
Synthesis, Characterization and Anti-Cancer Activity of Hydrazide Derivatives Incorporating a Quinoline Moiety



Abstract
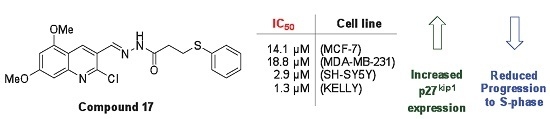
:
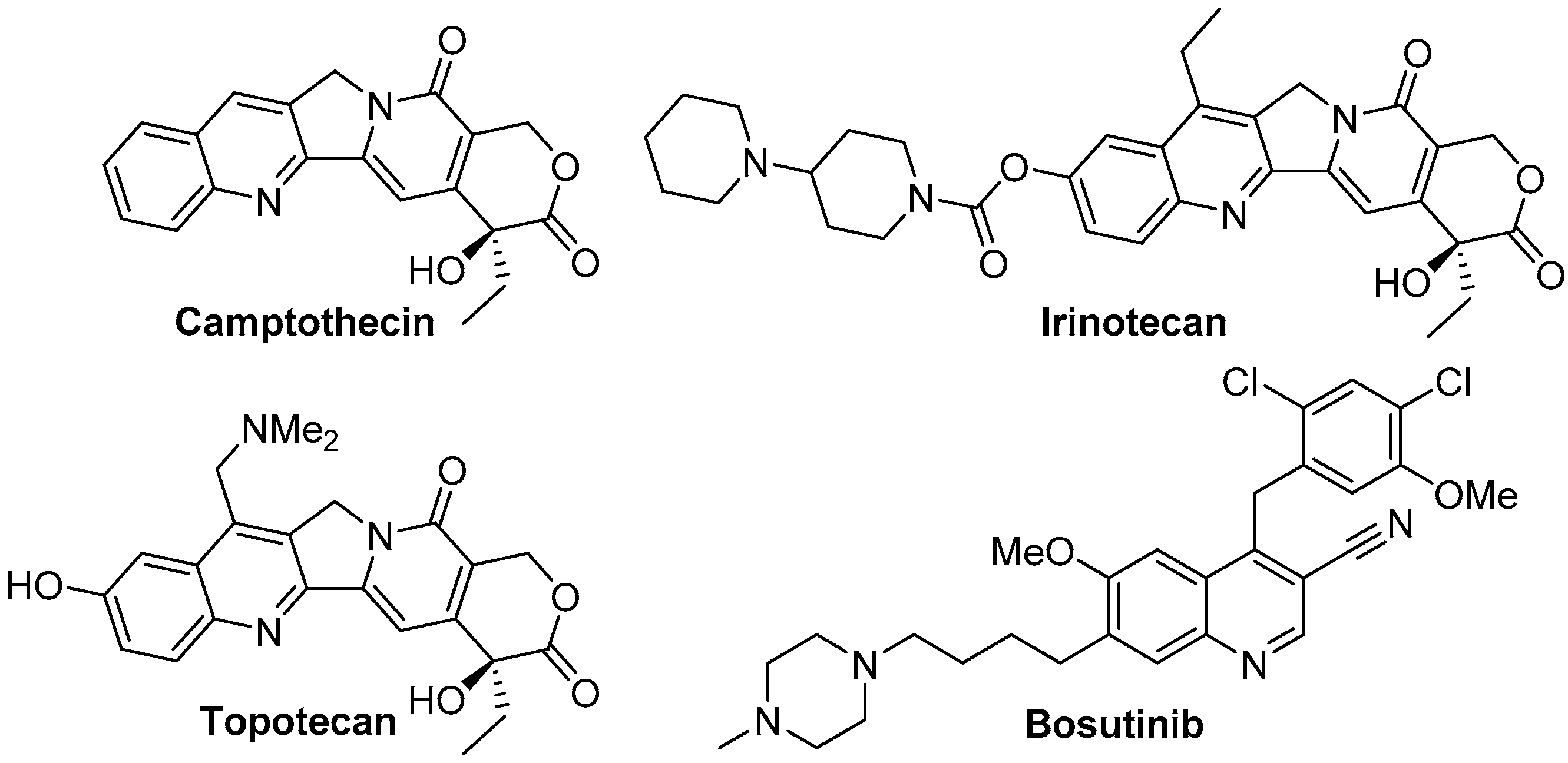
1. Introduction
2. Results and Discussion
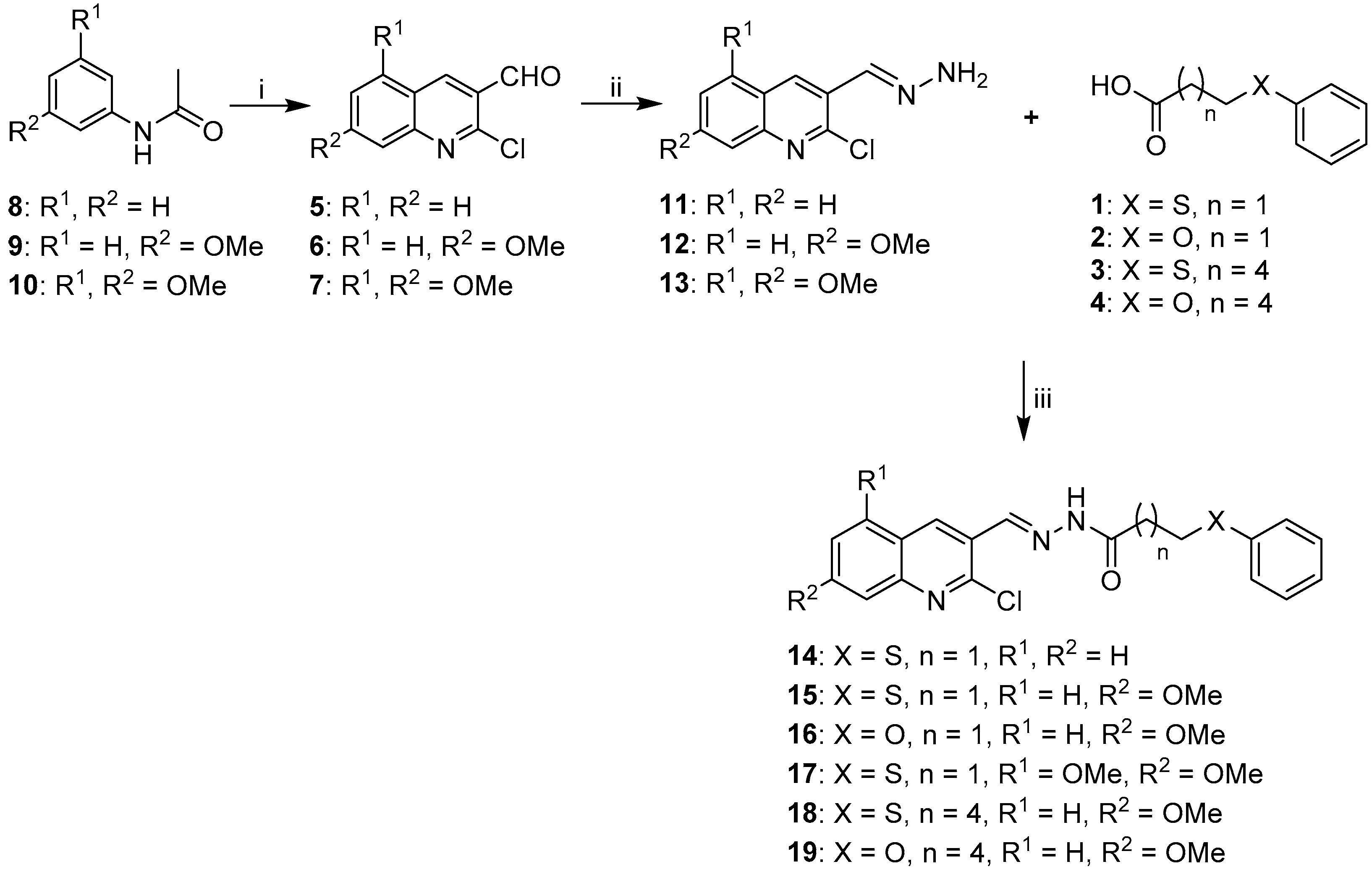
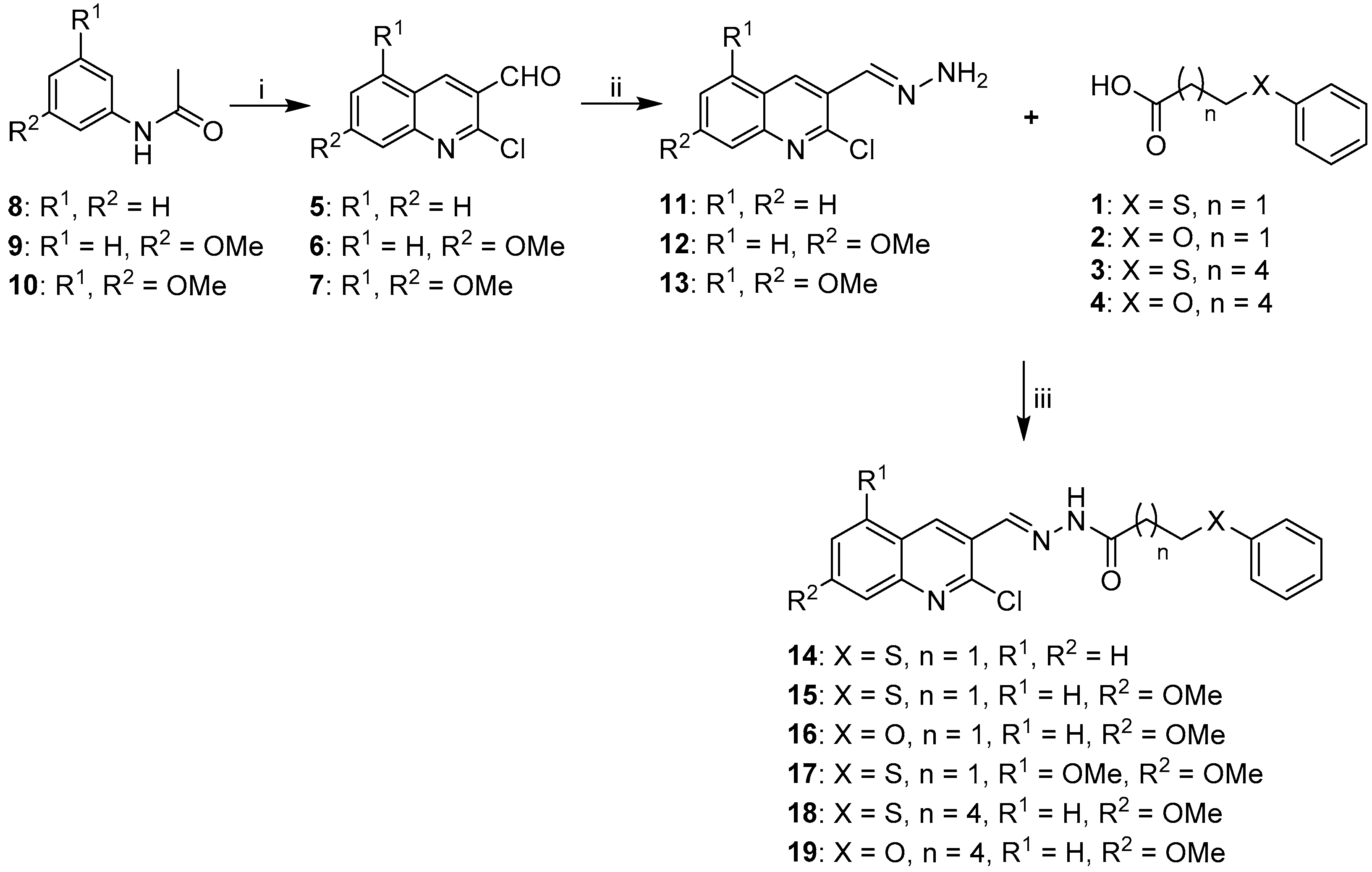
2.1. Synthesis of Quinoline-Based Hydrazide-Hydrazones
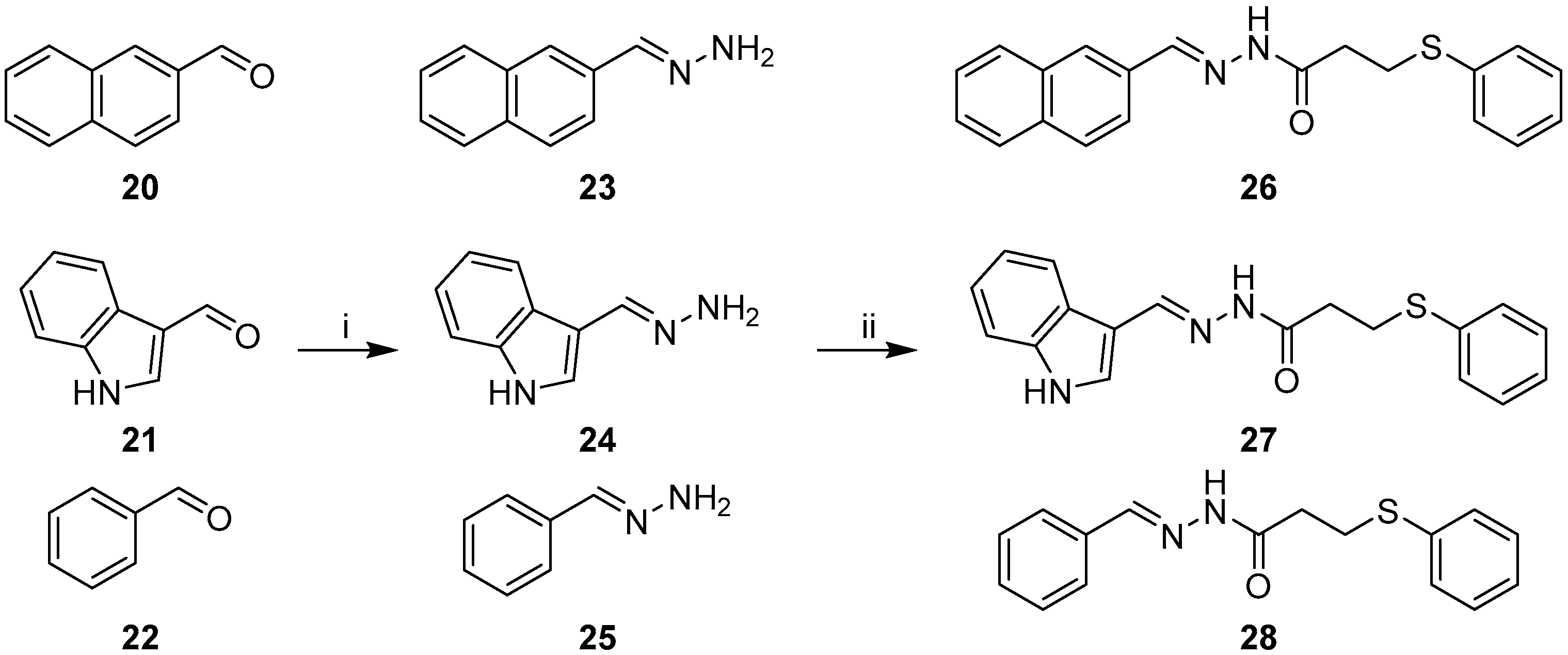
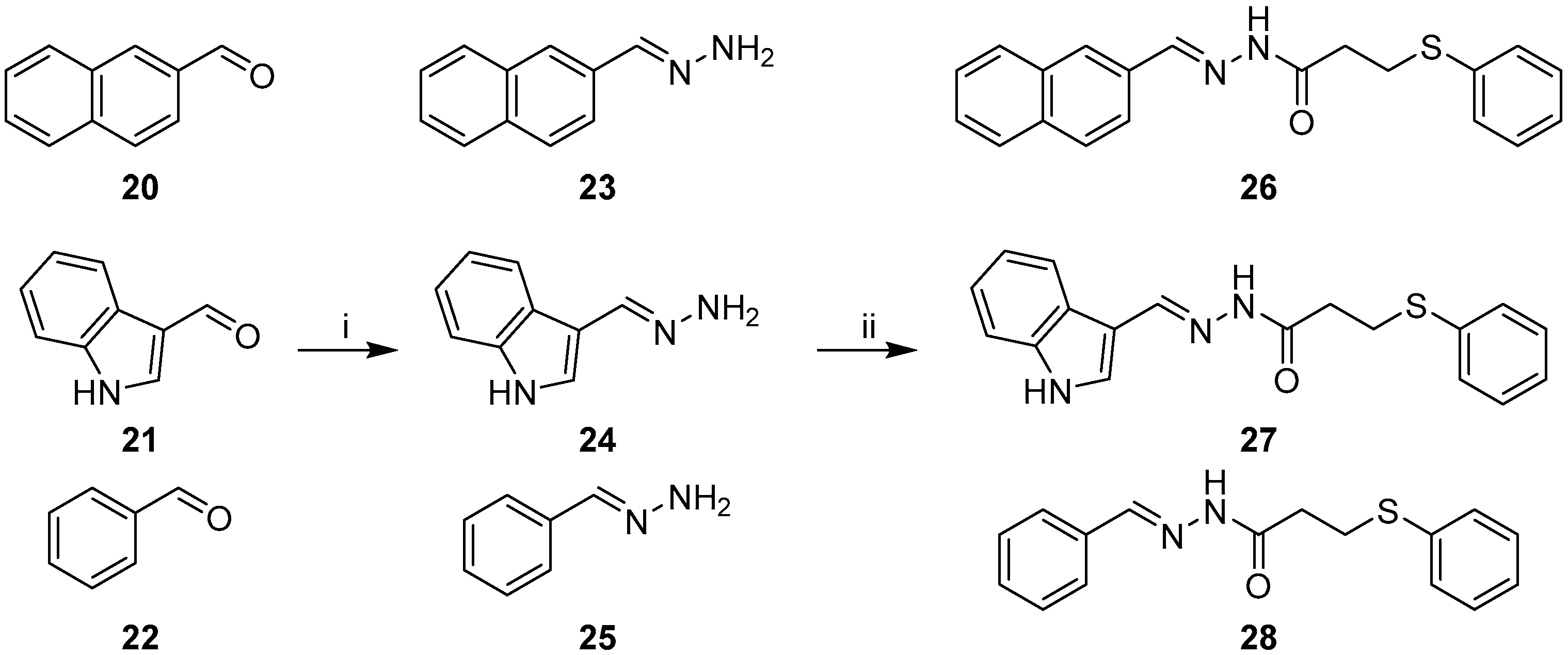
2.2. Synthesis of Non-Quinoline Hydrazide-Hydrazones
2.3. Biological Results
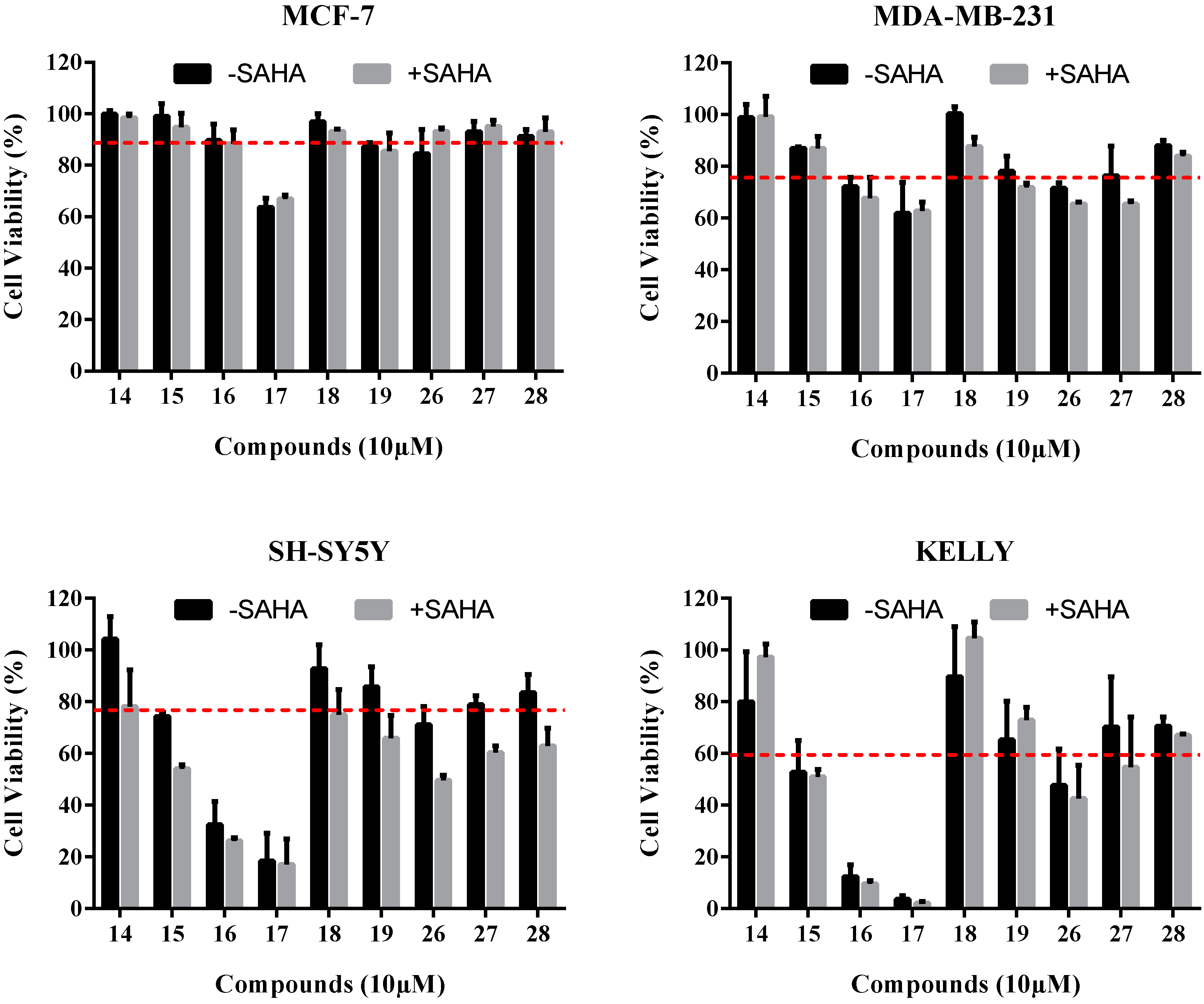
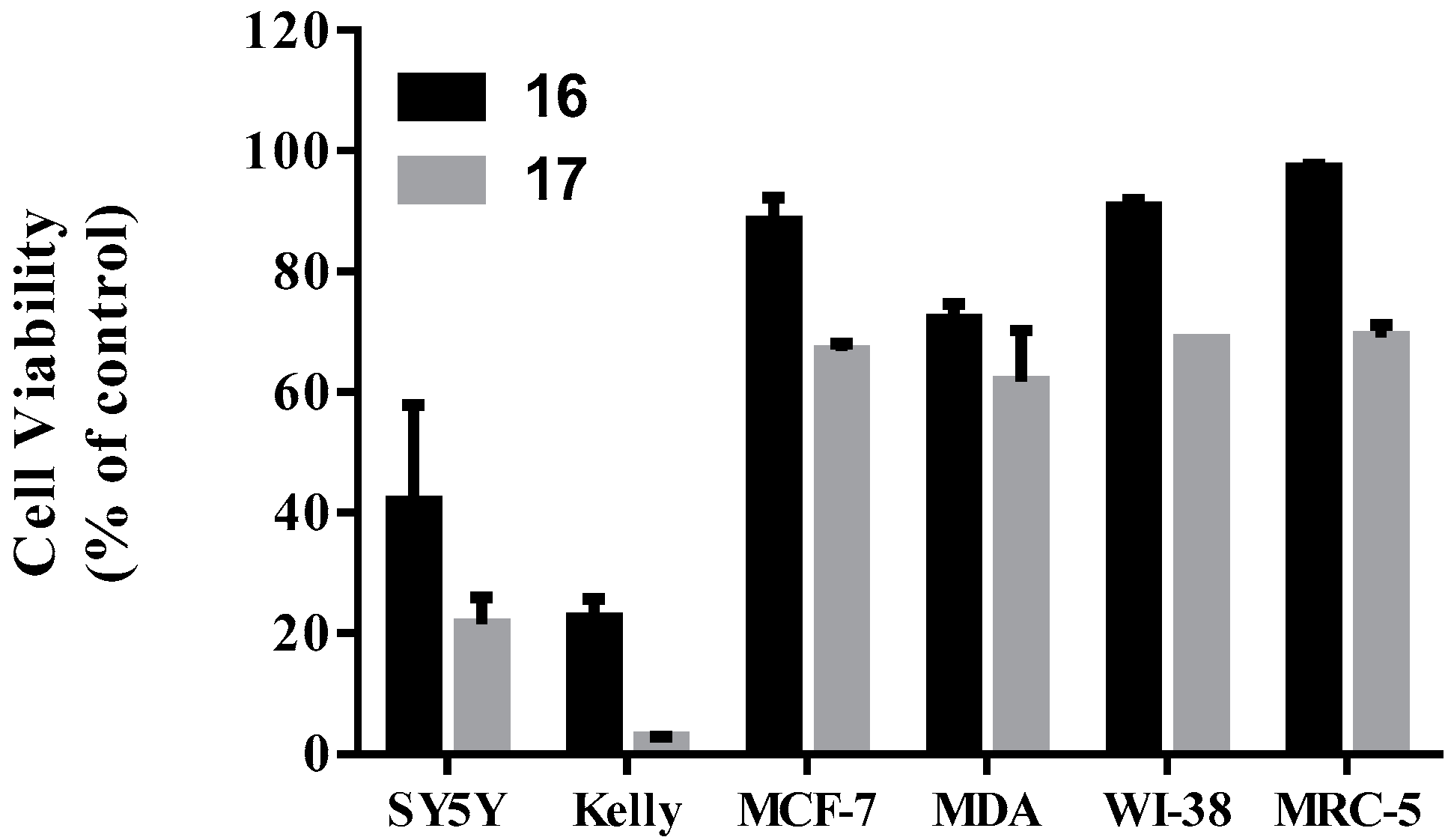
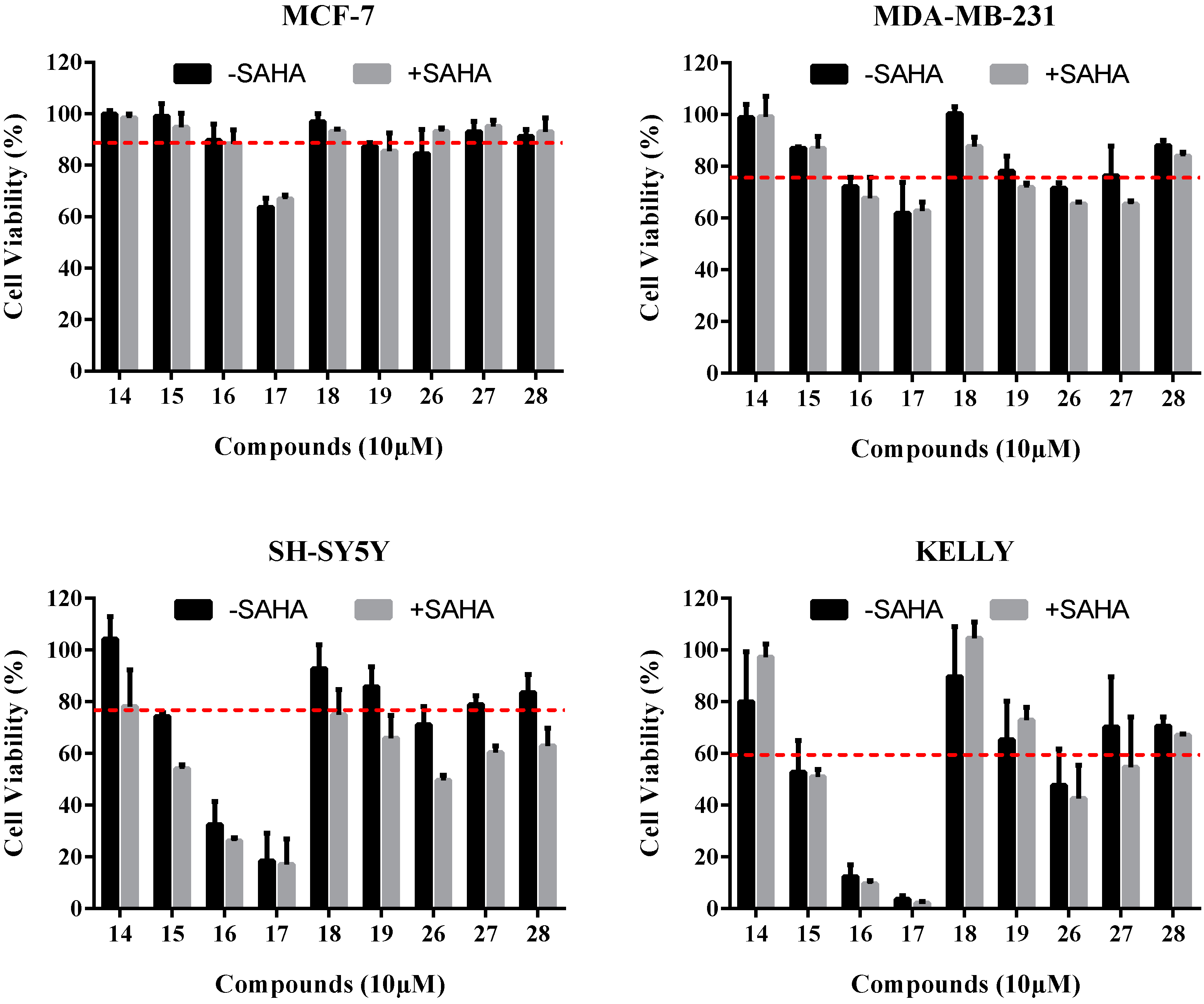
2.3.1. Cell Viability Assays
2.3.2. Toxicity Study on Normal Human Cells
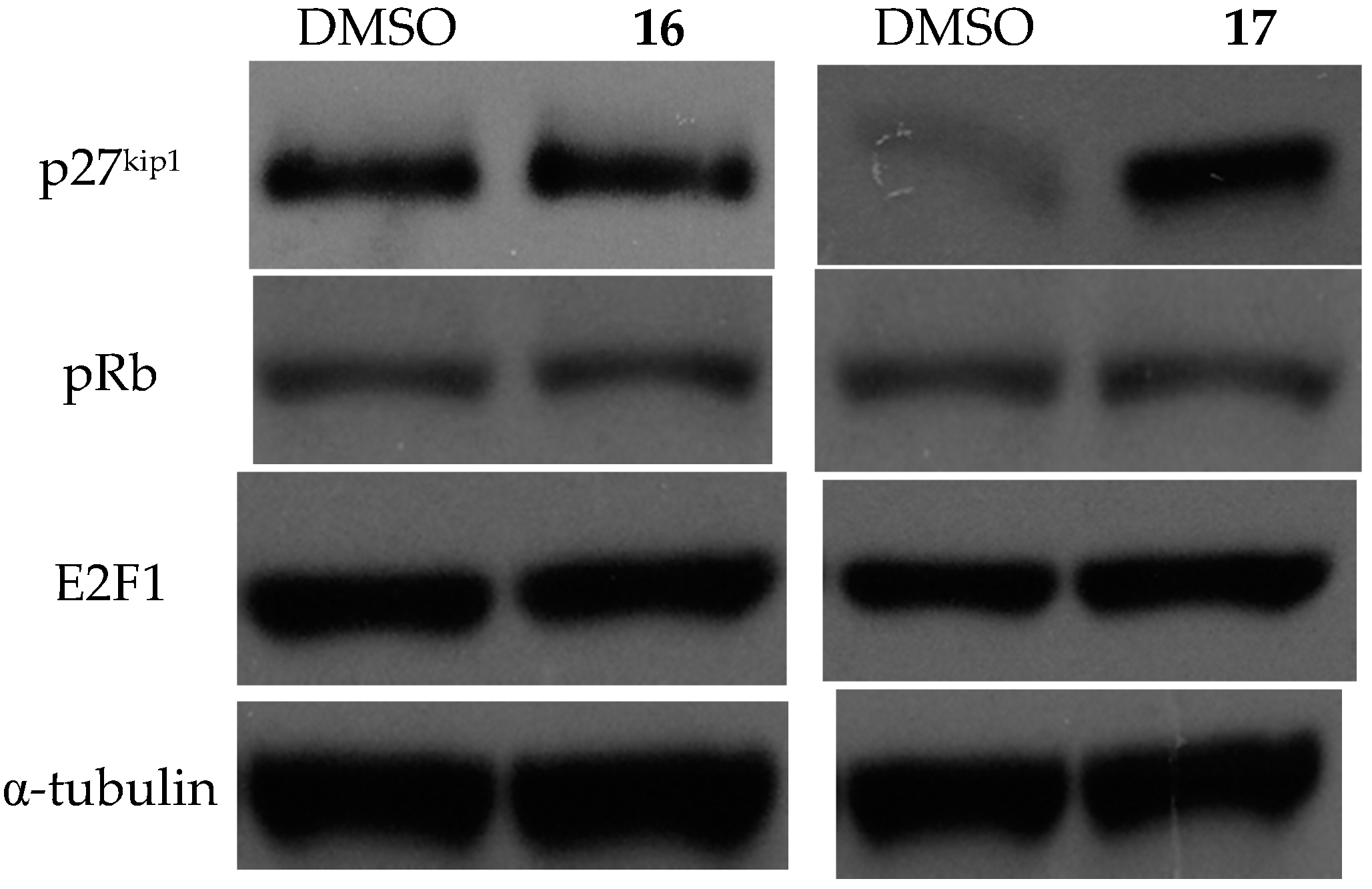
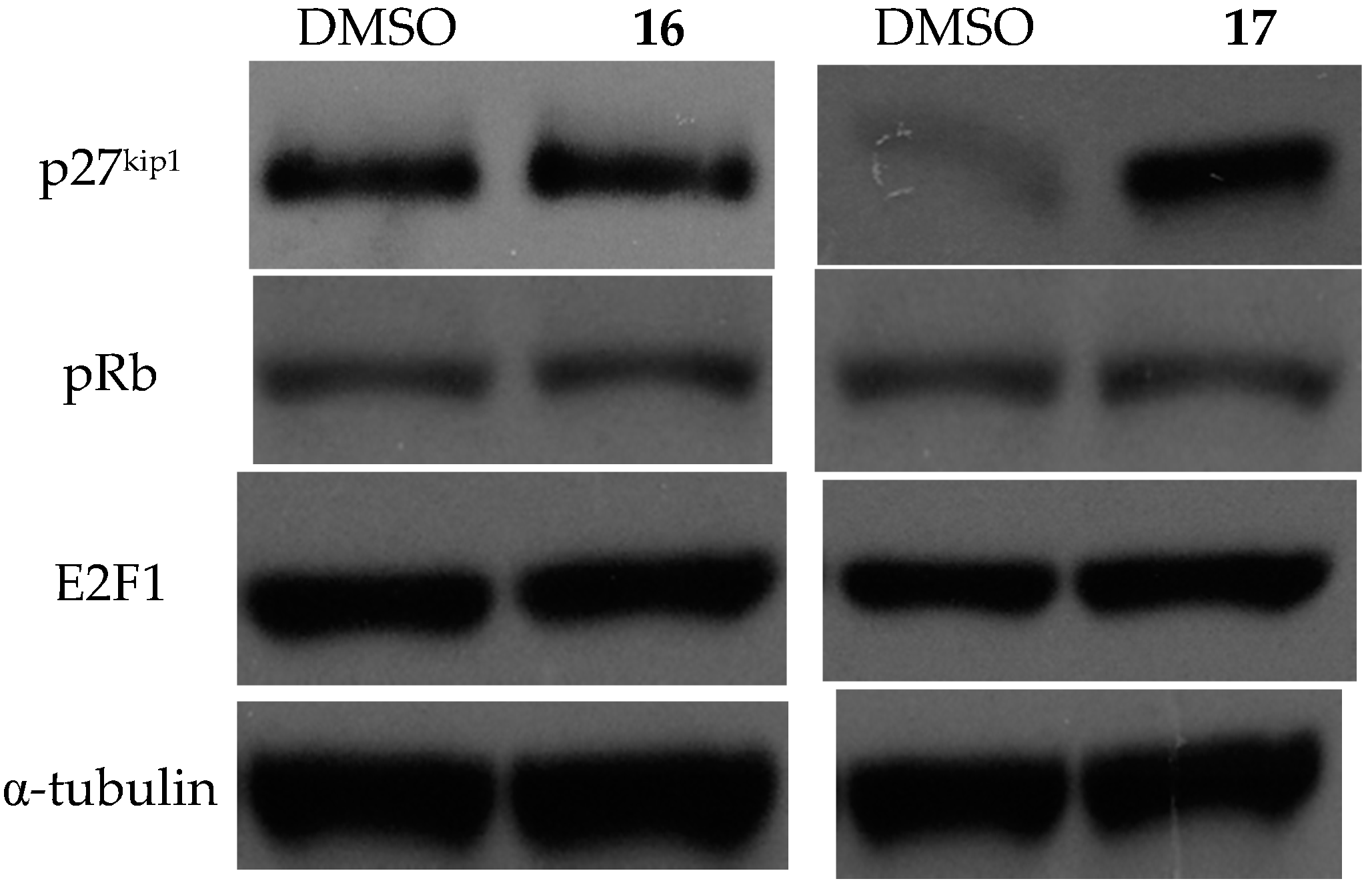
2.3.3. Effects on Cell Cycle and the Expression of Related Proteins
3. Materials and Methods
3.1. General Information
3.1.1. General Procedure for the Synthesis of Substituted Carboxylic Acids 1–4
3.1.2. General Procedure for the Synthesis of Acetylated Anilines 8–10
3.1.3. General Procedure for the Synthesis of 2-Chloro-3-Formylquinolines (5–7).
3.1.4. General Procedure for the Preparation of Hydrazones 11–13, 23–25
3.1.5. General Procedure for EDC Coupling (compounds 14–19, 26–28)
3.2. Cell Biology Techniques
3.2.1. Method for Cell Viability Assays
3.2.2. Cell Cycle Study by Propidium Iodide Staining
3.2.3. Immunoblotting of Cell Cycle Regulatory Protein Expression
3.3. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| HDACi | Histone deacetylase inhibitors |
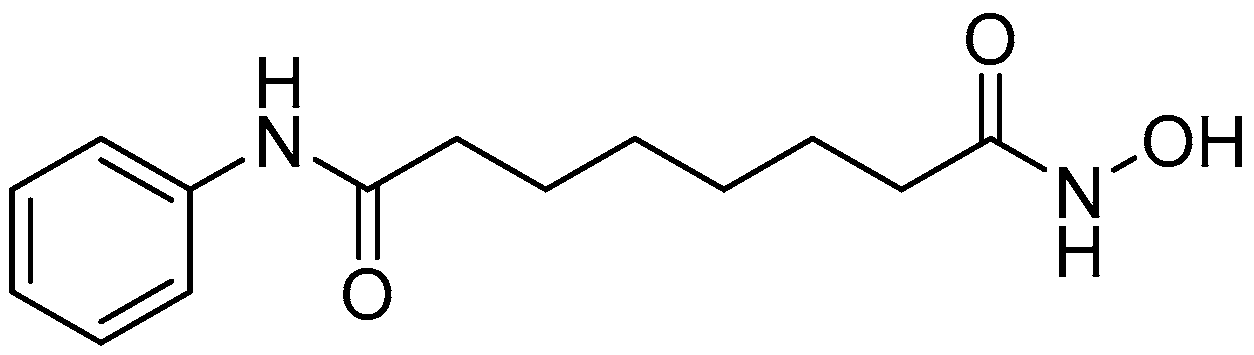
| SAHA | Suberoylanilide hydroxamic acid |
| WEHI | Walter & Eliza Hall Institute |
| DMF | Dimethylformamide |
| SD | Standart Deviation |
| DMSO | Dimethylsulfoxide |
| DMAP | Dimethoxyaminopyridine |
| KBr | Potassium Bromide |
| Et3N | Triethylamine |
| PI | Propidium iodide |
| SEM | Standart Error of Mean |
| ORTEP | Oak Ridge thermal ellipsoid |
References and Notes
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Lautz, T.B.; Jie, C.; Clark, S.; Naiditch, J.A.; Jafari, N.; Qiu, Y.Y.; Zheng, X.; Chu, F.; Madonna, M.B. The effect of vorinostat on the development of resistance to doxorubicin in neuroblastoma. PLoS ONE 2012, 7, e40816. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choy, M.L.; Marks, P.A. Mechanisms of Resistance to Histone Deacetylase Inhibitors. In Advances in Cancer Research; Steven, G., Ed.; Academic Press: San Diego, CA, USA, 2012; Volume 116, pp. 39–86. [Google Scholar]
- Richon, V.M. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, 2–6. [Google Scholar] [CrossRef]
- Basu, H.S.; Mahlum, A.; Mehraein-Ghomi, F.; Kegel, S.J.; Guo, S.; Peters, N.R.; Wilding, G. Pre-treatment with anti-oxidants sensitizes oxidatively stressed human cancer cells to growth inhibitory effect of suberoylanilide hydroxamic acid (SAHA). Cancer Chemother. Pharmacol. 2011, 67, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; O′Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef] [PubMed]
