
In Situ Investigation of a Self-Accelerated Cocrystal Formation by Grinding Pyrazinamide with Oxalic Acid

Abstract
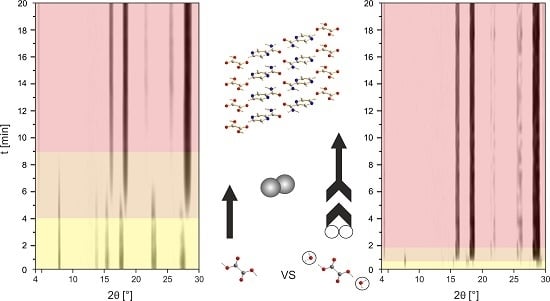
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
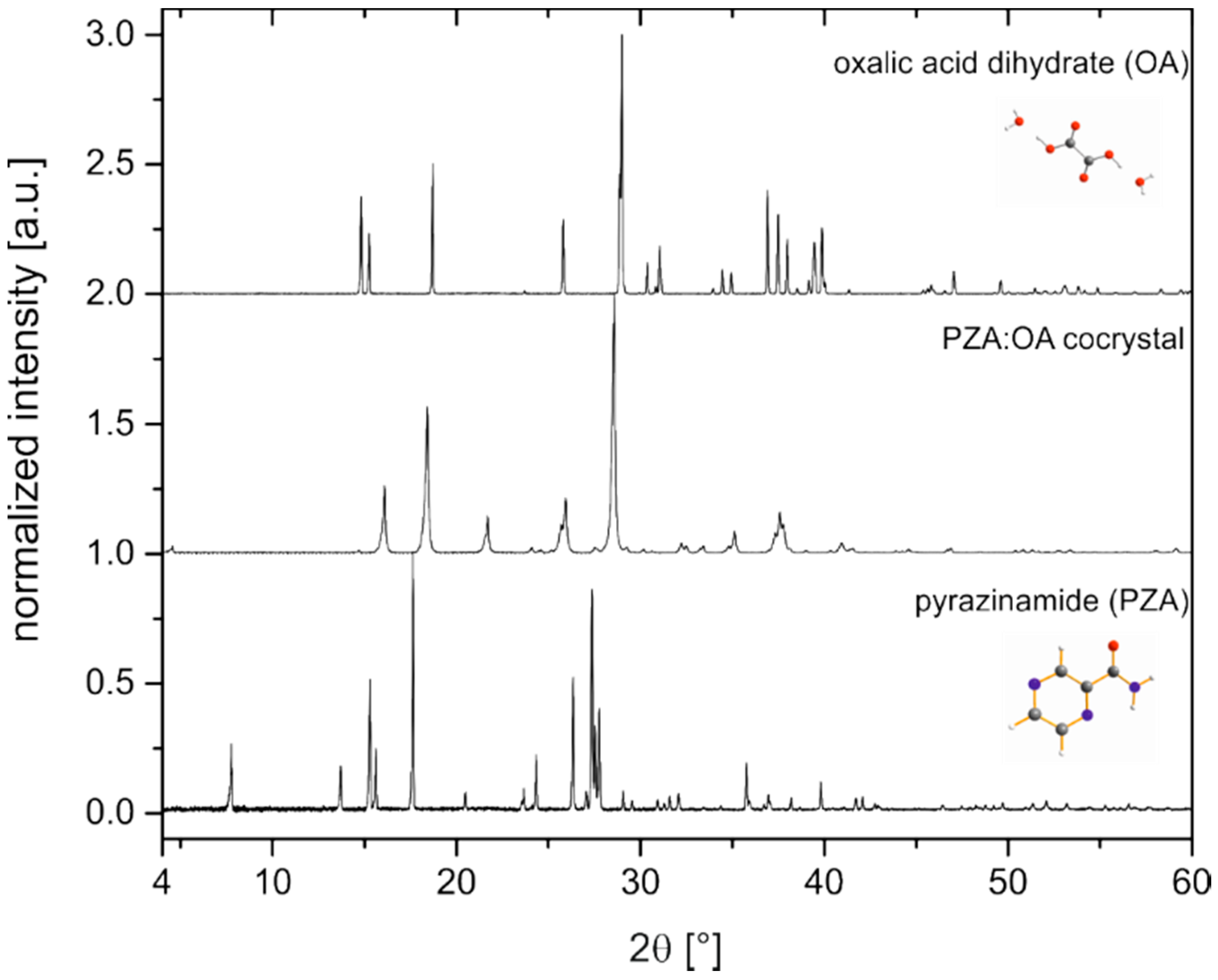
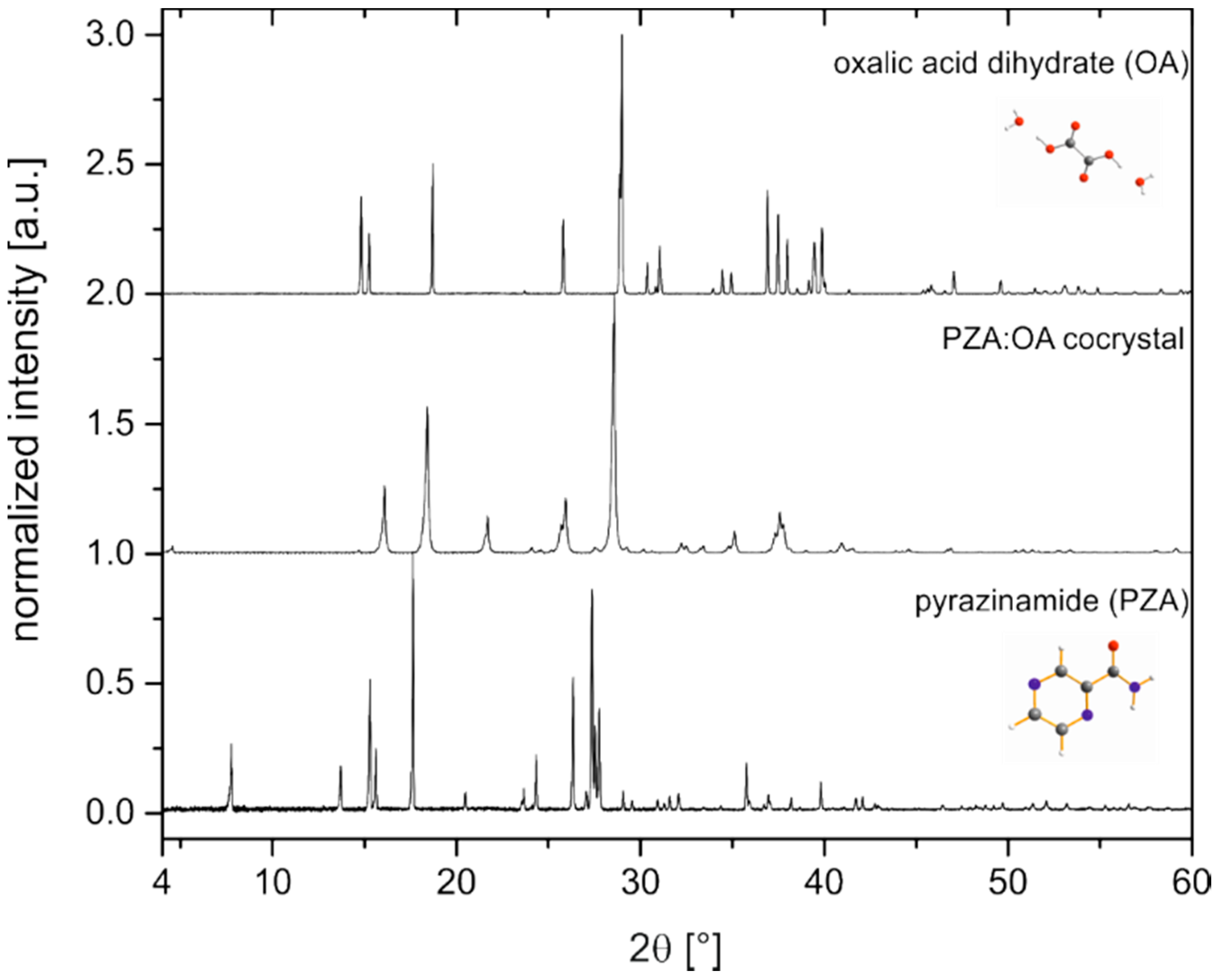
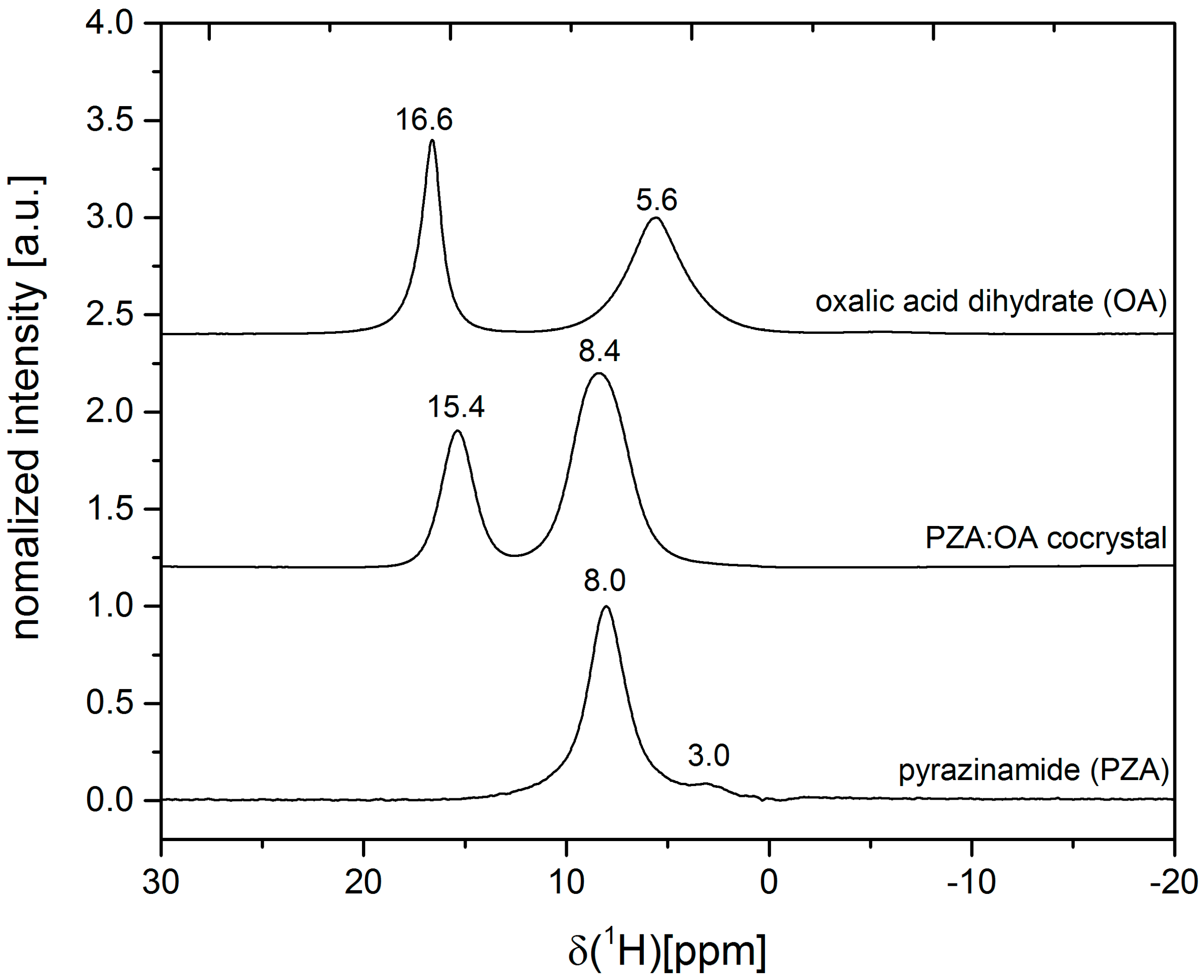
2.1. Characterization of the PZA:OA (1:1) Cocrystal
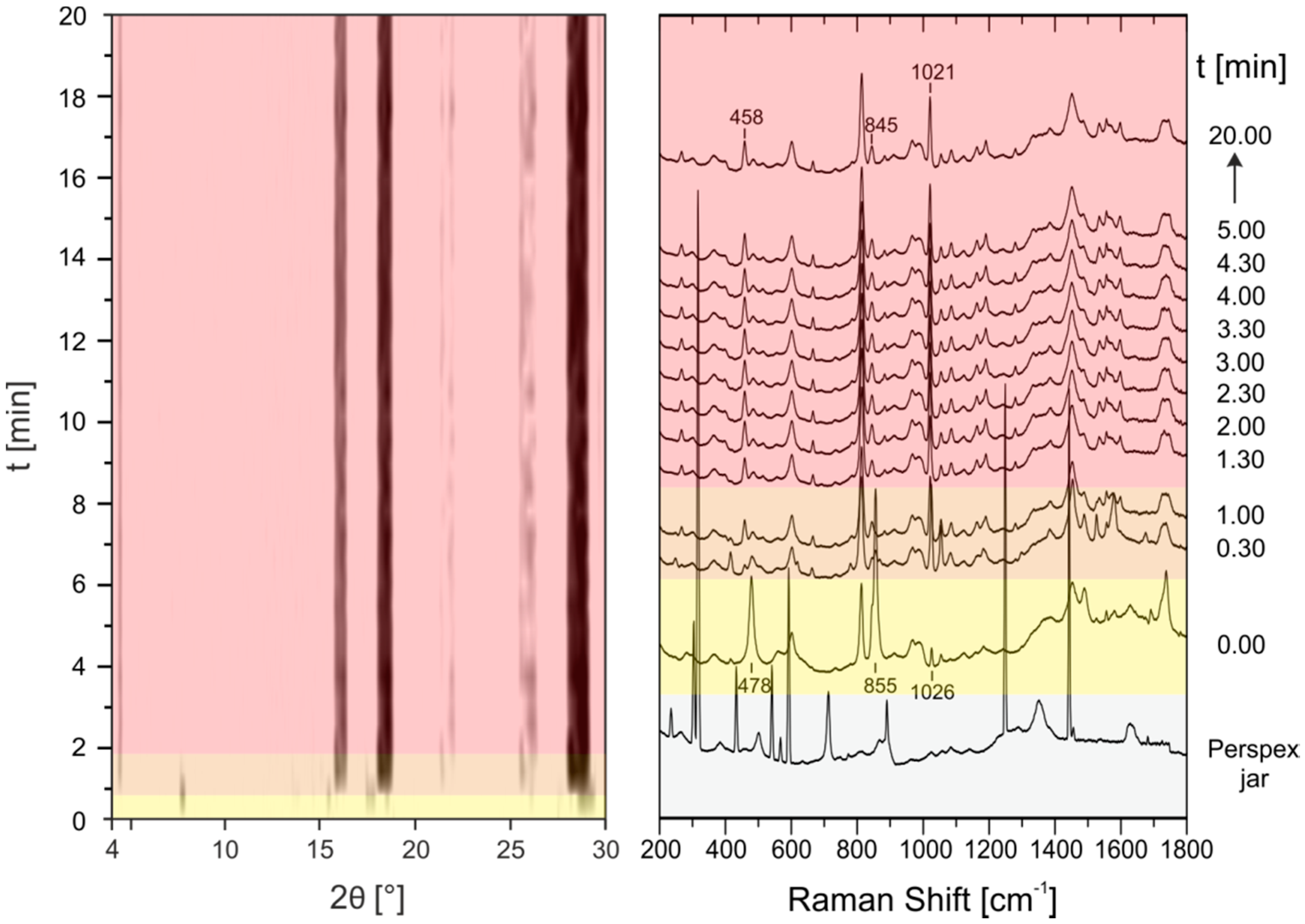
2.2. In Situ Investigation of the PZA:OA (1:1) Cocrystal
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Mechanochemical Synthesis
3.2.2. PXRD Measurements
3.2.3. In Situ Synchrotron XRD
3.2.4. Raman Spectroscopy
3.2.5. ssNMR Spectroscopy
3.2.6. SEM
3.2.7. DTA-TG Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| API | active pharmaceutical ingredient |
| CSD | Cambridge Structure Database |
| DTA-TG | differential thermal analysis with coupled thermogravimetric analysis |
| FDA | U.S. Food and Drug administration |
| LAG | liquid-assisted grinding |
| MAS | magic angle spinning |
| NIR | near infrared |
| OA | oxalic acid |
| PXRD | powder X-ray diffraction |
| PZA | pyrazinamide |
| SEM | scanning electron microscopy |
| ssNMR | solid-state NMR spectroscopy |
References
- Guidance for Industry, ANDA: Pharmaceutical Solid Polymorphism; Food and Drug Adminstration: Silver Spring, MD, USA, 2007.
