
Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis sydowiana and Their 20S Proteasome Inhibitory Activities

Abstract
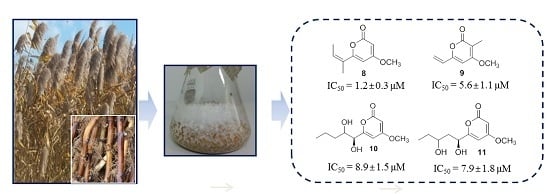
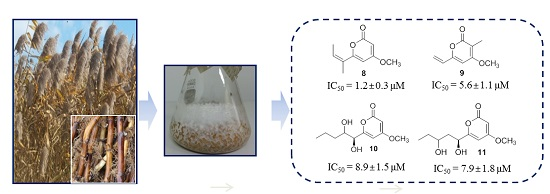
:
1. Introduction
2. Results and Discussion
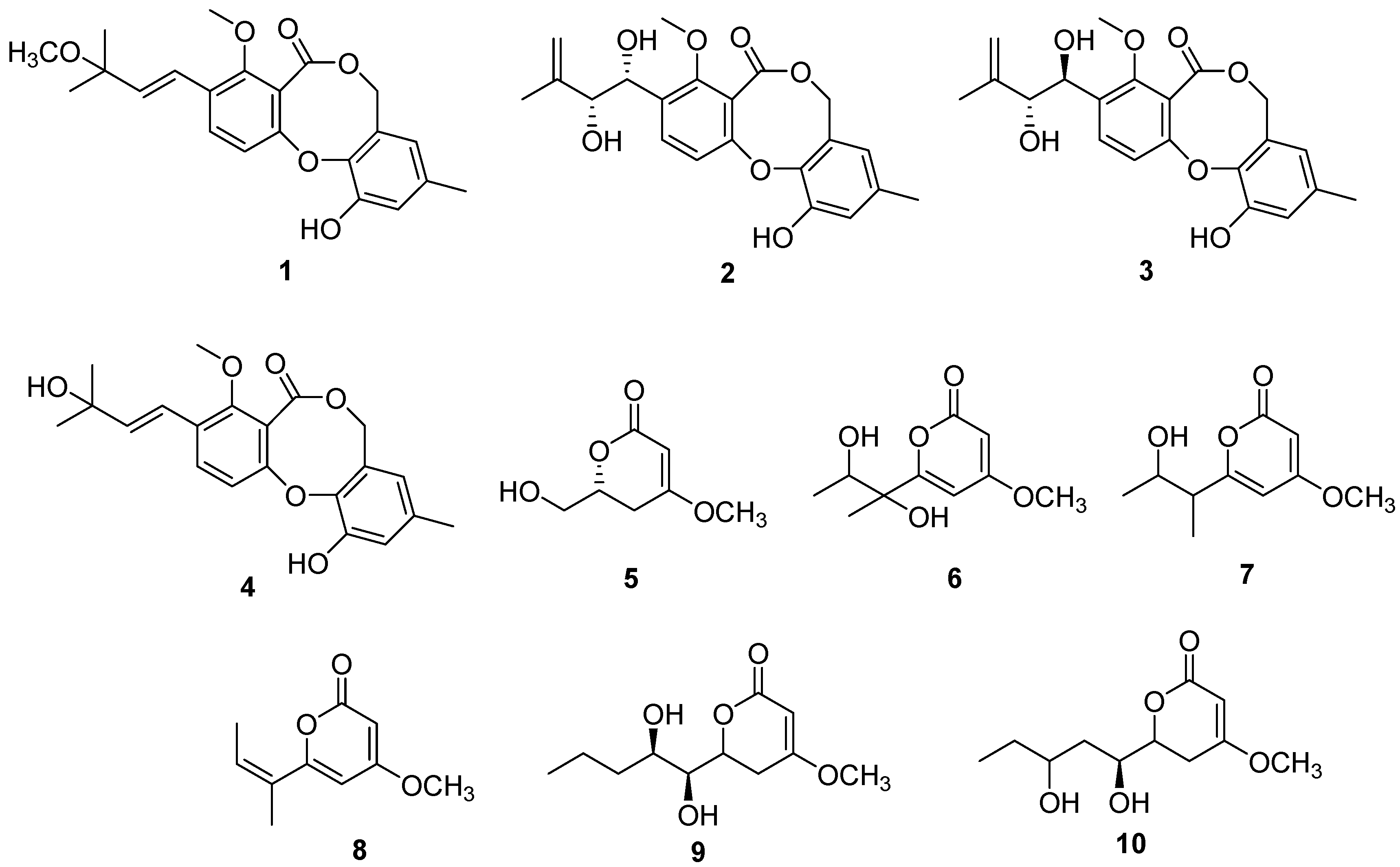
2.1. Structure Determination
2.2. 20S Proteasome Inhibitory Activities
3. Experimental Section
3.1. General Methods
3.2. Cultures
3.3. Extraction and Isolation
3.4. Proteasome Inhibition Assay
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2012, 16, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Franke, W.W.; Kleinschmidt, J.A. Distinct 19 and 20S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 1994, 269, 7709–7718. [Google Scholar] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Gille, C.; Goede, A.; Schlöetelburg, C.; Preissner, R.; Kloetzel, P.M.; Göbel, U.B.; Frömmel, C. A comprehensive view on proteasomal sequences: Implications for the evolution of the proteasome. J. Mol. Biol. 2003, 326, 1437–1448. [Google Scholar] [CrossRef]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004, 5, 417–421. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug. Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H. 20S proteasome inhibitory activity of flavonoids isolated from Spatholobus suberectus. Phytother. Res. 2011, 25, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.X.; Zou, W.X. Endophytes: A rich source of functional metabolites. Nat. Prod. Rep. 2001, 18, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.; Daisy, B.; Castillo, U.; Harper, J. Natural products from endophytic microorganisms. J. Nat. Prod. 2004, 67, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.; Daisy, B. Bioprospecting for microbial endophytes and their natural products. Microbiol. Mol. Biol. Rev. 2003, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ebada, S.S.; Proskach, E.P. Pestalotiopsis, a highly creative genus: Chemistry and bioactivity of secondary metabolites. Fungal Divers. 2010, 44, 15–31. [Google Scholar] [CrossRef]
- Strobel, G.; Yang, X.; Sears, J.; Kramer, R.; Sidhu, R.S.; Hess, W.M. Taxol from Pestalotiopsis microspora, an endophytic fungus of Taxus wallachiana. Microbiology 1996, 142, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Strobel, G.A.; Sidhu, R.; Hess, W.M.; Ford, F. Endophytic taxol-producing fungi from bald cypress, Taxodium distichum. J. Microbiol. 1996, 142, 2223–2226. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Kaneko, T.; Koshino, H.; Esumi, Y.; Uzawa, J.; Sugawara, F. Penicillides from Penicillium sp. isolated from Taxus cuspidata. Nat. Prod. Lett. 2000, 14, 477–484. [Google Scholar] [CrossRef]