- Rundall, B.K.; Denlinger, C.E.; Jones, D.R. Combined histone deacetylase and NF-κB inhibition sensitizes non-small cell lung cancer to cell death. Surgery 2005, 138, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, J.; Gange, J.; Kumar, K.S.; Muller, C.; Bader, P.; Beck, J.F. Histone deacetylase inhibitors interact synergistically with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to induce apoptosis in carcinoma cell lines. Investig. New Drug 2005, 23, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, C.E.; Rundall, B.K.; Jones, D.R. Proteasome inhibition sensitizes non-small cell lung cancer to histone deacetylase inhibitor-induced apoptosis through the generation of reactive oxygen species. J Thorac. Cardiov. Sur. 2004, 128, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Arndt, G.; (Children’s Cancer Institute, Sydney, NSW, Australia). Personal Communication, 2009.
- Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp. 316–336. [Google Scholar]
- Chen, Y.L.; Huang, C.J.; Huang, Z.Y.; Tseng, C.H.; Chang, F.S.; Yang, S.H.; Lin, S.R.; Tzeng, C.C. Synthesis and antiproliferative evaluation of certain 4-anilino-8-methoxy-2-phenylquinoline and 4-anilino-8-hydroxy-2-phenylquinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3098–3105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Zhao, Y.L.; Lu, C.M.; Tzeng, C.C.; Wang, J.P. Synthesis, cytotoxicity, and anti-inflammatory evaluation of 2-(furan-2-yl)-4-(phenoxy)quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 4373–4378. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Lau, E.; Scortegagna, M.; Ruller, C.; De, S.K.; Barile, E.; Krajewski, S.; Aza-Blanc, P.; Williams, R.; Pinkerton, A.B.; et al. Inhibition of melanoma development in the Nras(Q61K): Ink4a(−/−) mouse model by the small molecule BI-69A11. Pigm. Cell Melanoma Res. 2013, 26, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Gholap, A.R.; Toti, K.S.; Shirazi, F.; Kumari, R.; Bhat, M.K.; Deshpande, M.V.; Srinivasan, K.V. Synthesis and evaluation of antifungal properties of a series of the novel 2-amino-5-oxo-4-phenyl-5,6,7,8-tetrahydroquinoline-3-carbonitrile and its analogues. Bioorg. Med. Chem. 2007, 15, 6705–6715. [Google Scholar] [CrossRef] [PubMed]
- Krafts, K.; Hempelmann, E.; Skorska-Stania, A. From methylene blue to chloroquine: A brief review of the development of an antimalarial therapy. Parasitol. Res. 2012, 111, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.P.; Domagala, J.M.; Hagen, S.E.; Heifetz, C.L.; Hutt, M.P.; Nichols, J.B.; Trehan, A.K. Quinolone antibacterial agents. Synthesis and structure-activity relationships of 8-substituted quinoline-3-carboxylic acids and 1,8-naphthyridine-3-carboxylic acids. J. Med. Chem. 1988, 31, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Adsule, S.; Barve, V.; Chen, D.; Ahmed, F.; Dou, Q.P.; Padhye, S.; Sarkar, F.H. Novel Schiff base copper complexes of quinoline-2 carboxaldehyde as proteasome inhibitors in human prostate cancer cells. J. Med. Chem. 2006, 49, 7242–7246. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Oka, M.; Aizawa, K.; Soda, H.; Fukuda, M.; Terashi, K.; Ikeda, K.; Mizuta, Y.; Noguchi, Y.; Kimura, Y.; et al. Direct interaction between a quinoline derivative, MS-209, and multidrug resistance protein (MRP) in human gastric cancer cells. Biochem. Bioph. Res. Co. 1999, 255, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Rivenson, A. Inhibitory effect of Bifidobacterium longum on colon, mammary, and liver carcinogenesis induced by 2-amino-3-methylimidazo[4,5-f]quinoline, a food mutagen. Cancer Res. 1993, 53, 3914–3918. [Google Scholar] [PubMed]
- Shi, A.; Nguyen, T.A.; Battina, S.K.; Rana, S.; Takemoto, D.J.; Chiang, P.K.; Hua, D.H. Synthesis and anti-breast cancer activities of substituted quinolones. Bioorg. Med. Chem. Lett. 2008, 18, 3364–3368. [Google Scholar] [CrossRef] [PubMed]
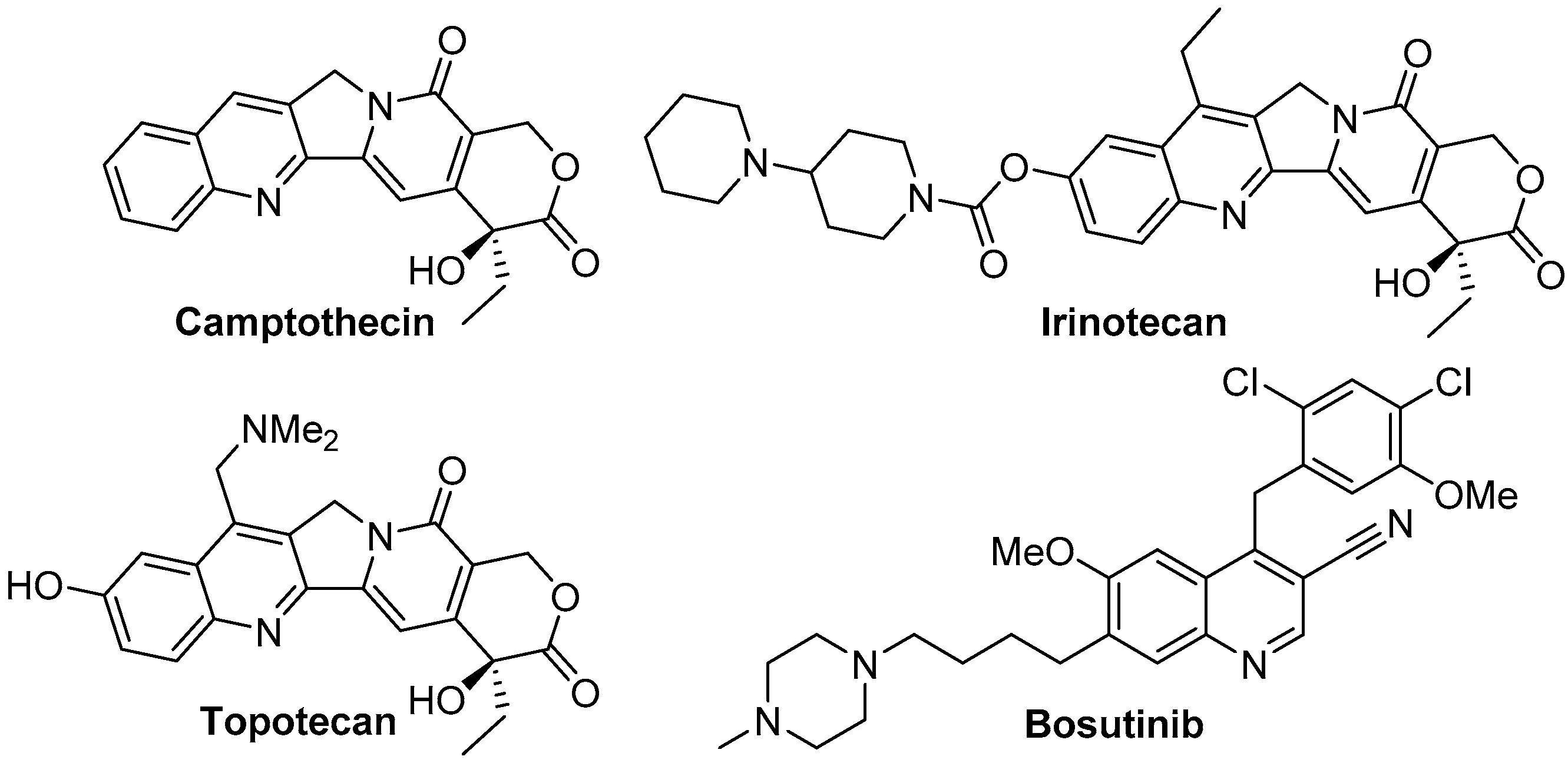
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata1,2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Sawada, S.; Okajima, S.; Aiyama, R.; Nokata, K.I.; Furuta, T.; Yokokura, T.; Sugino, E.; Yamaguchi, K.; Miyasaka, T. Synthesis and antitumor activity of 20(S)-camptothecin derivatives: Carbamate-linked, water-soluble derivaties of 7-ethyl-10-hydroxycamptothecin. Chem. Pharm. Bull. 1991, 39, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kantarjian, H.M.; Brummendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.-W.; Kantarjian, H.M.; Brümmendorf, T.H.; Dyagil, I.; Griskevicius, L.; Malhotra, H.; Powell, C.; Gogat, K.; Countouriotis, A.M.; et al. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J. Clin. Oncol. 2012, 30, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Obafemi, C.A.; Nwinyi, O.C.; Akinpelu, D.A. Microwave-assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg. Med. Chem. 2010, 18, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, P.; Leroux, V.; Sergheraert, C.; Grellier, P. Design, synthesis and in vitro antimalarial activity of an acylhydrazone library. Bioorg. Med. Chem. Lett. 2006, 16, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.W.; Wu, L.L.; Zhao, B.X.; Dong, W.L.; Miao, J.Y. Synthesis of novel substituted pyrazole-5-carbohydrazide hydrazone derivatives and discovery of a potent apoptosis inducer in A549 lung cancer cells. Bioorg. Med. Chem. 2009, 17, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Metwally, K.A.; Abdel-Aziz, L.M.; Lashine, E.S.M.; Husseiny, M.I.; Badawy, R.H. Hydrazones of 2-aryl-quinoline-4-carboxylic acid hydrazides: Synthesis and preliminary evaluation as antimicrobial agents. Bioorg. Med. Chem. 2006, 14, 8675–8682. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, S.; Adhikari, A.V.; Pal, N.K.; Chowdhury, I.H. Design and synthesis of some new quinoline-3-carbohydrazone derivatives as potential antimycobacterial agents. Bioorg. Med. Chem. Lett. 2010, 20, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, A.; Monga, V.; Malde, A.; Coutinho, E.; Jain, R. Synthesis, anti-tuberculosis activity, and 3D-QSAR study of 4-(adamantan-1-yl)-2-substituted quinolines. Bioorg. Med. Chem. 2007, 15, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.D.; Adhikari, A.V.; Telkar, S.; Chowdhury, I.H.; Mahmood, R.; Pal, N.K.; Row, G.; Sumesh, E. Design, synthesis and docking studies of new quinoline-3-carbohydrazide derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2011, 46, 5283–5292. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bawa, S.; Drabu, S.; Kumar, R.; Machawal, L. Synthesis and in vivo anti-convulsant evaluation of 2-chloroquinolinyl hydrazone derivatives. Acta. Pol. Pharmaceutica 2010, 67, 567–573. [Google Scholar]
- Reddy, L.V.; Nallapati, S.B.; Beevi, S.S.; Mangamoori, L.N.; Mukkanti, K.; Pal, S. A “green” synthesis of N-(quinoline-3-ylmethylene)benzohydrazide derivatives and their cytotoxicity activities. J. Brazil. Chem. Soc. 2011, 22, 1742–1749. [Google Scholar] [CrossRef]
- You, Q.D.; Li, Z.Y.; Huang, C.H.; Yang, Q.; Wang, X.J.; Guo, Q.L.; Chen, X.G.; He, X.G.; Li, T.K.; Chern, J.W. Discovery of a novel series of quinolone and naphthyridine derivatives as potential topoisomerase I inhibitors by scaffold modification. J. Med. Chem. 2009, 52, 5649–5661. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.M.; Harmange, J.C.; Polverino, A.J.; Bauer, D.; Berry, L.; Berry, V.; Borg, G.; Bready, J.; Chen, D.; Choquette, D.; et al. Evaluation of a series of naphthamides as potent, orally active vascular endothelial growth factor receptor-2 tyrosine kinase inhibitors. J. Med. Chem. 2008, 51, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Moffat, D.; Patel, S.; Day, F.; Belfield, A.; Donald, A.; Rowlands, M.; Wibawa, J.; Brotherton, D.; Stimson, L.; Clark, V.; et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl] amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J. Med. Chem. 2010, 53, 8663–8678. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fujiwara, Y.; Osawa, T.; Sakai, T.; Kubo, K.; Kubo, K.; Nishitoba, T.; Kimura, K.; Senga, T.; Murooka, H.; et al. Orally active anti-proliferation agents: Novel diphenylamine derivatives as FGF-R2 auto-phosphorylation inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Catoen-Chackal, S.; Facompré, M.; Houssin, R.; Pommery, N.; Goossens, J.-F.; Colson, P.; Bailly, C.; Hénichart, J.-P.; Catoen-Chackal, S.; Facompre, M.; et al. DNA Binding To Guide the Development of Tetrahydroindeno[1,2-b]pyrido[4,3,2-de]quinoline Derivatives as Cytotoxic Agents. J. Med. Chem. 2004, 47, 3665–3673. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, V.; Castagliuolo, I.; Ferlin, M.G. 3-substituted 7-phenyl-pyrroloquinolinones show potent cytotoxic activity in human cancer cell lines. J. Med. Chem. 2007, 50, 5509–5513. [Google Scholar] [CrossRef] [PubMed]