- Aakeroy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. Crystengcomm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Fischer, F.; Heidrich, A.; Greiser, S.; Benemann, S.; Rademann, K.; Emmerling, F. Polymorphism of Mechanochemically Synthesized Cocrystals: A Case Study. Cryst. Growth Des. 2016, 16, 1701–1707. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Solvent-drop grinding: Green polymorph control of cocrystallisation. Chem. Commun. 2004, 7, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Friscic, T.; Jones, W.; Motherwell, W.D.S. Screening for pharmaceutical cocrystal hydrates via neat and liquid-assisted grinding. Mol. Pharm. 2007, 4, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Toda, F.; Jones, W. Mechanochemistry and co-crystal formation: Effect of solvent on reaction kinetics. Chem. Commun. 2002. [Google Scholar] [CrossRef]
- Friscic, T.; Jones, W. Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding. Cryst. Growth Des. 2009, 9, 1621–1637. [Google Scholar] [CrossRef]
- Tumanov, I.A.; Achkasov, A.F.; Boldyreva, E.V.; Boldyrev, V.V. Following the products of mechanochemical synthesis step by step. CrystEngComm 2011, 13, 2213–2216. [Google Scholar] [CrossRef]
- Jayasankar, A.; Somwangthanaroj, A.; Shao, Z.J.; Rodríguez-Hornedo, N. Cocrystal Formation during Cogrinding and Storage is Mediated by Amorphous Phase. Pharm. Res. 2006, 23, 2381–2392. