136. [Google Scholar] [CrossRef]
- Zucca, P.; Cocco, G.; Pintus, M.; Rescigno, A.; Sanjust, E. Biomimetic sulfide oxidation by the means of immobilized Fe(III)-5,10,15,20-tetrakis(pentafluorophenyl)porphin under mild experimental conditions. J. Chem. 2013, 2013. [Google Scholar] [CrossRef]
- Zucca, P.; Mocci, G.; Rescigno, A.; Sanjust, E. 5,10,15,20-Tetrakis(4-sulfonato-phenyl)porphine-Mn(III) immobilized on imidazole activacted silica as a novel lignin-peroxidase-like biomimetic catalyst. J. Mol. Catal. A Chem. 2007, 278, 220–227. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Rinaldi, A.C.; Sanjust, E. Biomimetic metalloporphines and metalloporphyrins as potential tools for delignification: molecular mechanisms and application perspectives. J. Mol. Catal. A Chem. 2014, 388, 2–34. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Sanjust, E. Ligninolytic peroxidase-like activity of a synthetic metalloporphine immobilized onto mercapto-grafted crosslinked PVA inspired by the active site of cytochrome P450. Chin. J. Catal. 2011, 32, 1663–1666. [Google Scholar] [CrossRef]
- Zucca, P.; Sollai, F.; Garau, A.; Rescigno, A.; Sanjust, E. Fe(III)-5,10,15,20-Tetrakis(pentafluorophenyl)porphine supported on pyridyl-functionalized, crosslinked poly(vinylalcohol) as a biomimetic versatile-peroxidase-like catalyst. J. Mol. Catal. A Chem. 2009, 306, 89–96. [Google Scholar] [CrossRef]
- Huang, G.; Mo, L.Q.; Cai, J.L.; Cao, X.; Peng, Y.; Guo, Y.A.; Wei, S.J. Environmentally friendly and efficient catalysis of cyclohexane oxidation by iron meso-tetrakis(pentafluorophenyl)porphyrin immobilized on zinc oxide. Appl. Catal. B 2015, 162, 364–371. [Google Scholar] [CrossRef]
- Simoes, M.M.Q.; Neves, C.M.B.; Pires, S.M.G.; Graca, M.; Neves, M.S.; Cavaleiro, J.A.S. Mimicking P450 processes and the use of metalloporphyrins. Pure Appl. Chem. 2013, 85, 1671–1681. [Google Scholar] [CrossRef]
- Rebelo, S.; Silva, A.; Medforth, C.; Freire, C. Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Auwarter, W.; Ecija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at interfaces. Nat Chem 2015, 7, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, L.R. The colours of life: An introduction to the chemistry of porphyrins and related compounds. Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Springer International Publishing: Berlin, Germany, 2015. [Google Scholar]
- Urbani, M.; Grätzel, M.; Nazeeruddin, M.K.; Torres, T. Meso-substituted porphyrins for dye-sensitized solar cells. Chem. Rev. 2014, 114, 12330–12396. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine. World Scientific Publishing Company Pte Limited: Singapore, 2012. [Google Scholar]
- Kärkäs, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef] [PubMed]
- Vasapollo, G.; Mele, G.; Sole, R.D.; Pio, I.; Li, J.; Mazzetto, S.E. Use of Novel Cardanol-Porphyrin Hybrids and Their TiO2-Based Composites for the Photodegradation of 4-Nitrophenol in Water. Molecules 2011, 16, 5769–5784. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.Y.; Dolphin, D. Metalloporphyrins Catalyzed Oxidations; Montanari, F., Casella, L., Eds.; Kluwer Academics Publishers: Dordrecth, The Netherlands, 1994; pp. 269–306. [Google Scholar]
- Suslick, K.S. The Porphyrin Handbook. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 4. [Google Scholar]
- Sheldon, R.A. Metalloporphyrins in Catalytic Oxidations; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Montanari, F.; Casella, L. Metalloporphyrins Catalyzed Oxidations; Kluwer: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Mansuy, D.; Battioni, P. Metalloporphyrins in Catalytic Oxidations; Sheldon, R.A., Ed.; Marcell Dekker Inc.: New York, NY, USA, 1994; pp. 99–132. [Google Scholar]
- Mansuy, D. The great diversity of reactions catalyzed by cytochromes P450. Comp. Biochem. Physiol. C: Pharmacol. Toxicol. Endocrinol. 1998, 121, 5–14. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; Bernadou, J. Metal-oxo species in P450 enzymes and biomimetic models. Oxo-hydroxo tautomerism with water-soluble metalloporphyrins. Top. Catal. 2002, 21, 47–54. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; Springer Berlin Heidelberg: Berlin, Germany, 2003. [Google Scholar]
- Dau, H.A.; Ullrich, R.; Benndorf, D.; Svatoś, A.; Muck, A.; Hofrichter, M. The coprophilous mushroom Coprinus radians secretes a haloperoxidase that catalyzes aromatic peroxygenation. Appl. Environ. Microbiol. 2007, 73, 5477–5485. [Google Scholar]
- Kinne, M.; Zeisig, C.; Ullrich, R.; Kayser, G.; Hammel, K.E.; Hofrichter, M. Stepwise oxygenations of toluene and 4-nitrotoluene by a fungal peroxygenase. Biochem. Biophys. Res. Commun. 2010, 397, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, R.; Dolge, C.; Kluge, M.; Hofrichter, M. Pyridine as novel substrate for regioselective oxygenation with aromatic peroxygenase from Agrocybe aegerita. FEBS Lett. 2008, 582, 4100–4106. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, R.; Nüske, J.; Scheibner, K.; Spantzel, J.; Hofrichter, M. Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Appl. Environ. Microbiol. 2004, 70, 4575–4581. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, M.; Ullrich, R.; Pecyna, M.J.; Liers, C.; Lundell, T. New and classic families of secreted fungal heme peroxidases. Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [Google Scholar] [CrossRef] [PubMed]
- Yarman, A.; Gröbe, G.; Neumann, B.; Kinne, M.; Gajovic-Eichelmann, N.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. The aromatic peroxygenase from Marasmius rutola—A new enzyme for biosensor applications. Anal. Bioanal. Chem. 2012, 402, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Bernadou, J.; Meunier, B. Biomimetic chemical catalysts in the oxidative activation of drugs. Adv. Synth. Catal. 2004, 346, 171–184. [Google Scholar] [CrossRef]
- Mansuy, D. A brief history of the contribution of metallporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C. R. Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for hydrocarbon oxygenations and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Meunier, B. Models of Heme Peroxidases and Catalases. In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier, B., Ed.; Imperial College Press: London, UK, 2000; pp. 171–214. [Google Scholar]
- Simões, M.M.Q.; De Paula, R.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Metalloporphyrins in the biomimetic oxidative valorization of natural and other organic substrates. J. Porphyrins Phthalocyanines 2009, 13, 589–596. [Google Scholar] [CrossRef]
- Lohmann, W.; Karst, U. Biomimetic modeling of oxidative drug metabolism: Strategies, advantages and limitations. Anal. Bioanal. Chem. 2008, 391, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Neves, C.M.B.; Simões, M.M.Q.; Domíngues, M.R.M.; Santos, I.C.M.S.; Neves, M.G.P.M.S.; Paz, F.A.A.; Silva, A.M.S.; Cavaleiro, J.A.S. Oxidation of diclofenac catalyzed by manganese porphyrins: Synthesis of novel diclofenac derivatives. RSC Adv. 2012, 2, 7427–7438. [Google Scholar] [CrossRef]
- Dos Santos, J.S.; Faria, A.L.; da Silva Amorin, P.M.; La Luna, F.M.; Caiado, K.L.; E Silva, D.O.C.; Sartoratto, P.P.C.; Assis, M.D. Iron(III) porphyrin covalently supported onto magnetic amino-functionalized nanospheres as catalyst for hydrocarbon and herbicide oxidations. J. Brazil. Chem. Soc. 2012, 23, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Rothemund, P. Formation of porphyrins from pyrrole and aldehydes. J. Am. Chem. Soc. 1935, 57, 2010–2011. [Google Scholar] [CrossRef]
- Rothemund, P. A new porphyrin synthesis. The synthesis of porphin. J. Am. Chem. Soc. 1936, 58, 625–627. [Google Scholar] [CrossRef]
- Rothemund, P. Porphyrin studies. III. The structure of the porphine ring system. J. Am. Chem. Soc. 1939, 61, 2912–2915. [Google Scholar] [CrossRef]
- Aronoff, S.; Calvin, M. The porphyrin-like products of the reaction of pyrrole with benzaldehyde. J. Org. Chem. 1943, 8, 205–223. [Google Scholar] [CrossRef]