- Pospíšil, J.; Markó, I.E. Metathesis-based synthesis of 3-methoxy α,β-unsaturated lactones: Total synthesis of (R)-kavain and of the C1-C6 fragment of jerangolid D. Tetrahedron Lett. 2008, 49, 1523–1526. [Google Scholar] [CrossRef]
- Xu, J.; Aly, A.H.; Wray, V.; Proksch, P. Polyketide derivatives of endophytic fungus Pestalotiopsis sp. isolated from the Chinese mangrove plant Rhizophora mucronata. Tetrahedron Lett. 2011, 52, 21–25. [Google Scholar]
- Mcgahren, W.J.; Ellestad, G.A.; Morton, G.O.; Kunstmann, M.P. A new fungal lactone, LL-P88Ob, and a New Pyrone, LL-P880g, from a Penicillium sp. J. Org. Chem. 1973, 38, 3542–3544. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Qi, Y.X.; Liu, S.C.; Guo, L.D.; Chen, X.L. Photipyrones A and B, new pyrone derivatives from the plant endophytic fungus Pestalotiopsis photiniae. J. Antibiot. 2012, 65, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Hirota, H.; Imachi, M.; Fujimuro, M.; Onuki, H.; Ohta, T.; Yokosawa, H. Himeic acid A: A new ubiquitin-activating enzyme inhibitor isolated from a marine-derived fungus, Aspergillus sp. Bioorg. Med. Chem. Lett. 2005, 15, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Gräwert, M.A.; Groll, M. Exploiting nature’s rich source of proteasome inhibitors as starting points in drug development. Chem. Commun. 2012, 48, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.

{kind=link}
{kind=link}
| Compound | IC50 a (μM) | Compound | IC50 a (μM) |
|---|---|---|---|
| 1 | 30.5 ± 1.5 | 7 | >100 |
| 2 | 18.5 ± 4.2 | 8 | 1.2 ± 0.3 |
| 3 | 12.4 ± 1.1 | 9 | 8.9 ± 1.5 |
| 4 | >100 | 10 | 7.9 ± 1.8 |
| 5 | 20.5 ± 2.3 | Epoxomicin b | 0.072 |
| 6 | >100 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X.; Kim, S.; Liu, C.; Shim, S.H. Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis sydowiana and Their 20S Proteasome Inhibitory Activities. Molecules 2016, 21, 944. https://doi.org/10.3390/molecules21070944
Xia X, Kim S, Liu C, Shim SH. Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis sydowiana and Their 20S Proteasome Inhibitory Activities. Molecules. 2016; 21(7):944. https://doi.org/10.3390/molecules21070944
Chicago/Turabian StyleXia, Xuekui, Soonok Kim, Changheng Liu, and Sang Hee Shim. 2016. "Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis sydowiana and Their 20S Proteasome Inhibitory Activities" Molecules 21, no. 7: 944. https://doi.org/10.3390/molecules21070944
APA StyleXia, X., Kim, S., Liu, C., & Shim, S. H. (2016). Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis sydowiana and Their 20S Proteasome Inhibitory Activities. Molecules, 21(7), 944. https://doi.org/10.3390/molecules21070944