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Hubert, M.; Saville, D.; Rades, T.; Aaltonen, J. Formation Kinetics and Stability of Carbamazepine-Nicotinamide Cocrystals Prepared by Mechanical Activation. Cryst. Growth Des. 2009, 9, 2377–2386. [Google Scholar] [CrossRef]
- Losev, E.A.; Boldyreva, E.V. The role of a liquid in “dry” co-grinding: A case study of the effect of water on mechanochemical synthesis in a “l-serine-oxalic acid” system dagger. Crystengcomm 2014, 16, 3857–3866. [Google Scholar] [CrossRef]
- Uzarevic, K.; Strukil, V.; Mottillo, C.; Julien, P.A.; Puskaric, A.; Friscic, T.; Halasz, I. Exploring the Effect of Temperature on a Mechanochemical Reaction by in Situ Synchrotron Powder X-ray Diffraction. Cryst. Growth Des. 2016, 16, 2342–2347. [Google Scholar] [CrossRef]
- Fischer, F.; Scholz, G.; Benemann, S.; Rademann, K.; Emmerling, F. Evaluation of the formation pathways of cocrystal polymorphs in liquid-assisted syntheses. Crystengcomm 2014, 16, 8272–8278. [Google Scholar] [CrossRef]
- Hasa, D.; Rauber, G.S.; Voinovich, D.; Jones, W. Cocrystal Formation through Mechanochemistry: From Neat and Liquid-Assisted Grinding to Polymer-Assisted Grinding. Angew. Chem. Int. Ed. 2015, 54, 7371–7375. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C.; Reutzel, S.M. Hydrogen-bond directed cocrystallization and molecular recognition properties of acyclic imides. J. Am. Chem. Soc. 1991, 113, 2586–2598. [Google Scholar] [CrossRef]
