
Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Prediction of Drug-Likeness
2.2. Docking Assessment
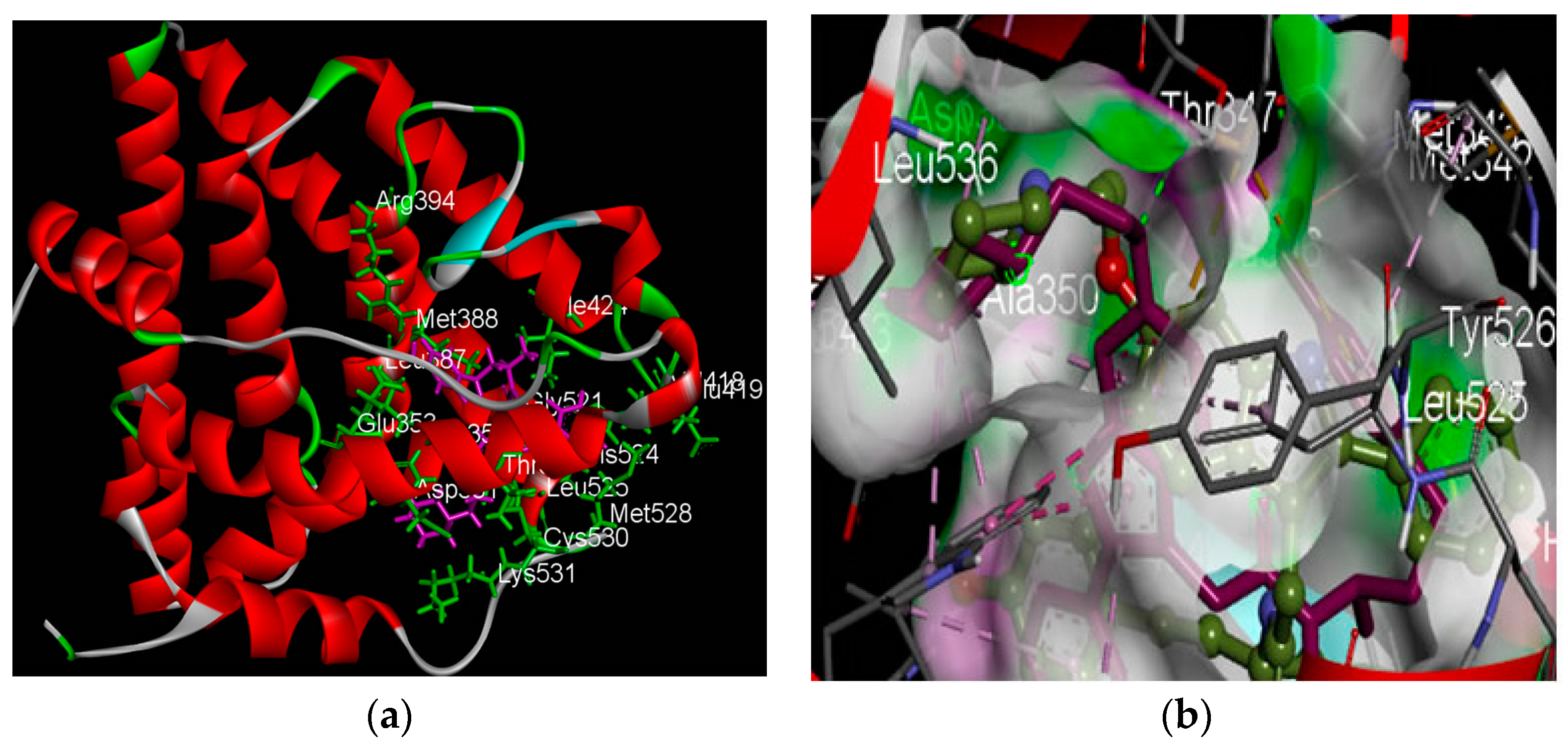
2.2.1. Structure of the Human Estrogen