- Calvin, M.; Ball, R.H.; Aronoff, S. α,β,γ,δ-Tetraphenylchlorin. J. Am. Chem. Soc. 1943, 65, 2259–2259. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A simplified synthesis for meso-tetraphenylporphin. J. Org. Chem. 1967, 32, 476–476. [Google Scholar] [CrossRef]
- Barnett, G.H.; Hudson, M.F.; Smith, K.M. Concerning meso-tetraphenylporphyrin purification. J. Chem. Soc. Perkin Trans. 1 1975, 1401–1403. [Google Scholar] [CrossRef]
- Gonsalves, A.; Varejão, J.M.; Pereira, M.M. Some new aspects related to the synthesis of meso-substituted porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Gonsalves, A.D.A.R.; Pereira, M.M. A new look into the rothemund meso-tetraalkyl and tetraarylporphyrin synthesis. J. Heterocycl. Chem. 1985, 22, 931–933. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Hsu, H.C.; Schreiman, I.C. Synthesis of tetraphenylporphyrins under very mild conditions. Tetrahedron Lett. 1986, 27, 4969–4970. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo reactions revisited: Synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Sharghi, H.; Nejad, A.H. Phosphorus pentachloride (PCl5) mediated synthesis of tetraarylporphyrins. Helv. Chim. Acta 2003, 86, 408–414. [Google Scholar] [CrossRef]
- Sharghi, H.; Nejad, A.H. Novel synthesis of meso-tetraarylporphyrins using CF3SO 2Cl under aerobic oxidation. Tetrahedron 2004, 60, 1863–1868. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Tomé, A.C.; Neves, M.G.P.M.S. The Porphyrin Handbook. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 4. [Google Scholar]
- Chauhan, S.; Sahoo, B.; Srinivas, K. Microwave-assisted synthesis of 5,10,15,20-tetraaryl porphyrins. Synth. Commun. 2001, 31, 33–37. [Google Scholar] [CrossRef]
- De Paula, R.; Faustino, M.A.; Pinto, D.C.; Neves, M.G.; Cavaleiro, J.A. Kinetic study of meso-tetraphenylporphyrin synthesis under microwave irradiation. J. Heterocycl. Chem. 2008, 45, 453–459. [Google Scholar] [CrossRef]
- Liu, M.O.; Tai, C.-H.; Wang, W.-Y.; Chen, J.-R.; Hu, A.T.; Wei, T.-H. Microwave-assisted synthesis and reverse saturable absorption of phthalocyanines and porphyrins. J. Organomet. Chem. 2004, 689, 1078–1084. [Google Scholar] [CrossRef]
- Nascimento, B.F.; Pineiro, M.; Rocha Gonsalves, A.M.d.A.; Ramos Silva, M.; Matos Beja, A.; Paixão, J.A. Microwave-assisted synthesis of porphyrins and metalloporphyrins: A rapid and efficient synthetic method. J. Porphyrins Phthalocyanines 2007, 11, 77–84. [Google Scholar] [CrossRef]
- Henriques, C.A.; Pinto, S.; Aquino, G.L.; Pineiro, M.; Calvete, M.J.; Pereira, M.M. Ecofriendly porphyrin synthesis by using water under microwave irradiation. ChemSusChem 2014, 7, 2821–2824. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, J.F.; Battioni, P.; de Foor, W.R.; Mansuy, D. Synthesis and remarkable properties of iron β-polynitroporphyrins as catalysts for monooxygenation reactions. J. Chem. Soc. Chem. Commun. 1994, 23–24. [Google Scholar] [CrossRef]
- Bartoli, J.F.; Mouries-Mansuy, V.; le Barch-Ozette, K.; Palacio, M.; Battioni, P.; Mansuy, D. New manganese β-polynitroporphyrins as particularly efficient catalysts for biomimetic hydroxylation of aromatic compounds with H2O2. Chem. Commun. 2000, 827–828. [Google Scholar] [CrossRef]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual Ligands for Metals and Metal-Catalyzed Oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- Bäckvall, J.E. Modern Oxidation Methods; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Beller, M. The Current Status and Future Trends in Oxidation Chemistry. Adv. Synth. Catal. 2004, 346, 107–108. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent advances in heterogeneous selective oxidation catalysis for sustainable chemistry. Chem. Soc. Rev. 2014, 43, 3480–3524. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Aoki, M.; Sato, K. Green oxidation with aqueous hydrogen peroxide. Chem. Commun. 2003, 1977–1986. [Google Scholar] [CrossRef]
- Sanderson, W.R. Cleaner industrial processes using hydrogen peroxide. Pure Appl. Chem. 2000, 72, 1289–1304. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Wu, W.; Jiang, H. Palladium-Catalyzed Oxidation of Unsaturated Hydrocarbons Using Molecular Oxygen. Acc. Chem. Res. 2012, 45, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Fundamentals of green chemistry: efficiency in reaction design. Chem. Soc. Rev. 2012, 41, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Dan-Hua, S.; Lin-Tao, J.; Zhi-Gang, L.; Wen-Bin, S.; Can-Cheng, G. Ethylbenzene oxidation over hybrid metalloporphyrin@silica nanocomposite microspheres. J. Mol. Catal. A Chem. 2013, 379, 15–20. [Google Scholar] [CrossRef]
- Cai, J.H.; Huang, J.W.; Yu, H.C.; Ji, L.N. Manganeseporphyrins immobilized on silica microspheres as biomimetic catalysts hydroxylating cyclohexane with molecular oxygen. J. Sol-Gel Sci. Technol. 2011, 58, 698–704. [Google Scholar] [CrossRef]
- Adam, F.; Ooi, W.T. Selective oxidation of benzyl alcohol to benzaldehyde over Co-metalloporphyrin supported on silica nanoparticles. Appl. Catal. A. 2012, 445–446, 252–260. [Google Scholar] [CrossRef]
- Faria, A.L.; Mac Leod, T.O.C.; Barros, V.P.; Assis, M.D. Hydrocarbon oxidation catalyzed by iron and manganese porphyrins anchored on aminofunctionalized supports. J. Brazil. Chem. Soc. 2009, 20, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Mesbahi, E.; Safari, N.; Gheidi, M. Investigation of axial ligand effects on catalytic activity of manganese porphyrin, evidence for the importance of hydrogen bonding in cytochrome-P450 model reactions. J. Porphyrins Phthalocyanines 2014, 18, 354–365. [Google Scholar] [CrossRef]
- Maraval, V.; Ancel, J.E.; Meunier, B. Manganese(III) porphyrin catalysts for the oxidation of terpene derivatives: A comparative study. J. Catal. 2002, 206, 349–357. [Google Scholar] [CrossRef]
- Meunier, B.; Guilmet, E.; Carvalho, M.E.D.; Poilblanc, R. Sodium hypochlorite: a convenient oxygen source for olefin epoxidation catalyzed by (porphyrinato)manganese complexes. J. Am. Chem. Soc. 1984, 106, 6668–6676. [Google Scholar] [CrossRef]
- Martins, R.R.L.; Neves, M.G.P.M.S.; Silvestre, A.J.D.; Simões, M.M.Q.; Silva, A.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Tagliatesta, P.; Crestini, C. Oxidation of unsaturated monoterpenes with hydrogen peroxide catalysed by manganese (III) porphyrin complexes. J. Mol. Catal. A Chem. 2001, 172, 33–42. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Nascimento, G.M.S.F.C.; Neves, M.G.P.M.S.; Pinto, M.T.; Silvestre, A.J.D.; Vicente, M.G.H. Oxidation of natural compounds catalyzed by Mn(III) porphyrin complexes. Tetrahedron Lett. 1996, 37, 1893–1896. [Google Scholar] [CrossRef]
- Martins, R.R.L.; Neves, M.G.P.M.S.; Silvestre, A.J.D.; Silva, A.M.S.; Cavaleiro, J.A.S. Oxidation of aromatic monoterpenes with hydrogen peroxide catalysed by Mn (III) porphyrin complexes. J. Mol. Catal. A Chem. 1999, 137, 41–47. [Google Scholar] [CrossRef]
- Belvedere, S.; Breslow, R. Regioselective Oxidation of Steroids by a Manganese Porphyrin Carrying Metal Coordinating Groups. Bioorg. Chem. 2001, 29, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Gabriele, B.; Yang, J. Geometrically directed selective steroid hydroxylation with high turnover by a fluorinated artificial cytochrome P-450. Tetrahedron Lett. 1998, 39, 2887–2890. [Google Scholar] [CrossRef]
- Fang, Z.; Breslow, R. Metal Coordination-Directed Hydroxylation of Steroids with a Novel Artificial P-450 Catalyst. Org. Lett. 2006, 8, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Ogawa, S.; Hosoi, K.; Makino, M.; Fujimoto, Y.; Goto, T.; Mano, N.; Goto, J.; Hofmann, A.F. Regioselective oxyfunctionalization of unactivated carbons in steroids by a model of cytochrome P-450: Osmiumporphyrin complex/tert-butyl hydroperoxide system. J. Org. Chem. 2007, 72, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Lida, T.; Ogawa, S.; Miyata, S.; Goto, T.; Mano, N.; Goto, J.; Nambara, T. Biomimetic oxidation of unactivated carbons in steroids by a model of cytochrome P-450, oxorutheniumporphyrinate complex. Lipids 2004, 39, 873–880. [Google Scholar] [PubMed]