- Hollingsworth, M.D.; Brown, M.E.; Santarsiero, B.D.; Huffman, J.C.; Goss, C.R. Template-directed synthesis of 1–1 layered complexes of alpha, omega-dinitriles and urea-packing efficiency versus specific functional-group interactions. Chem. Mater. 1994, 6, 1227–1244. [Google Scholar] [CrossRef]
- Karki, S.; Fabian, L.; Friscic, T.; Jones, W. Powder x-ray diffraction as an emerging method to structurally characterize organic solids. Org. Lett. 2007, 9, 3133–3136. [Google Scholar] [CrossRef] [PubMed]
- Trobs, L.; Emmerling, F. Mechanochemical synthesis and characterisation of cocrystals and metal organic compounds. Faraday Discuss. 2014, 170, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Cincic, D.; Friscic, T.; Jones, W. A stepwise mechanism for the mechanochemical synthesis of halogen-bonded cocrystal architectures. J. Am. Chem. Soc. 2008, 130, 7524–7525. [Google Scholar] [CrossRef] [PubMed]
- Halasz, I.; Friscic, T.; Kimber, S.A.J.; Uzarevic, K.; Puskaric, A.; Mottillo, C.; Julien, P.; Strukil, V.; Honkimaki, V.; Dinnebier, R.E. Quantitative in situ and real-time monitoring of mechanochemical reactions. Faraday Discuss. 2014, 170, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Batzdorf, L.; Fischer, F.; Wilke, M.; Wenzel, K.