Receptor α, 2IOG
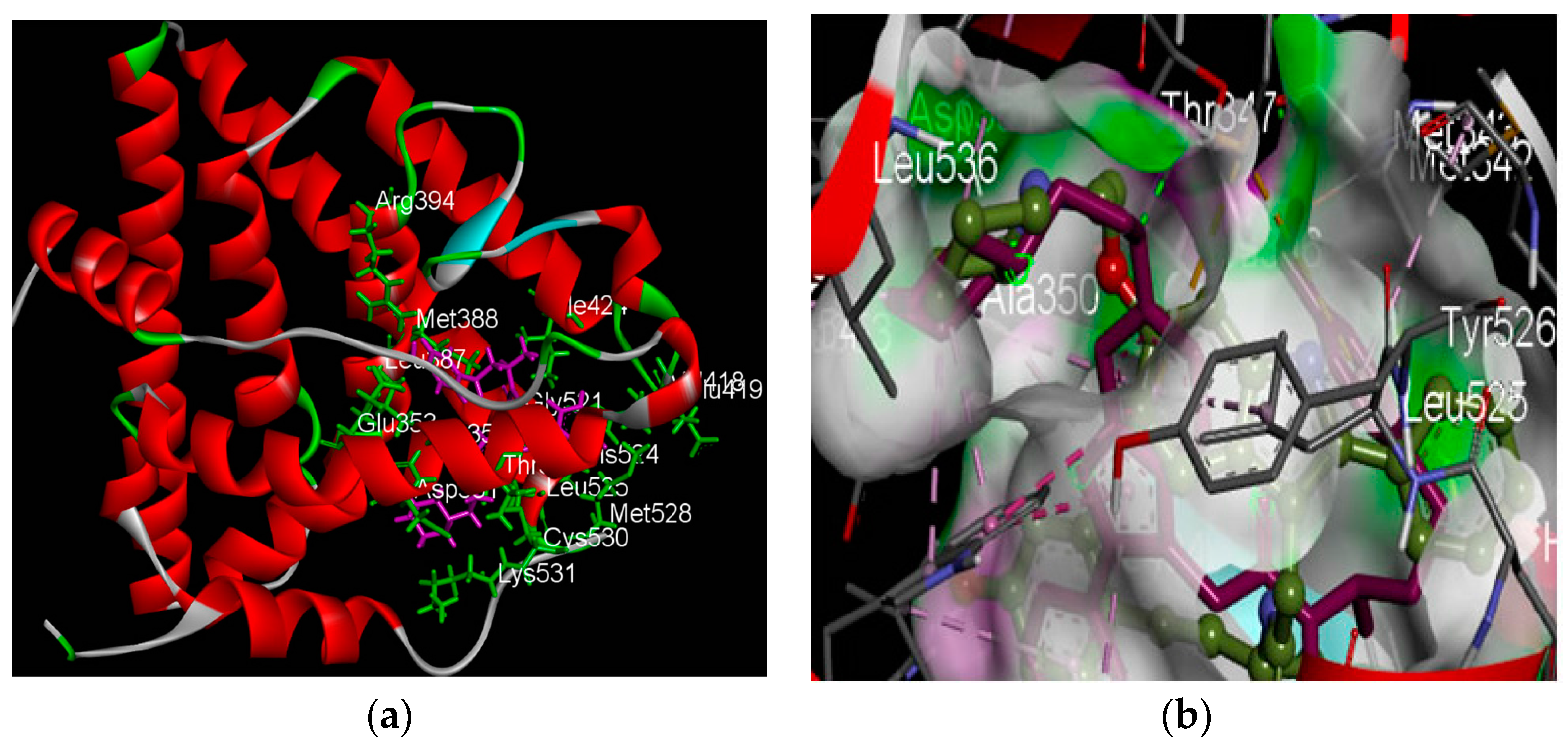
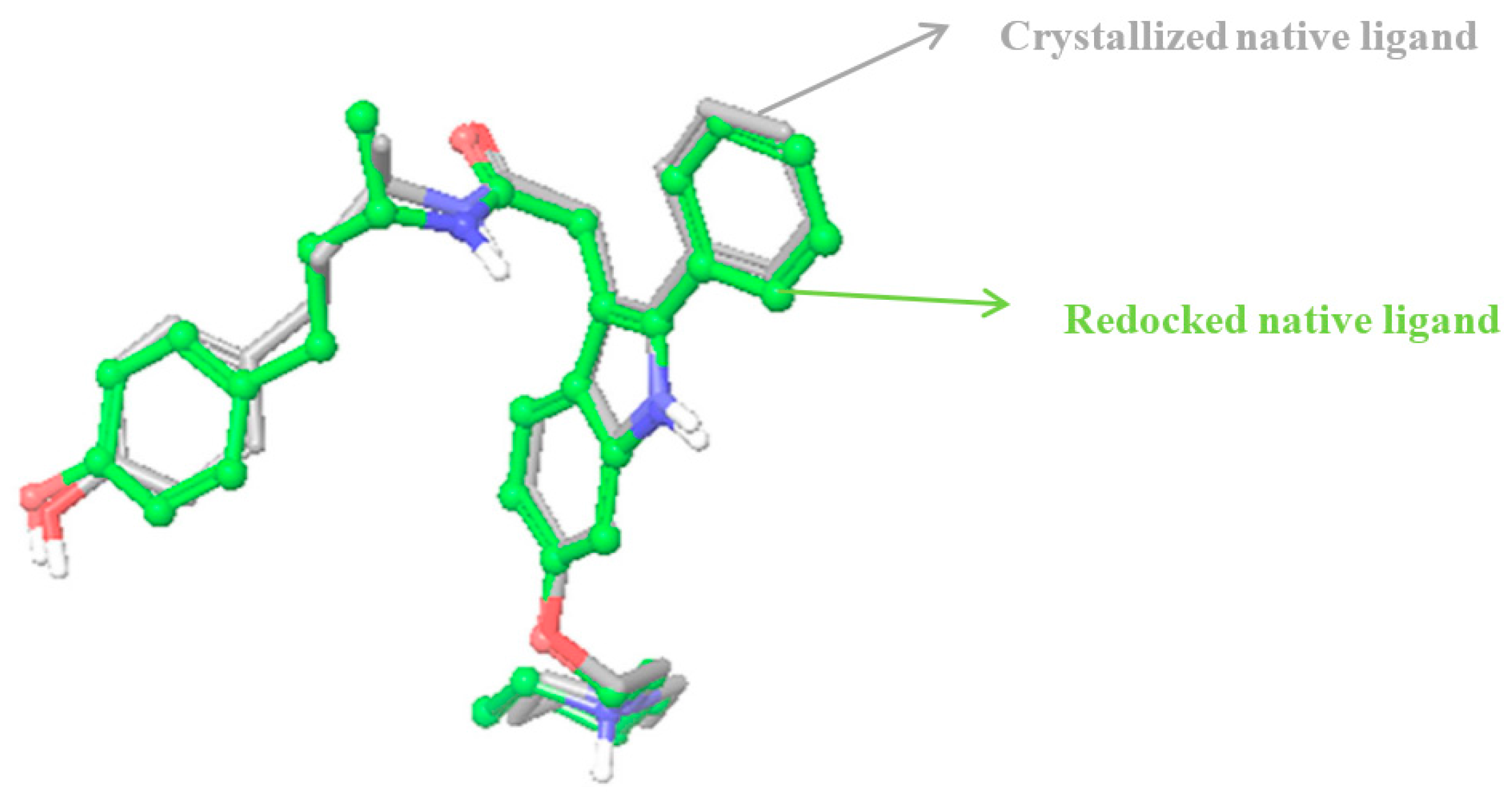