- Ogawa, S.; Hosoi, K.; Iida, T.; Wakatsuki, Y.; Makino, M.; Fujimoto, Y.; Hofmann, A.F. Osmiumporphyrin-Catalyzed Oxyfunctionalization and Isomerization of Natural (5β)-Bile Acids with tert-Butyl Hydroperoxide. Eur. J. Org. Chem. 2007, 2007, 3555–3563. [Google Scholar] [CrossRef]
- Ogawa, S.; Iida, T.; Goto, T.; Mano, N.; Goto, J.; Nambara, T. The remote-oxyfunctionalization of unactivated carbons in (5β)-3-oxobile acids by 2,6-dichloropyridine N-oxide catalyzed by ruthenium-porphyrin and HBr: A direct lactonization at C-20. Org. Biomol. Chem. 2004, 2, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Shingaki, T.; Miura, K.; Higuchi, T.; Hirobe, M.; Nagano, T. Regio- and stereo-selective oxidation of steroids using 2,6-dichloropyridine N-oxide catalysed by ruthenium porphyrins. Chem. Commun. 1997, 861–862. [Google Scholar] [CrossRef]
- Yang, J.; Gabriele, B.; Belvedere, S.; Huang, Y.; Breslow, R. Catalytic oxidations of steroid substrates by artificial cytochrome P-450 enzymes. J. Org. Chem. 2002, 67, 5057–5067. [Google Scholar] [CrossRef] [PubMed]
- Mansuy, D. Biocatalysis and substrate chemodiversity: Adaptation of aerobic living organisms to their chemical environment. Catal. Today 2008, 138. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Capel-Sanchez, M.C.; Perez-Presas, P.; Fierro, J.L.G. Oxidative processes of desulfurization of liquid fuels. J. Chem. Technol. Biotechnol. 2010, 85, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhou, A.; Song, C. A novel method for oxidative desulfurization of liquid hydrocarbon fuels based on catalytic oxidation using molecular oxygen coupled with selective adsorption. Catal. Today 2007, 123, 276–284. [Google Scholar] [CrossRef]
- Pires, S.M.G.; Simões, M.M.Q.; Santos, I.C.M.S.; Rebelo, S.L.H.; Pereira, M.M.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Biomimetic oxidation of organosulfur compounds with hydrogen peroxide catalyzed by manganese porphyrins. Appl. Catal. A. 2012, 439–440, 51–56. [Google Scholar] [CrossRef]
- Pires, S.M.G.; Simões, M.M.Q.; Santos, I.C.M.S.; Rebelo, S.L.H.; Paz, F.A.A.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of organosulfur compounds using an iron(III) porphyrin complex: An environmentally safe and efficient approach. Appl. Catal. B. 2014, 160–161, 80–88. [Google Scholar] [CrossRef]
- Fontaine, B.; Piccolo, A. Co-polymerization of penta-halogenated phenols in humic substances by catalytic oxidation using biomimetic catalysis. Environ. Sci. Pollut. Res. 2012, 19, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, A.; Piccolo, A. Enhanced catechol oxidation by heterogeneous biomimetic catalysts immobilized on clay minerals. J. Mol. Catal. A Chem. 2013, 371, 8–14. [Google Scholar] [CrossRef]
- Nuzzo, A.; Piccolo, A. Oxidative and photoxidative polymerization of humic suprastructures by heterogeneous biomimetic catalysis. Biomacromolecules 2013, 14, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Sannino, F.; Spaccini, R.; Savy, D.; Piccolo, A. Remediation of highly contaminated soils from an industrial site by employing a combined treatment with exogeneous humic substances and oxidative biomimetic catalysis. J. Hazard. Mater. 2013, 261C, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Shigetatsu, S.; Fukushima, M.; Nagao, S. Oxidative degradation of 2,6-dibromophenol using anion-exchange resin supported supramolecular catalysts of iron(III)-5,10,15,20-tetrakis (p-hydroxyphenyl)porphyrin bound to humic acid prepared via formaldehyde and urea-formaldehyde polycondensation. J. Environ. Sci. Health. A Tox. Hazard. Subst. Environ. Eng. 2010, 45, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Díaz, G.; Blanco-López, M.C.; Lobo-Castañón, M.J.; Miranda-Ordieres, A.J.; Tuñón-Blanco, P. Hemo-acrylic polymers as catalyst for the oxidative dehalogenation of 2,4,6-trichlorophenol. Chloroperoxidase's mimic imprinting effects. J. Mol. Catal. A Chem. 2012, 353–354, 117–121. [Google Scholar] [CrossRef]
- Zhu, Q.; Mizutani, Y.; Maeno, S.; Fukushima, M. Oxidative debromination and degradation of tetrabromo-bisphenol A by a functionalized silica-supported iron(III)-tetrakis(p-sulfonatophenyl)porphyrin catalyst. Molecules 2013, 18, 5360–5372. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Mizutani, Y.; Maeno, S.; Zhu, Q.; Kuramitz, H.; Nagao, S. Influence of Halogen Substituents on the Catalytic Oxidation of 2,4,6-Halogenated Phenols by Fe(III)-Tetrakis(p-hydroxyphenyl) porphyrins and Potassium Monopersulfate. Molecules 2011, 17, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Zhu, Q.; Igrashi, M.; Kodama, R.; Maeno, S.; Fukushima, M. Catalytic oxidation of tetrabromobisphenol A by iron(III)-tetrakis(p-sulfonatephenyl)porphyrin catalyst supported on cyclodextrin polymers with potassium monopersulfate. J. Mol. Catal. B Enzym. 2015, 119, 64–70. [Google Scholar] [CrossRef]
- Van Pée, K.H.; Unversucht, S. Biological dehalogenation and halogenation reactions. Chemosphere 2003, 52, 299–312. [Google Scholar] [CrossRef]
- Crestini, C.; Pastorini, A.; Tagliatesta, P. Metalloporphyrins immobilized on montmorillonite as biomimetic catalysts in the oxidation of lignin model compounds. J. Mol. Catal. A Chem. 2004, 208, 195–202. [Google Scholar] [CrossRef]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef] [Green Version]
- Linhares, M.; Rebelo, S.L.H.; Simões, M.M.Q.; Silva, A.M.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Freire, C. Biomimetic oxidation of indole by Mn(III)porphyrins. Appl. Catal. A. 2014, 470, 427–433. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Linhares, M.; Simões, M.M.Q.; Silva, A.M.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Freire, C. Indigo dye production by enzymatic mimicking based on an iron(III)porphyrin. J. Catal. 2014, 315, 33–40. [Google Scholar] [CrossRef]
- Friedermann, G.R.; Halma, M.; de Freitas Castro, K.A.D.; Benedito, F.L.; Doro, F.G.; Drechsel, S.M.; Mangrich, A.S.; Assis, M.d.D.; Nakagaki, S. Intermediate species generated from halogenated manganese porphyrins electrochemically and in homogeneous catalysis of alkane oxidation. Appl. Catal., A. 2006, 308, 172–181. [Google Scholar] [CrossRef]
- Intrieri, D.; Caselli, A.; Ragaini, F.; Cenini, S.; Gallo, E. Ruthenium porphyrins-catalyzed atom-efficient amination of C-H bonds by arylazides. J. Porphyrins Phthalocyanines 2010, 14, 732–740. [Google Scholar] [CrossRef]
- Saha, T.K.; Frauendorf, H.; John, M.; Dechert, S.; Meyer, F. Efficient Oxidative Degradation of Azo Dyes by a Water-Soluble Manganese Porphyrin Catalyst. ChemCatChem 2013, 5, 796–805. [Google Scholar] [CrossRef]
- Barros, V.P.; Assis, M.D. Iron porphyrins as biomimetical models for disperse azo dye oxidation. J. Brazil. Chem. Soc. 2013, 24, 830–836. [Google Scholar] [CrossRef]
- Tatsumi, T.; Nakamura, M.; Tominaga, H.O. Hydroxylation of alkanes catalyzed by manganese tetraphenylporphyrin immobilized on imidazole-modified silica gel. Catal. Today 1989, 6, 163–170. [Google Scholar] [CrossRef]
- Zucca, P.; Vinci, C.; Rescigno, A.; Dumitriu, E.; Sanjust, E. Is the bleaching of phenosafranine by hydrogen peroxide oxidation catalyzed by silica-supported 5,10,15,20-tetrakis-(sulfonatophenyl)porphine-Mn(III) really biomimetic? J. Mol. Catal. A Chem. 2010, 321, 27–33. [Google Scholar] [CrossRef]
- Zucca, P.; Vinci, C.; Sollai, F.; Rescigno, A.; Sanjust, E. Degradation of Alizarin Red S under mild experimental conditions by immobilized 5,10,15,20-tetrakis(4-sulfonatophenyl)porphine-Mn(III) as a biomimetic peroxidase-like catalyst. J. Mol. Catal. A Chem. 2008, 288, 97–102. [Google Scholar] [CrossRef]