-J.; Emmerling, F. Direct In Situ Investigation of Milling Reactions Using Combined X-ray Diffraction and Raman Spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Friscic, T.; Halasz, I.; Beldon, P.J.; Belenguer, A.M.; Adams, F.; Kimber, S.A.J.; Honkimaki, V.; Dinnebier, R.E. Real-time and in situ monitoring of mechanochemical milling reactions. Nat. Chem. 2013, 5, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Gracin, D.; Strukil, V.; Friscic, T.; Halasz, I.; Uzarevic, K. Laboratory Real-Time and In Situ Monitoring of Mechanochemical Milling Reactions by Raman Spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 6193–6197. [Google Scholar] [CrossRef] [PubMed]
- Užarević, K.; Halasz, I.; Friščić, T. Real-Time and In Situ Monitoring of Mechanochemical Reactions: A New Playground for All Chemists. J. Phys. Chem. Lett. 2015, 6, 4129–4140. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Scholz, G.; Batzdorf, L.; Wilke, M.; Emmerling, F. Synthesis, structure determination, and formation of a theobromine:oxalic acid 2:1 cocrystal. Crystengcomm 2015, 17, 824–829. [Google Scholar] [CrossRef]
- McMahon, J.A.; Bis, J.A.; Vishweshwar, P.; Shattock, T.R.; McLaughlin, O.L.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. 3. Primary amide supramolecular heterosynthons and their role in the design of pharmaceutical co-crystals. Z. Krist. 2005, 220, 340–350. [Google Scholar] [CrossRef]