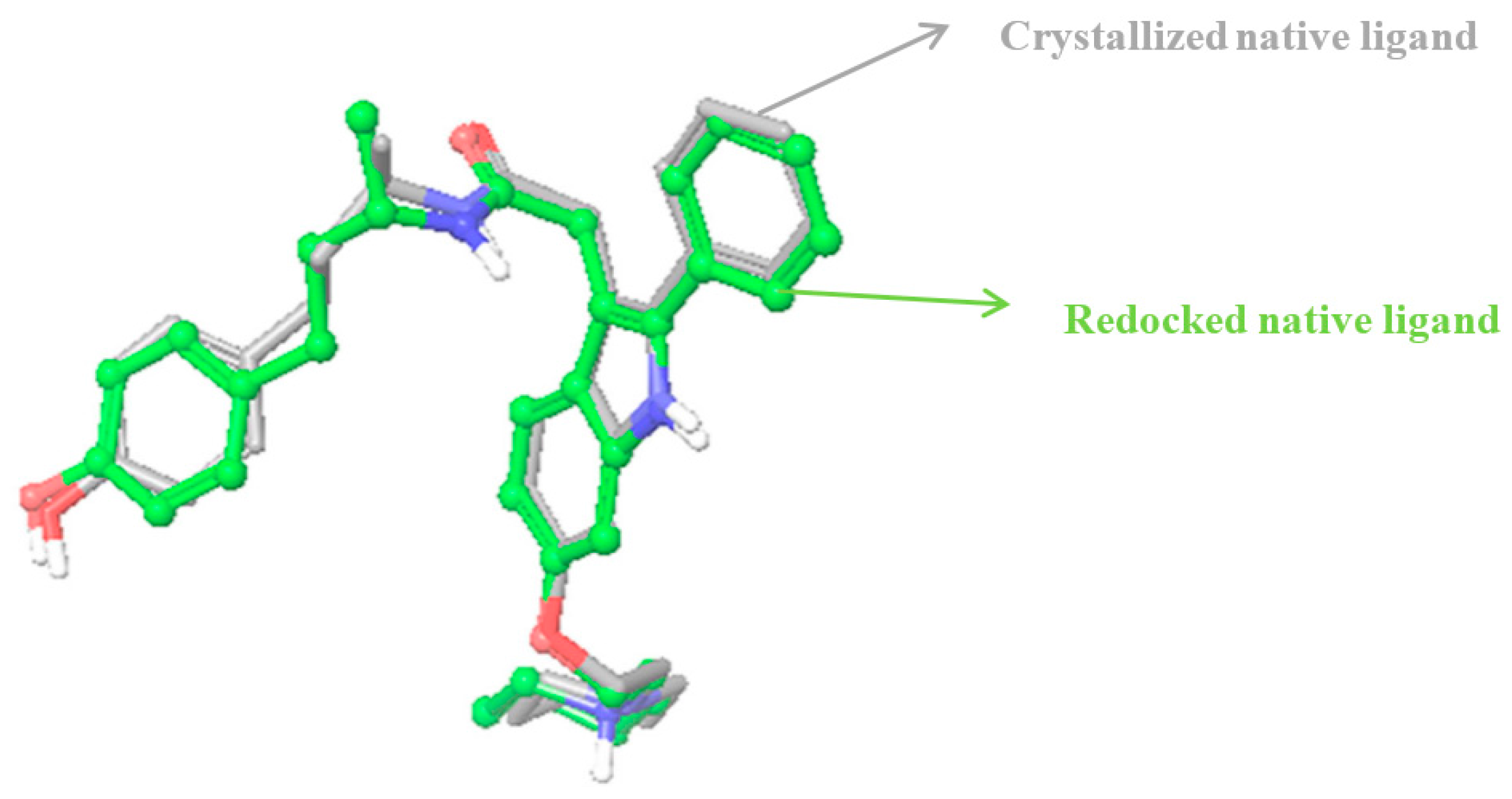
2.2.2. Identification of Estrogen Receptor-Binding