- Magedov, I.V.; Manpadi, M.; Ogasawara, M.A.; Dhawan, A.S.; Rogelj, S.; van slambrouck, S.; Steelant, W.F.A.; Evdokimov, N.M.; Uglinskii, P.Y.; Elias, E.M.; et al. Structural Simplification of Bioactive Natural Products with Multicomponent Synthesis. 2. Antiproliferative and Antitubulin Activities of Pyrano[3,2-c]pyridones and Pyrano[3,2-c]quinolones. J. Med. Chem. 2008, 51, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Chen, Y.-L.; Hsu, C.-Y.; Chen, T.-C.; Cheng, C.-M.; Tso, H.-C.; Lu, Y.-J.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of 3-phenylquinolinylchalcone derivatives against non-small cell lung cancers and breast cancers. Eur. J. Med. Chem. 2013, 59, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Chen, Y.-L.; Yang, C.-L.; Cheng, C.-M.; Han, C.-H.; Tzeng, C.-C. Synthesis of 6-substituted-9-methoxy-11H-indeno[1,2-c]quinoline-11-one derivatives as potential anticancer agents. Bioorg. Med. Chem. 2012, 20, 4397–4404. [Google Scholar] [CrossRef] [PubMed]
- Santosa, R.C.; Salvadora, J.A.R.; Cortésb, R.; Pachónd, G.; Marinb, S.; Cascanteb, M. New betulinic acid derivatives induce potent and selective antiproliferative activity through cell cycle arrest at the S phase and caspase dependent apoptosis in human cancer cells. Biochimie 2011, 93, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Szulawska-Mroczek, A.; Koprowska, K.; Stasiak, M.; Lewgowd, W.; Stanczak, A.; Czyz, M. Synthesis and in vitro biological evaluation of new polyamine conjugates as potential anticancer drugs. Eur. J. Med. Chem. 2010, 45, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Chen, Y.-L.; Lu, P.-J.; Yang, C.-N.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of certain indeno[1,2-c]quinoline derivatives. Bioorg. Med. Chem. 2008, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Chen, Y.-L.; Wang, C.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of certain pyrido[3,2-g]quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 7370–7376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Melkoumian, Z.K.; Lucktong, A.; Moniwa, M.; Davie, J.R.; Strobl, J.S. Rapid induction of histone hyperacetylation and cellular differentiation in human breast tumor cell lines following degradation of histone deacetylase-1. J. Biol. Chem. 2000, 275, 35256–35263. [Google Scholar] [CrossRef] [PubMed]