- Abourahma, H.; Shah, D.D.; Melendez, J.; Johnson, E.J.; Holman, K.T. A Tale of Two Stoichiometrically Diverse Cocrystals. Cryst. Growth Des. 2015, 15, 3101–3104. [Google Scholar] [CrossRef]
- Adalder, T.K.; Sankolli, R.; Dastidar, P. Homo- or Heterosynthon? A Crystallographic Study on a Series of New Cocrystals Derived from Pyrazinecarboxamide and Various Carboxylic Acids Equipped with Additional Hydrogen Bonding Sites. Cryst. Growth Des. 2012, 12, 2533–2542. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Sun, B.-W. Pharmaceutical Co-Crystals of Pyrazinecarboxamide (PZA) with Various Carboxylic Acids: Crystallography, Hirshfeld Surfaces, and Dissolution Study. Cryst. Growth Des. 2013, 13, 2098–2106. [Google Scholar] [CrossRef]
- Wang, J.-R.; Ye, C.; Zhu, B.; Zhou, C.; Mei, X. Pharmaceutical cocrystals of the anti-tuberculosis drug pyrazinamide with dicarboxylic and tricarboxylic acids. Crystengcomm 2015, 17, 747–752. [Google Scholar] [CrossRef]
- Ebisuzaki, Y.; Angel, S.M. Raman study of hydrogen bonding in α and β-oxalic acid dihydrate. J. Raman Spectrosc. 1981, 11, 306–311. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: a program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Topas version 5; Bruker AXS: Karlsruhe, Germany, 2014.
- Paris, O.; Li, C.; Siegel, S.; Weseloh, G.; Emmerling, F.; Riesemeier, H.; Erko, A.; Fratzl, P. A new experimental station for simultaneous X-ray microbeam scanning for small- and wide-angle scattering and fluorescence at BESSY II. J. Appl. Crystallogr. 2007, 40, 466–470. [Google Scholar] [CrossRef]