- Iamamoto, Y.; Idemori, Y.M.; Nakagaki, S. Cationic ironporphyrins as catalyst in comparative oxidation of hydrocarbons: homogeneous and supported on inorganic matrices systems. J. Mol. Catal. A Chem. 1995, 99, 187–193. [Google Scholar] [CrossRef]
- Zucca, P.; Sanjust, E. Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms. Molecules 2014, 19, 14139–14194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the Support Properties for Immobilization or Purification of Enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Nakagaki, S.; Mantovani, K.M.; Machado, S.G.; Castro, K.A.D.F.; Wypych, F. Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results. Molecules 2016, 21, 291. [Google Scholar] [CrossRef] [PubMed]
- Assis, M.d.D.; Smith, J.R.L. Hydrocarbon oxidation with iodosylbenzene catalysed by the sterically hindered iron(III) 5-(pentafluorophenyl)-10,15,20-tris(2,6-dichlorophenyl)porphyrin in homogeneous solution and covalently bound to silica. J. Chem. Soc. Perkin Trans. 2 1998, 2221–2226. [Google Scholar] [CrossRef]
- Benedito, F.L.; Nakagaki, S.; Saczk, A.A.; Peralta-Zamora, P.G.; Costa, C.M.M. Study of metalloporphyrin covalently bound to silica as catalyst in the ortho-dianisidine oxidation. Appl. Catal. A 2003, 250. [Google Scholar] [CrossRef]
- Ghiaci, M.; Molaie, F.; Sedaghat, M.E.; Dorostkar, N. Metalloporphyrin covalently bound to silica. Preparation, characterization and catalytic activity in oxidation of ethylbenzene. Catal. Commun. 2010, 11, 694–699. [Google Scholar] [CrossRef]
- Gao, B.; Wang, R.; Zhang, Y. Immobilization of manganoporphyrin on a novel polymeric support and catalytic oxidation characteristic of supported catalyst. J. Appl. Polym. Sci. 2009, 112, 2764–2772. [Google Scholar] [CrossRef]
- Martinez-Lorente, M.A.; Battioni, P.; Kleemiss, W.; Bartoli, J.F.; Mansuy, D. Manganese porphyrins covalentely bound to silica and montmorillonite K10 as efficient catalysts for alkene and alkane oxidation by hydrogen peroxide. J. Mol. Catal. A Chem. 1996, 113, 343–353. [Google Scholar] [CrossRef]
- Nishimoto, R.; Zhu, Q.; Miyamoto, T.; Sato, T.; Tu, X.; Aneksampant, A.; Fukushima, M. Monopersulfate oxidation of Acid Orange 7 with an iron(III)-tetrakis(N-methylpyridinium-4-yl)porphyrin intercalated into the layers of montmorillonite and pillared clay. J. Mol. Catal. A Chem. 2014, 396, 84–89. [Google Scholar] [CrossRef]
- Kitamura, Y.; Mifune, M.; Takatsuki, T.; Iwasaki, T.; Kawamoto, M.; Iwado, A.; Chikuma, M.; Saito, Y. Ion-exchange resins modified with metal-porphyrin as a catalysis for oxidation of epinephrine (adrenaline). Catal. Commun. 2008, 9, 224–228. [Google Scholar] [CrossRef]
- Farzaneh, F.; Poorkhosravani, M.; Ghandi, M. Utilization of immobilized biomimetic iron complexes within nanoreactors of Al-MCM-41 as cyclohexane oxidation catalyst. J. Mol. Catal. A Chem. 2009, 308, 108–113. [Google Scholar] [CrossRef]
- Karimipour, G.; Rezaei, M.; Ashouri, D. Zeolite encapsulated Fe-poprhyrin for catalytic oxidation with iodobenzene diacetate (PhI(OAc)2). J. Mex. Chem. Soc. 2013, 57, 276–282. [Google Scholar]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Axial ligand effect on the rate constant of aromatic hydroxylation by iron(IV)-Oxo complexes mimicking cytochrome P450 enzymes. J. Phys. Chem. B 2012, 116, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.M.; Pruneau, J.M.; Laverack, C.A.; Dutton, A.S.; Jones, G.B. Biomimetic oxidation of acetaminophen prodrugs catalyzed by iron porphyrins: Effect of nitrogen and thiolate axial ligands on drug and metabolite formation. Appl. Catal. A. 2016, 510, 204–215. [Google Scholar] [CrossRef]
- Maeno, S.; Zhu, Q.; Sasaki, M.; Miyamoto, T.; Fukushima, M. Monopersulfate oxidation of tetrabromobisphenol A by an iron(III)-phthalocyaninetetrasulfate catalyst coordinated to imidazole functionalized silica particles. J. Mol. Catal. A Chem. 2015, 400, 56–63. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Mortazavi-Manesh, A. Immobilized manganese porphyrin on functionalized magnetic nanoparticles via axial ligation: Efficient and recyclable nanocatalyst for oxidation reactions. J. Coord. Chem. 2015, 1–14. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Pintus, M.; Rinaldi, A.C.; Sanjust, E. Degradation of textile dyes using immobilized lignin peroxidase-like metalloporphines under mild experimental conditions. Chem. Cent. J. 2012, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagaki, S.; Ferreira, G.; Ucoski, G.; Dias de Freitas Castro, K. Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules 2013, 18, 7279–7308. [Google Scholar] [CrossRef] [PubMed]
- Guedes, A.A.; Smith, J.R.L.; Nascimento, O.R.; Guedes, D.F.C.; Assis, M.d.D. Catalytic activity of halogenated iron porphyrins in alkene and alkane oxidations by iodosylbenzene and hydrogen peroxide. J. Brazil. Chem. Soc. 2005, 16, 835–843. [Google Scholar] [CrossRef]
- Karimipour, G. Manganese Porphyrin Supported on Multiwalled Carbon Nanotube (MWCN) as Solid Catalyst for Alkene Epoxidation. Int. J. Chem. Eng. Appl. 2014, 5, 194–197. [Google Scholar] [CrossRef]
- Zollinger, H. Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments; Wiley: New York, NY, USA, 2003. [Google Scholar]
- Carliell, C.M.; Barclay, S.J.; Naidoo, N.; Buckley, C.A.; Mulholland, D.A.; Senior, E. Anaerobic decolorisation of reactive dyes in conventional sewage treatment processes. Water SA 1994, 20, 341–344. [Google Scholar]
- Walthall, W.K.; Stark, J.D. The acute and chronic toxicity of two xanthene dyes, fluorescein sodium salt and phloxine B, to Daphnia pulex. Environ. Pollut. 1999, 104, 207–215. [Google Scholar] [CrossRef]
- Zucca, P.; Cocco, G.; Sollai, F.; Sanjust, E. Fungal laccases as tools for biodegradation of industrial dyes. Biocatalysis 2015, 1, 82–108. [Google Scholar] [CrossRef] [Green Version]
- Platzek, T.; Lang, C.; Grohmann, G.; Gi, U.S.; Baltes, W. Formation of a carcinogenic aromatic amine from an azo dye by human skin bacteria in vitro. Hum. Exp. Toxicol. 1999, 18, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Vandevivere, P.C.; Bianchi, R.; Verstraete, W. Treatment and reuse of wastewater from the textile wet-processing industry: Review of emerging technologies. J. Chem. Technol. Biotechnol. 1998, 72, 289–302. [Google Scholar] [CrossRef]
- Robinson, T.; McMullan, G.; Marchant, R.; Nigam, P. Remediation of dyes in textile effluent: A critical review on current treatment technologies with a proposed alternative. Bioresour. Technol. 2001, 77, 247–255. [Google Scholar] [CrossRef]
- Rai, H.S.; Bhattacharyya, M.S.; Singh, J.; Bansal, T.K.; Vats, P.; Banerjee, U.C. Removal of dyes from the effluent of textile and dyestuff manufacturing industry: A review of emerging techniques with reference to biological treatment. Crit. Rev. Environ. Sci. Technol. 2005, 35, 219–238. [Google Scholar] [CrossRef]
- Waring, D.R.; Hallas, G. The Chemistry and Application of Dyes; Springer: Berlin, Germany, 2013. [Google Scholar]
- Ţurcaş, C.V.; Sebe, I. Azo dyes complexes. Synthesis and tinctorial properties. UPB Sci. Bull. Ser. B Chem. Mater. Sci. 2012, 74, 109–118. [Google Scholar]
- DeVito, S.C. Predicting Azo Dye Toxicity. Crit. Rev. Environ. Sci. Technol. 1993, 23, 249–324. [Google Scholar]
- Popli, S.; Patel, U.D. Destruction of azo dyes by anaerobic-aerobic sequential biological treatment: A review. Int. J. Environ. Sci. Technol. 2015, 12, 405–420. [Google Scholar] [CrossRef]
- Sendelbach, L.E. A review of the toxicity and carcinogenicity of anthraquinone derivatives. Toxicology 1989, 57, 227–240. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar, N.; Nam, S.; Skaltsounis, A.L. A colorful history: the evolution of indigoids. Prog. Chem. Org. Nat. Prod. 2014, 99, 69–145. [Google Scholar] [PubMed]
- Cooksey, C. Tyrian purple: The first four thousand years. Sci. Prog. 2013, 96, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Colour Index, 3rd ed.; The Society of Dyers and Colourists: Bradford, UK, 1971; Volume 4.