- Hengst, L.; Reed, S.I. Inhibitors of the Cip/Kip family. Curr. Top. Microbiol. Immunol. 1998, 227, 25–41. [Google Scholar] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, J.; Pagano, M. Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 2000, 183, 10–17. [Google Scholar] [CrossRef]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Catzavelos, C.; Bhattacharya, N.; Ung, Y.C.; Wilson, J.A.; Roncari, L.; Sandhu, C.; Shaw, P.; Yeger, H.; Morava-Protzner, I.; Kapusta, L.; et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat. Med. 1997, 3, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.; Baldi, A.; de Luca, A.; Groger, A.M.; Loda, M.; Giordano, G.G.; Caputi, M.; Baldi, F.; Pagano, M.; Giordano, A. Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res. 1997, 57, 3381–3385. [Google Scholar] [PubMed]
- Loda, M.; Cukor, B.; Tam, S.W.; Lavin, P.; Fiorentinc, M.; Draetta, G.F.; Jessup, J.M.; Pagano, M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med. 1997, 3, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Masciullo, V.; Sgambato, A.; Pacilio, C.; Pucci, B.; Ferrandina, G.; Palazzo, J.; Carbone, A.; Cittadini, A.; Mancuso, S.; Scambia, G.; et al. Frequent loss of expression of the cyclin-dependent kinase inhibitor p27 in epithelial ovarian cancer. Cancer Res. 1999, 59, 3790–3794. [Google Scholar] [PubMed]
- Porter, P.L.; Barlow, W.E.; Yeh, I.-T.; Lin, M.G.; Yuan, X.P.; Donato, E.; Sledge, G.W.; Shapiro, C.L.; Ingle, J.N.; Haskell, C.M.; et al. p27Kip1 and cyclin E expression and breast cancer survival after treatment with adjuvant chemotherapy. J. Natl. Cancer Inst. 2006, 98, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Tsihlias, J.; Kapusta, L.R.; DeBoer, G.; Morava-Protzner, I.; Zbieranowski, I.; Bhattacharya, N.; Catzavelos, G.C.; Klotz, L.H.; Slingerland, J.M. Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res. 1998, 58, 542–548. [Google Scholar] [PubMed]
- Barata, J.T.; Cardoso, A.A.; Nadler, L.M.; Boussiotis, V.A. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 2001, 98, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Hoque, M. O.; Bando, T.; Yoshida, H.; Sato, M. Over expression of p27Kip1 induces growth arrest and apoptosis in an oral cancer cell line. Oral Oncol. 2002, 38, 730–736. [Google Scholar]
- Jia, W.; Liu, Y.; Li, W.; Liu, Y.; Zhang, D.; Zhang, P.; Gong, P. Synthesis and in vitro anti-hepatitis B virus activity of 6H-[1]benzothiopyrano[4,3-b]quinolin-9-ols. Bioorg. Med. Chem. 2009, 17, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Meth-Cohn, O.; Narine, B.; Tarnowski, B. A versatile new synthesis of quinolines and related fused pyridines, Part 5. The synthesis of 2-chloroquinoline-3-carbaldehydes. J. Chem. Soc. Perkin. Trans. 1 1981, 1, 1520–1530. [Google Scholar] [CrossRef]
- French, H.E.; Wirtel, A.F. Alpha-naphthylisocynate as a regaent for phenols and aliphatic amines. J. Am. Chem. Soc. 1926, 48, 1736–1739. [Google Scholar] [CrossRef]