- Hammersley, A.P.; Brown, K.; Burmeister, W.; Claustre, L.; Gonzalez, A.; McSweeney, S.; Mitchell, E.; Moy, J.-P.; Svensson, S.O.; Thompson, A.W. Calibration and Application of an X-ray Image Intensifier/Charge-Coupled Device Detector for Monochromatic Macromolecular Crystallography. J. Synchrotron Radiat. 1997, 4, 67–77. [Google Scholar] [CrossRef] [PubMed]
- DIFFRAC.EVA; AXS Bruker: Karlsruhe, Germany, 2015.
- Sample Availability: Not available.



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulla, H.; Greiser, S.; Benemann, S.; Rademann, K.; Emmerling, F. In Situ Investigation of a Self-Accelerated Cocrystal Formation by Grinding Pyrazinamide with Oxalic Acid. Molecules 2016, 21, 917. https://doi.org/10.3390/molecules21070917
Kulla H, Greiser S, Benemann S, Rademann K, Emmerling F. In Situ Investigation of a Self-Accelerated Cocrystal Formation by Grinding Pyrazinamide with Oxalic Acid. Molecules. 2016; 21(7):917. https://doi.org/10.3390/molecules21070917
Chicago/Turabian StyleKulla, Hannes, Sebastian Greiser, Sigrid Benemann, Klaus Rademann, and Franziska Emmerling. 2016. "In Situ Investigation of a Self-Accelerated Cocrystal Formation by Grinding Pyrazinamide with Oxalic Acid" Molecules 21, no. 7: 917. https://doi.org/10.3390/molecules21070917