- Pardal, A.C.; Ramos, S.S.; Santos, L.; Almeida, P. Synthesis and fixation of aminocyanines to microcrystalline cellulose using cyanuric chloride as a cross-linking agent. Color. Technol. 2001, 117, 43–48. [Google Scholar] [CrossRef]
- Mujumdar, R.B.; Ernst, L.A.; Mujumdar, S.R.; Lewis, C.J.; Waggoner, A.S. Cyanine dye labeling reagents: Sulfoindocyanine succinimidyl esters. Bioconj. Chem. 1993, 4, 105–111. [Google Scholar] [CrossRef]
- Griffiths, J. Colour and Constitution of Organic Molecules; Academic Press: San Diego, CA, USA, 1976. [Google Scholar]
- Rayati, S.; Sheybanifard, Z. Catalytic activity of Mn(III) and Fe(III) porphyrins supported onto multi-walled carbon nanotubes in the green oxidation of organic dyes with hydrogen peroxide: A comparative study. J. Iran. Chem. Soc. 2016, 13, 541–546. [Google Scholar] [CrossRef]
- Agnemo, R.; Gellersted, G. The Reactions of Lignin with Alkaline Hydrogen Peroxide. Part II. Factors Influencing the Decomposition of Phenolic Structures. Acta Chem. Scand. 1979, 33B, 337–342. [Google Scholar] [CrossRef]
- Rao, P.; Hayon, E. Redox potentials of free radicals. IV. Superoxide and hydroperoxy radicals. O2− and HO2. J. Phys. Chem. 1975, 79, 397–402. [Google Scholar] [CrossRef]
- Wang, S.-S.; Yang, G.-Y. Recent Advances in Polyoxometalate-Catalyzed Reactions. Chem. Rev. 2015, 115, 4893–4962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, M.; Jiang, Y.; Hu, M.; Li, S.; Zhai, Q. Efficient decolorization/degradation of aqueous azo dyes using buffered H2O2 oxidation catalyzed by a dosage below ppm level of chloroperoxidase. Chem. Eng. J. 2012, 191, 236–242. [Google Scholar] [CrossRef]
- Hodges, G.R.; Lindsay Smith, J.R.; Oakes, J. Mechanism of oxidation of azo dyes by a sterically hindered anionic oxoiron(IV) porphyrin in aqueous solution. J. Chem. Soc. Perkin Trans. 2 1998, 617–627. [Google Scholar] [CrossRef]
- Hodges, G.R.; Smith, J.R.L.; Oakes, J. The oxidation of azo dyes by peroxy acids and tert-butyl hydroperoxide in aqueous solution catalysed by iron(III) 5,10,15,20-tetra(2,6-dichloro-3-sulfonatophenyl)porphyrin: Product studies and mechanism. J. Chem. Soc. Perkin Trans. 2 1999, 1943–1952. [Google Scholar] [CrossRef]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A model ‘rebound’ mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Pasti-Grigsby, M.B.; Paszczynski, A.; Goszczynski, S.; Crawford, D.L.; Crawford, R.L. Influence of aromatic substitution patterns on azo dye degradability by Streptomyces spp. and Phanerochaete chrysosporium. Appl. Environ. Microbiol. 1992, 58, 3605–3613. [Google Scholar] [PubMed]
- Jing, J.; Zhang, Y.; Feng, J.; Li, W.; Yu, W.W. Facile preparation and high performance of magnetically separable metalloporphyrin. Chem. Eng. J. 2015, 263, 385–391. [Google Scholar] [CrossRef]
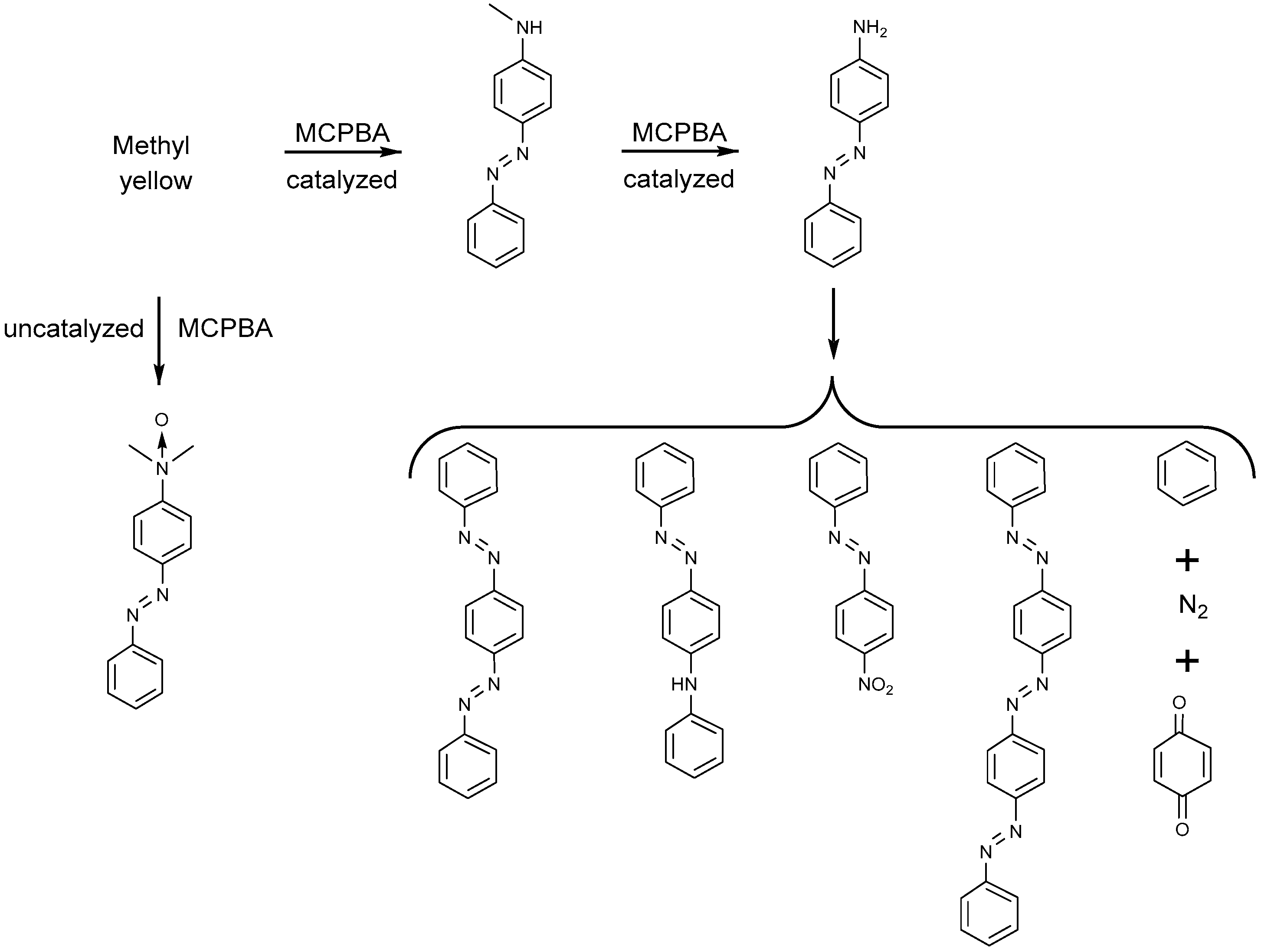
- Emmert Iii, F.L.; Thomas, J.; Hon, B.; Gengenbach, A.J. Metalloporphyrin catalyzed oxidation of methyl yellow and related azo compounds. Inorg. Chim. Acta 2008, 361, 2243–2251. [Google Scholar] [CrossRef]
- Ueno, K.; Akiyoshi, S. Polyazobenzenes. II. Synthesis and ultraviolet absorption spectra of polyazobenzenes containing nitro, amino and hydroxyl groups. J. Am. Chem. Soc. 1954, 76, 3667–3670. [Google Scholar] [CrossRef]
- Ueno, K.; Akiyoshi, S. Kinetic study on the condensation reaction of aniline and nitrosobenzenes. J. Am. Chem. Soc. 1954, 76, 3670–3672. [Google Scholar] [CrossRef]
- Ogata, Y.; Takagi, Y. Kinetics of the condensation of anilines with nitrosobenzenes to form azobenzenes. J. Am. Chem. Soc. 1958, 80, 3591–3595. [Google Scholar] [CrossRef]
- Yunes, R.A.; Terenzani, A.J.; Do Amaral, L. Kinetics and mechanism for azobenzene formation. J. Am. Chem. Soc. 1975, 97, 368–373. [Google Scholar] [CrossRef]
- Barros, V.P.; Faria, A.L.; MacLeod, T.C.O.; Moraes, L.A.B.; Assis, M.D. Ironporphyrin immobilized onto montmorillonite as a biomimetical model for azo dye oxidation. Int. Biodeterior. Biodegrad. 2008, 61, 337–344. [Google Scholar] [CrossRef]
- Zhao, X.; Hardin, I.R.; Hwang, H.M. Biodegradation of a model azo disperse dye by the white rot fungus Pleurotus ostreatus. Int. Biodeterior. Biodegrad. 2006, 57. [Google Scholar] [CrossRef]
- Bhirud, R.G.; Srisankar, E.V.; Narayan, K.S. Oxidation of dyes by manganese tetraphenyl porphyrin activated peroxy bleach. Proc. Indian Acad. Sci. Chem. Sci. 1991, 103, 83–93. [Google Scholar]
- Nango, M.; Iwasaki, T.; Takeuchi, Y.; Kurono, Y.; Tokuda, J.; Oura, R. Peroxide decoloration of azo dyes catalyzed by polyethylene glycol-linked manganese halogenated porphyrins. Langmuir 1998, 14, 3272–3278. [Google Scholar] [CrossRef]
- Nakamura, J.; Oura, R.; Nango, M. Peroxide decoloration of azo dye catalyzed by manganese porphyrin derivatives in non-aqueous solvent. Text. Res. J. 2008, 78, 1080–1086. [Google Scholar] [CrossRef]
- Tokuda, J.; Ohura, R.; Iwasaki, T.; Takeuchi, Y.; Kashiwada, A.; Nango, M. Decoloration of azo dyes by hydrogen peroxide catalyzed by water-soluble manganese porphyrins. Text. Res. J. 1999, 69, 956–960. [Google Scholar] [CrossRef]
- Serra, A.C.; Docal, C.; Gonsalves, A.M.D.A.R. Efficient azo dye degradation by hydrogen peroxide oxidation with metalloporphyrins as catalysts. J. Mol. Catal. A Chem. 2005, 238, 192–198. [Google Scholar] [CrossRef]
- Habibi, M.H.; Tangestaninejad, S.; Mirkhani, V. Efficient Catalytic Oxidation of Primary Aromatic Amines to Azo Derivatives by Manganese(III) Tetraphenylporphyrin. J. Chem. Res. 1998, 648–649. [Google Scholar] [CrossRef]