- Gligorijević, N.; Todorović, T.; Radulović, S.; Sladić, D.; Filipović, N.; Gođevac, D.; Jeremić, D.; Anđelković, K. Synthesis and characterization of new Pt(II) and Pd(II) complexes with 2-quinolinecarboxaldehyde seleno-semicarbazone: Cytotoxic activity evaluation of Cd(II), Zn(II), Ni(II), Pt(II) and Pd(II) complexes with heteroaromatic. Eur. J. Med. Chem. 2009, 44, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, E.; Thomas, S.P.; Poornima, P.; Kalaivani, P.; Prabhakaran, R.; Padma, V.V.; Natarajan, K. Evaluation of DNA binding, antioxidant and cytotoxic activity of mononuclear Co(III) complexes of 2-oxo-1,2-dihydrobenzo[H]quinoline-3-carbaldehyde thiosemicarbazones. Eur. J. Med. Chem. 2012, 50, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Serda, M.; Kalinowski, D.S.; Mrozek-Wilczkiewicz, A.; Musiol, R.; Szurko, A.; Ratuszna, A.; Pantarat, N.; Kovacevic, Z.; Merlot, A.M.; Richardson, D.R.; et al. Synthesis and characterization of quinoline-based thiosemicarbazones and correlation of cellular iron-binding efficacy to anti-tumor efficacy. Bioorg. Med. Chem. Lett. 2012, 22, 5527–5531. [Google Scholar] [CrossRef] [PubMed]
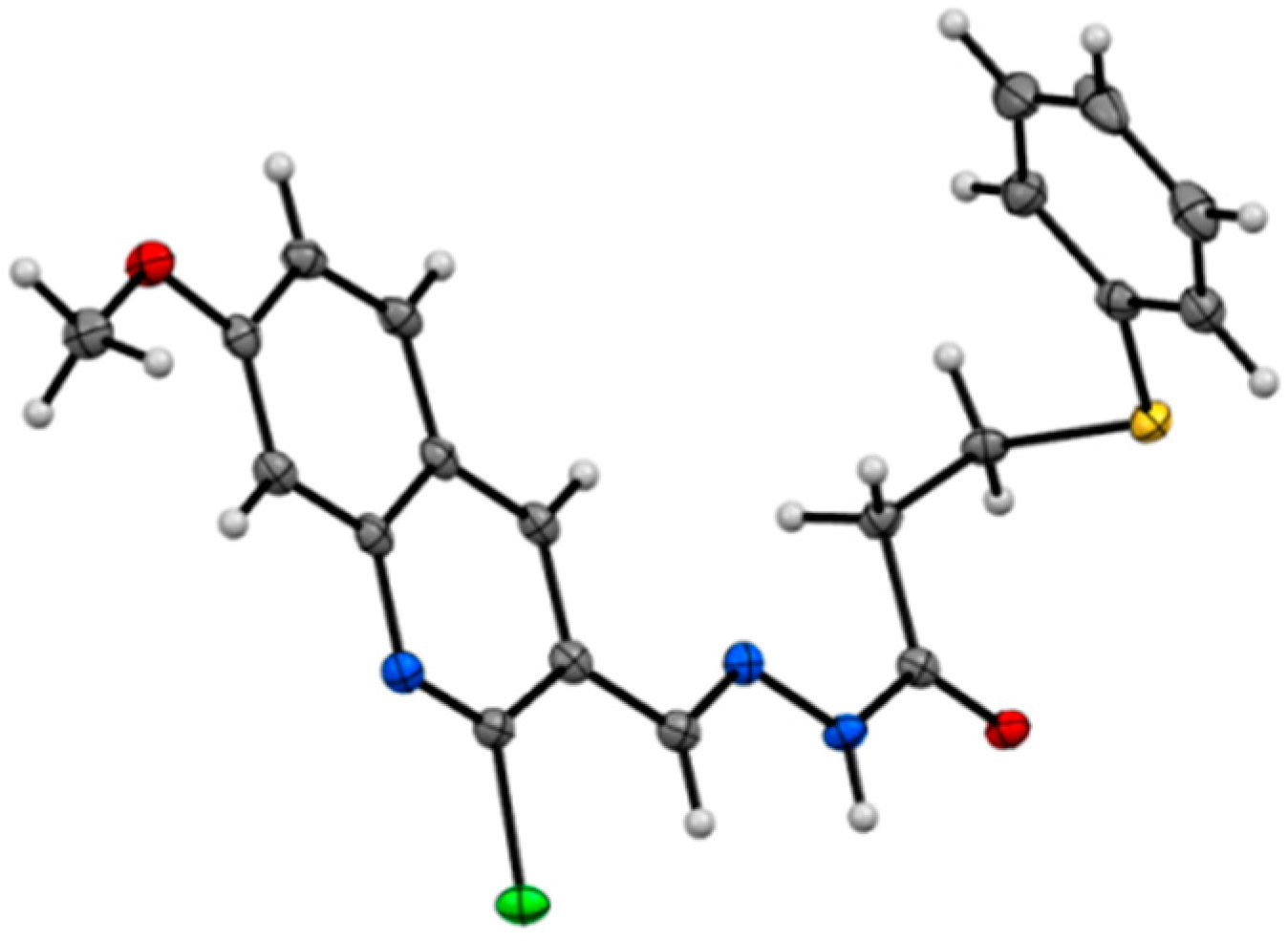
- CCDC 1436247 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk)
- Cheung, B.; Yan, J.; Smith, S.A.; Nguyen, T.; Lee, M.; Kavallaris, M.; Norris, M.D.; Haber, M.; Marshall, G.M. Growth inhibitory retinoid effects after recruitment of retinoid X receptor β to the retinoic acid receptor β promoter. Int. J. Cancer 2003, 105, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.M.F.; Orchison, J.J.A.; Whiting, D.A. A new synthetic approach to the rotenoid ring system. Tetrahedron 1989, 45, 5895–5906. [Google Scholar] [CrossRef]
- Hardouin, C.; Kelso, M.J.; Romero, F.A.; Rayl, T.J.; Leung, D.; Hwang, I.; Cravatt, B.F.; Boger, D.L. Structure-activity relationships of alpha-ketooxazole inhibitors of fatty acid amide hydrolase. J. Med. Chem. 2007, 50, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Harvill, E.K.; Herbst, R.M.; Schreiner, E.C.; Roberts, C.W. The Synthesis of 1,5-disubstituted tetrazoles. J. Org. Chem. 1950, 15, 662–670. [Google Scholar] [CrossRef]
- Ecke, G.G.; Napolitano, J.P.; Kolka, A.J. The ortho-alkylation of aromatic amines. J. Org. Chem. 1956, 21, 711–712. [Google Scholar] [CrossRef]
- Horning, E.C.; Horning, M.G. Aromatization Studies. V. Synthesis of alkylanilines from alkylcyclohexenones. J. Am. Chem. Soc. 1947, 69, 1907–1908. [Google Scholar] [CrossRef]
- Shvo, Y.; Arisha, A.H.I. Regioselective Catalytic dehydrogenation of aldehydes and ketones. J. Org. Chem. 1998, 63, 5640–5642. [Google Scholar] [CrossRef]
- Prakash Naik, H.R.; Bhojya Naik, H.S.; Ravikumar Naik, T.R.; Raghavendra, M.; Aravinda, T.; Lamani, D.S. Synthesis of quinoline-based thieno-seleno-phenylquinazolinones. Phosphorus Sulfur. 2009, 184, 460–470. [Google Scholar] [CrossRef]
- Doboszewski, B.; Herdewijn, P. Carbohydrate chiral-pool approach to four enantiomerically pure 2-naphthylmethyl 3-hydroxy-2-methylbutanoates. Tetrahedron 2008, 64, 5551–5562. [Google Scholar] [CrossRef]
- El-Rayyes, N.R.; Katrib, A.H. Synthesis and Xray photoelectron spectra of some azines. J. Chem. Eng. Data 1983, 28, 132–134. [Google Scholar] [CrossRef]
- Forbes, E.J.; Stacey, M.; Tatlow, J.C.; Wragg, R.T. The synthesis of 1-, 2- and 3-trifluoromethylcarbazoles by the fischer-indole method. Tetrahedron 1960, 8, 67–72. [Google Scholar] [CrossRef]
- Marshall, G.M.; Bell, J.L.; Koach, J.; Tan, O.; Kim, P.; Malyukova, A.; Thomas, W.; Sekyere, E.O.; Liu, T.; Cunningham, A.M.; et al. TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene 2010, 29, 6172–6183. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 14, 15, 16, 17, 18, 19, 26, 27 and 28 are available from the authors.
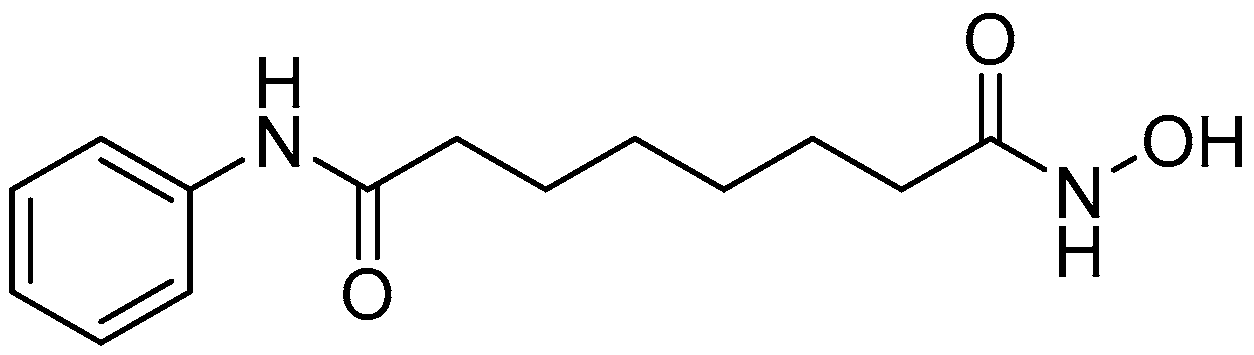
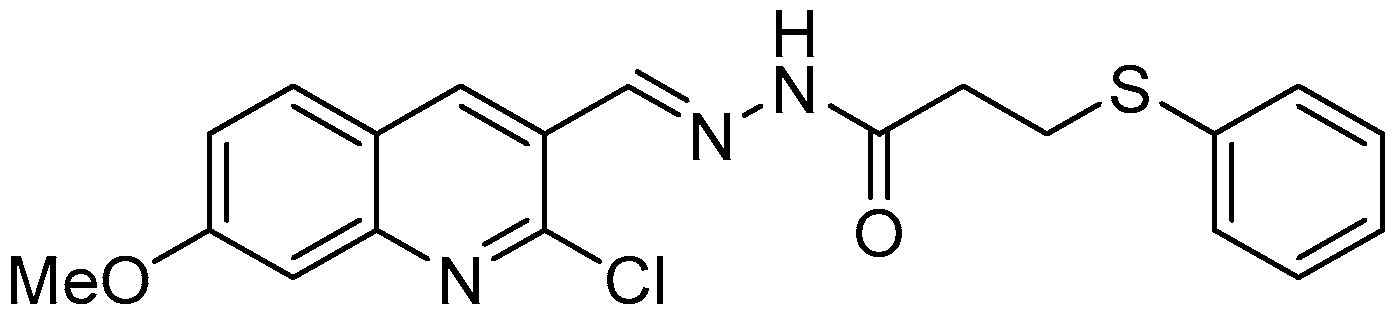


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Viability (% of Negative Control) | |||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | SH-SY5Y | Kelly | |
| 14 | 98.5 ± 1.0 | 93.8 ± 3.6 | 104.2 ± 6.2 | 80.0 ± 13.7 |
| 15 | 94.8 ± 3.8 | 86.9 ± 0.5 | 74.3 ± 1.0 | 52.7 ± 8.7 |
| 16 | 88.4 ± 3.8 | 72.0 ± 2.6 | 32.4 ± 6.4 | 12.4 ± 3.2 |
| 17 | 66.8 ± 1.2 | 61.8 ± 8.4 | 18.4 ± 7.0 | 3.7 ± 1.0 |
| 18 | 93.1 ± 0.6 | 100.2 ± 2.0 | 92.8 ± 6.6 | 89.7 ± 13.7 |
| 19 | 85.6 ± 4.9 | 78.0 ± 4.1 | 85.8 ± 5.5 | 65.2 ± 10.5 |
| 26 | 93.1 ± 1.0 | 71.5 ± 1.5 | 71.0 ± 5.0 | 47.6 ± 9.9 |
| 27 | 95.1 ± 1.7 | 76.4 ± 8.0 | 78.8 ± 2.5 | 70.2 ± 13.7 |
| 28 | 93.1 ± 3.8 | 87.9± 1.5 | 83.6 ± 4.9 | 70.6 ± 2.4 |
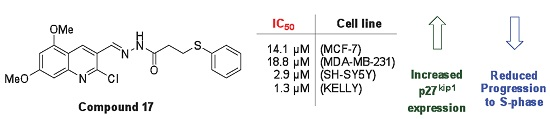
| Compound | IC50 (µM) a | |||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | SH-SY5Y | Kelly | |
| 16 | >25.0 | 22.1 | 5.7 | 2.4 |
| 17 | 14.1 | 18.8 | 2.9 | 1.3 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bingul, M.; Tan, O.; Gardner, C.R.; Sutton, S.K.; Arndt, G.M.; Marshall, G.M.; Cheung, B.B.; Kumar, N.; Black, D.S. Synthesis, Characterization and Anti-Cancer Activity of Hydrazide Derivatives Incorporating a Quinoline Moiety. Molecules 2016, 21, 916. https://doi.org/10.3390/molecules21070916
Bingul M, Tan O, Gardner CR, Sutton SK, Arndt GM, Marshall GM, Cheung BB, Kumar N, Black DS. Synthesis, Characterization and Anti-Cancer Activity of Hydrazide Derivatives Incorporating a Quinoline Moiety. Molecules. 2016; 21(7):916. https://doi.org/10.3390/molecules21070916
Chicago/Turabian StyleBingul, Murat, Owen Tan, Christopher R. Gardner, Selina K. Sutton, Greg M. Arndt, Glenn M. Marshall, Belamy B. Cheung, Naresh Kumar, and David StC. Black. 2016. "Synthesis, Characterization and Anti-Cancer Activity of Hydrazide Derivatives Incorporating a Quinoline Moiety" Molecules 21, no. 7: 916. https://doi.org/10.3390/molecules21070916
APA StyleBingul, M., Tan, O., Gardner, C. R., Sutton, S. K., Arndt, G. M., Marshall, G. M., Cheung, B. B., Kumar, N., & Black, D. S. (2016). Synthesis, Characterization and Anti-Cancer Activity of Hydrazide Derivatives Incorporating a Quinoline Moiety. Molecules, 21(7), 916. https://doi.org/10.3390/molecules21070916