- Häger, M.; Holmberg, K.; Rocha Gonsalves, A.M.d.A.; Serra, A.C. Oxidation of azo dyes in oil-in-water microemulsions catalyzed by metalloporphyrins in presence of lipophilic acids. Colloids Surf. Physicochem. Eng. Asp. 2001, 183–185, 247–257. [Google Scholar] [CrossRef]
- Thompson, K.M.; Griffith, W.P.; Spiro, M. Mechanism of bleaching by peroxides. Part 2—Kinetics of bleaching of alizarin and crocetin by hidrogen peroxide at high pH. J. Chem. Soc. Faraday Trans. 1993, 89, 4035–4043. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Q.Y.; Guo, Y.H.; Hu, C.W.; Wang, E. Efficient degradation of dye pollutants on nanoporous polyoxotungstate-anatase composite under visible-light irradiation. J. Mol. Catal. A Chem. 2005, 225, 203–212. [Google Scholar] [CrossRef]
- Cheng, C.; Li, X.; Ma, W.; Zhao, J.; Hidaka, H.; Serpone, N. Effect of transition metal ions on the TiO2-assisted photodegradation of dyes under visible irradiation: a probe for the interfacial electron transfer process and reaction mechanism. J. Phys. Chem. B 2002, 106, 318–324. [Google Scholar] [CrossRef]
- Sivalingam, G.; Nagaveni, K.; Hegde, M.S.; Madras, G. Photocatalytic degradation of various dyes by combustion synthesized nano anatase TiO2. Appl. Catal. B. 2003, 45, 23–38. [Google Scholar] [CrossRef]
- Gao, J.Z.; Yu, J.; Lu, Q.F.; He, X.Y.; Yang, W.; Li, Y.; Pu, L.M.; Yang, Z.M. Decoloration of alizarin red S in aqueous solution by glow discharge electrolysis. Dyes Pigments 2008, 76, 47–52. [Google Scholar] [CrossRef]
- Faouzi, A.M.; Nasr, B.; Abdellatif, G. Electrochemical degradation of anthraquinone dye Alizarin Red S by anodic oxidation on boron-doped diamond. Dyes Pigments 2007, 73, 86–89. [Google Scholar] [CrossRef]
- Córdoba, A.; Magario, I.; Ferreira, M.L. Modified chitosan as an economical support for hematin: Application in the decolorization of anthraquinone and azo dyes. J. Chem. Technol. Biotechnol. 2015, 90, 1665–1676. [Google Scholar] [CrossRef]
- Yao, Y.; Mao, Y.; Huang, Q.; Wang, L.; Huang, Z.; Lu, W.; Chen, W. Enhanced decomposition of dyes by hemin-ACF with significant improvement in pH tolerance and stability. J. Hazard. Mater. 2014, 264, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Ciric-Marjanovic, G.; Blinova, N.V.; Trchova, M.; Stejskal, J. Chemical oxidative polymerization of safranines. J. Phys. Chem. B 2007, 111, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Ucoski, G.M.; Machado, G.S.; Silva, G.D.F.; Nunes, F.S.; Wypych, F.; Nakagaki, S. Heterogeneous oxidation of the dye Brilliant Green with H2O2 catalyzed by supported manganese porphyrins. J. Mol. Catal. A Chem. 2015, 408, 123–131. [Google Scholar] [CrossRef]
- Archibald, F.S. A new assay for lignin-type peroxidases employing the dye Azure B. Appl. Environ. Microbiol. 1992, 58, 3110–3116. [Google Scholar] [PubMed]
- Su, R.; Sun, J.; Sun, Y.; Deng, K.; Cha, D.; Wang, D. Oxidative degradation of dye pollutants over a broad pH range using hydrogen peroxide catalyzed by FePz(dtnCl2)4. Chemosphere 2009, 77, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Umile, T.P.; Groves, J.T. Catalytic generation of chlorine dioxide from chlorite using a water-soluble manganese porphyrin. Angew. Chem. Int. Edit. 2011, 50, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, D.; Groves, J.T. Modeling the haloperoxidases: Reversible oxygen atom transfer between bromide ion and an oxo-Mn(V) porphyrin. J. Inorg. Biochem. 2007, 101, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento Brito, C.; Da Silva, D.R.; Garcia-Segura, S.; de Moura, D.C.; Martínez-Huitle, C.A. Indirect electrochemical oxidation of reactive blue 19 dye as a model organic substrate: Role of anode material and oxidants electrochemically generated. J. Electrochem. Soc. 2016, 163, E62–E69. [Google Scholar] [CrossRef]
- Hu, M.R.; Chao, Y.P.; Zhang, G.Q.; Xue, Z.Q.; Qian, S. Laccase-mediator system in the decolorization of different types of recalcitrant dyes. J. Ind. Microbiol. Biotechnol. 2009, 36, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Pardo, I.; Camarero, S. Laccase engineering by rational and evolutionary design. Cell. Mol. Life Sci. 2015, 72, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Kalsoom, U.; Ashraf, S.S.; Meetani, M.A.; Rauf, M.A.; Bhatti, H.N. Mechanistic study of a diazo dye degradation by Soybean Peroxidase. Chem. Cent. J. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Terres, J.; Battisti, R.; Andreaus, J.; de Jesus, P.C. Decolorization and degradation of Indigo Carmine dye from aqueous solution catalyzed by horseradish peroxidase. Biocatal. Biotransfor. 2014, 32, 64–73. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Olianas, A.; Maccioni, S.; Sollai, F.; Sanjust, E. Induction, purification, and characterization of a laccase isozyme from Pleurotus sajor-caju and the potential in decolorization of textile dyes. J. Mol. Catal. B Enzym. 2011, 68, 216–222. [Google Scholar] [CrossRef]
- Bilal, M.; Asgher, M.; Shahid, M.; Bhatti, H.N. Characteristic Features and Dye Degrading Capability of Agar-Agar gel Immobilized Manganese Peroxidase. Int. J. Biol. Macromol. 2016, 86, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.; Pickard, M.A.; Vazquez-Duhalt, R. Industrial dye decolorization by laccases from ligninolytic fungi. Curr. Microbiol. 1999, 38, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Littarru, M.; Rescigno, A.; Sanjust, E. Cofactor recycling for selective enzymatic biotrasformation of cinnamaldehyde to cinnamyl alcohol. Biosci. Biotechnol. Biochem. 2009, 73, 1224–1226. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Turano, P.; Tien, M.; Kirk, T.K. Proton NMR investigation into the basis for the relatively high redox potential of lignin peroxidase. Proc. Natl. Acad. Sci. USA 1991, 88, 6956–6960. [Google Scholar] [CrossRef] [PubMed]
- Kersten, P.J.; Kalyanaraman, B.; Hammel, K.E.; Reinhammars, B.; Kirk, T.K. Comparison of lignin peroxidase, horseradish peroxidase and laccase in the oxidation of methoxybenzenes. Biochem. J. 1990, 268, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Asgher, M.; Kamal, S.; Iqbal, H.M.N. Improvement of Catalytic Efficiency, Thermo-stability and Dye Decolorization Capability of Pleurotus ostreatus IBL-02 laccase by Hydrophobic Sol Gel Entrapment. Chem. Cent. J. 2012, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.; Sanromán, M.T.; Moldes, D. Recent developments and applications of immobilized laccase. Biotechnol. Adv. 2013, 31, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Sollai, F.; Zucca, P.; Sanjust, E.; Steri, D.; Rescigno, A. Umbelliferone and esculetin: Inhibitors or substrates for polyphenol oxidases? Biol. Pharm. Bull. 2008, 31, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Riva, S. Laccases: blue enzymes for green chemistry. Trends Biotechnol. 2006, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Salis, A.; Pisano, M.; Monduzzi, M.; Solinas, V.; Sanjust, E. Laccase from Pleurotus sajor-caju on functionalised SBA-15 mesoporous silica: Immobilisation and use for the oxidation of phenolic compounds. J. Mol. Catal. B Enzym. 2009, 58, 175–180. [Google Scholar] [CrossRef]
- Singh Arora, D.; Kumar Sharma, R. Ligninolytic fungal laccases and their biotechnological applications. Appl. Biochem. Biotechnol. 2010, 160, 1760–1788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Z.; Chen, H.; Zong, Y. Removal of 2,4-dichlorophenol by chitosan-immobilized laccase from Coriolus versicolor. Biochem. Eng. J. 2009, 45, 54–59. [Google Scholar] [CrossRef]
- Pereira, L.; Coelho, A.V.; Viegas, C.A.; Santos, M.M.C.d.; Robalo, M.P.; Martins, L.O. Enzymatic biotransformation of the azo dye Sudan Orange G with bacterial CotA-laccase. J. Biotechnol. 2009, 139, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.; Ragauskas, A. Laccase-mediator biobleaching applied to a direct yellow dyed paper. Biotechnol. Prog. 2004, 20, 1893–1896. [Google Scholar] [CrossRef] [PubMed]
- Bayramoǧlu, G.; Yilmaz, M.; Arica, M.Y. Reversible immobilization of laccase to poly(4-vinylpyridine) grafted and Cu(II) chelated magnetic beads: Biodegradation of reactive dyes. Bioresour. Technol. 2010, 101, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.P.; Ramsay, J.A. Dye decolorization and detoxification by laccase immobilized on porous glass beads. Bioresour. Technol. 2010, 101, 2230–2235. [Google Scholar] [CrossRef] [PubMed]
- Chander, M.; Arora, D.S.; Bath, H.K. Biodecolourisation of some industrial dyes by white-rot fungi. J. Ind. Microbiol. Biotechnol. 2004, 31, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, R.O.; Tavares, A.P.M.; Ferreira, L.A.; Loureiro, J.M.; Boaventura, R.A.R.; Macedo, E.A. Modeling the discoloration of a mixture of reactive textile dyes by commercial laccase. Bioresour. Technol. 2009, 100, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Milagres, A.M.F.; Mussatto, S.I. Reactive dyes and textile effluent decolorization by a mediator system of salt-tolerant laccase from Peniophora cinerea. Sep. Purif. Technol. 2014, 135, 183–189. [Google Scholar] [CrossRef]
- Wells, A.; Teria, M.; Eve, T. Green oxidations with laccase-mediator systems. Biochem. Soc. Trans. 2006, 34, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; da Costa, R.S.; Yokoyama, L.; Alhadeff, E.M.; Teixeira, L.A.C. Evaluation of Textile Dye Degradation Due to the Combined Action of Enzyme Horseradish Peroxidase and Hydrogen Peroxide. Appl. Biochem. Biotechnol. 2014, 174, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, S.; García Einschlag, F.S.; Rueda, E.H.; Ferreira, M.L. Horseradish peroxidase and hematin as biocatalysts for alizarin degradation using hydrogen peroxide. Ind. Eng. Chem. Res. 2010, 49, 6745–6752. [Google Scholar] [CrossRef]
- Kobayashi, S.; Nakano, M.; Kimura, T.; Schaap, A.P. On the mechanism of the peroxidase-catalyzed oxygen-transfer reaction. Biochemistry 1987, 26, 5019–5022. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Valade, A.G.; Combourieu, B.; Mielgo, I.; Bouchon, B.; Lema, J.M. Mechanism of enzymatic degradation of the azo dye Orange II determined by ex situ 1H nuclear magnetic resonance and electrospray ionization-ion trap mass spectrometry. Anal. Biochem. 2004, 335, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Lutton, J.D.; Abraham, N.G.; Drummond, G.S.; Levere, R.D.; Kappas, A. Zinc porphyrins: Potent inhibitors of hematopoieses in animal and human bone marrow. Proc. Natl. Acad. Sci. USA 1997, 94, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Muttaqin, F.Z.; Ibrahim, S.; Muthalib, A.; Tjahjono, D.H. Toxicity Prediction of meso-5,15-di[3,4-bis(carboxymethylenoxy)phenyl]porphyrin and meso-5,15-di[3,4-bis(carboxymethylenoxy)phenyl],10,20-diphenyl porphyrin. In Proceedings of the 3rd International Conference on Computation for Science and Technology, Bali, Indonesia, 23–25 September 2014; Volume 2.
- Nyarko, E.; Hara, T.; Grab, D.J.; Habib, A.; Kim, Y.; Nikolskaia, O.; Fukuma, T.; Tabata, M. In vitro toxicity of palladium(II) and gold(III) porphyrins and their aqueous metal ion counterparts on Trypanosoma brucei brucei growth. Chem. Biol. Interact. 2004, 148, 19–25. [Google Scholar] [CrossRef] [PubMed]














| Parameter | Range |
|---|---|
| pH | 5–13 |
| Temperature (°C) | 20–60 |
| Conductivity (mS/cm) | 0.1–120 |
| Biological Oxygen Demand (ppm) | 14–6000 |
| Chemical Oxygen Demand (ppm) | 150–90,000 |
| Total Suspended Solids (ppm) | 100–25,000 |
| Total Dissolved Solids (ppm) | 1800–30,000 |
| Total Kjeldahl Nitrogen (ppm) | 70–160 |
| Turbidity (NTU) | 0–200 |
| Color (Pt-Co) | 50–2500 |
| Absorbance | 0.9–200 |
| Acronyms | Designations | Ar | R |
|---|---|---|---|
| TPP | 5,10,15,20-tetraphenylporphyrin |  | 8 H |
| TDCPP | 5,10,15,20-tetrakis(2,6-dichlorophenyl)porphyrin |  | 8 H |
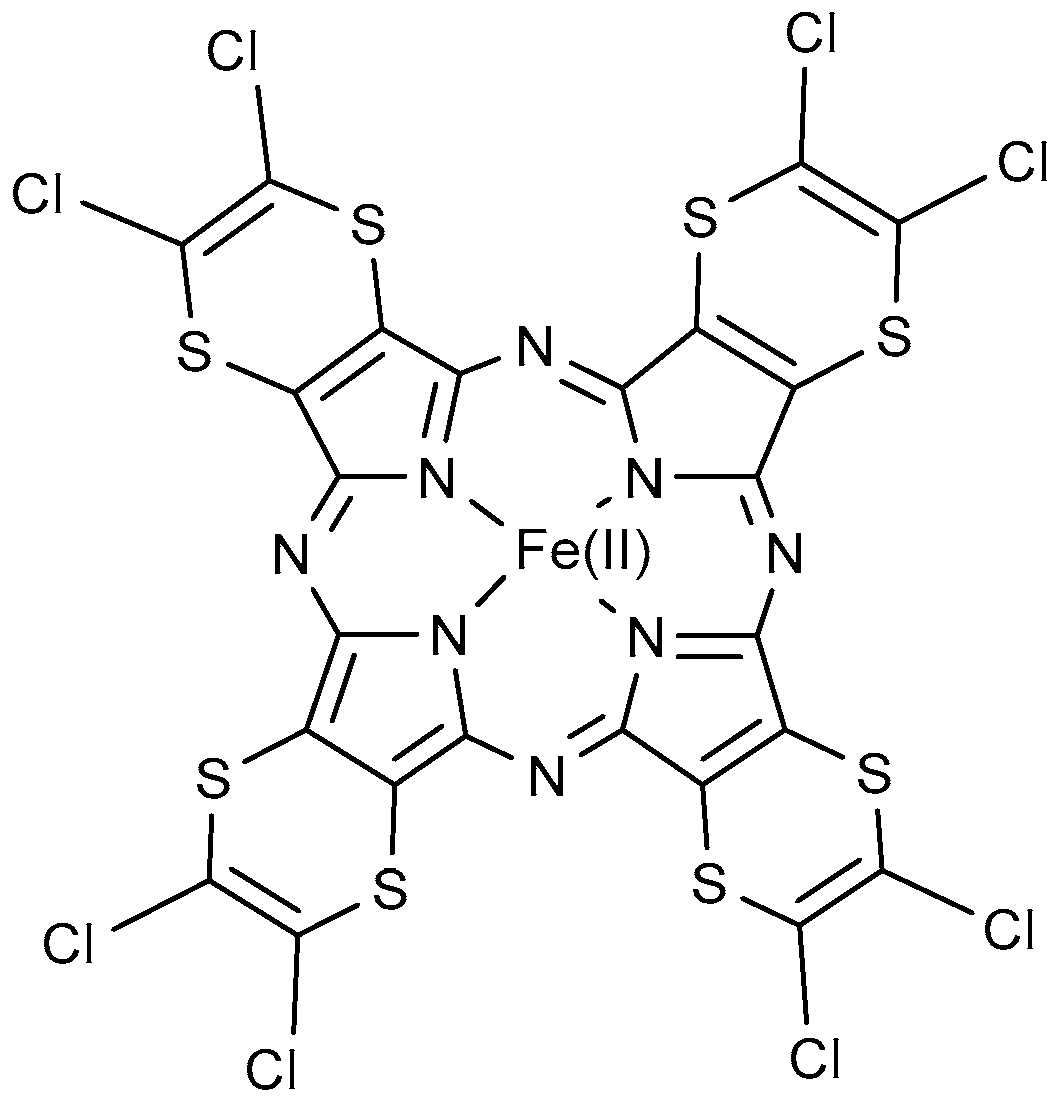
| TDCPCl8P | β-octachloro-5,10,15,20-tetrakis(2,6-dichlorophenyl)porphyrin | | 8 Br |
| TPFPP | 5,10,15,20-tetrakis(pentafluoro-phenyl)porphyrin |  | 8 H |
| TPFPF8P | β-octafluoro-5,10,15,20-tetrakis(pentafluorophenyl)porphyrin | | 8 Br |
| TCPP | 5,10,15,20-tetrakis(4-carboxy-phenyl)porphyrin |  | 8 H |
| TMCPP | 5,10,15,20-tetrakis(4-methoxy-carbonylphenyl)porphyrin |  | 8 H |
| THPP | 5,10,15,20-tetrakis(4-hydroxy-phenyl)porphyrin |  | 8H |
| TDCSPP | 5,10,15,20-tetrakis(2,6-dichloro-3-sulfonatophenyl)porphyrin |  | 8 H |
| T4MPyP | 5,10,15,20-tetrakis(N-methyl-pyridinium-4-yl)porphyrin |  | 8 H |
| TBr3MPP | 5,10,15,20-tetrakis(2,4,6-tribromo-3-methoxyphenyl)porphyrin |  | 8 H |
| TSPP | 5,10,15,20-tetrakis(4-sulfonato-phenyl)porphyrin |  | 8 H |
| Br8T3MPyP | β-octabromo-5,10,15,20-tetrakis-(N-methylpyridinium-3-yl)porphyrin |  | 8 Br |
| TDMImP | 5,10,15,20-tetrakis(1,3-dimethyl-imidazolium-2-yl)porphyrin |  | 8 H |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Interaction | Method of Immobilization | Advantages | Disadvantages |
|---|---|---|---|
| Physical inclusion | Encapsulation/entrapment | Minimization of chemical modification of the catalyst | Leakage |
| No emulation of peroxidases active site | |||
| Involvement of toxic and costly reagents | |||
| Weak chemical interaction | Adsorption | Minimization of chemical modification of the catalyst | Usually the interaction catalyst/support is weak |
| Easy method | Leakage | ||
| No emulation of peroxidases active site | |||
| Ion exchange | Minimization of chemical modification of the catalyst | Usually the interaction catalyst/support is weak | |
| Leakage | |||
| No emulation of peroxidases active site | |||
| Strong chemical interaction | Covalent binding | The interaction catalyst/support is very strong | Chemical modification of the catalyst is possible |
| Minimal catalyst leakage | No emulation of peroxidases active site | ||
| Axial coordination | Real emulation of peroxidases active site | Axial bis-ligation of the catalysts | |
| Increased stability and activity |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zucca, P.; Neves, C.M.B.; Simões, M.M.Q.; Neves, M.D.G.P.M.S.; Cocco, G.; Sanjust, E. Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes. Molecules 2016, 21, 964. https://doi.org/10.3390/molecules21070964
Zucca P, Neves CMB, Simões MMQ, Neves MDGPMS, Cocco G, Sanjust E. Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes. Molecules. 2016; 21(7):964. https://doi.org/10.3390/molecules21070964
Chicago/Turabian StyleZucca, Paolo, Cláudia M. B. Neves, Mário M. Q. Simões, Maria Da Graça P. M. S. Neves, Gianmarco Cocco, and Enrico Sanjust. 2016. "Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes" Molecules 21, no. 7: 964. https://doi.org/10.3390/molecules21070964