Pockets and Validation of Docking Protocol
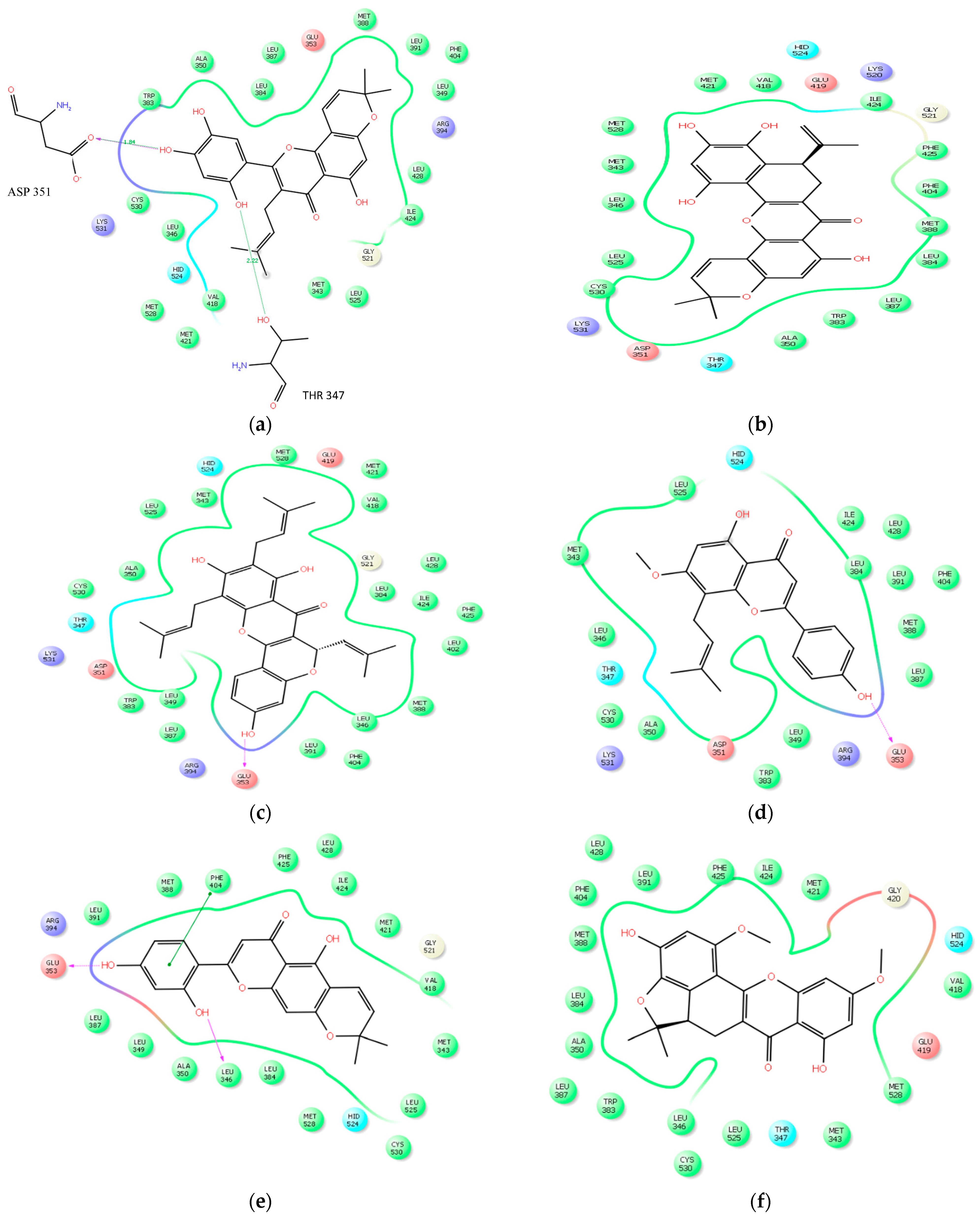
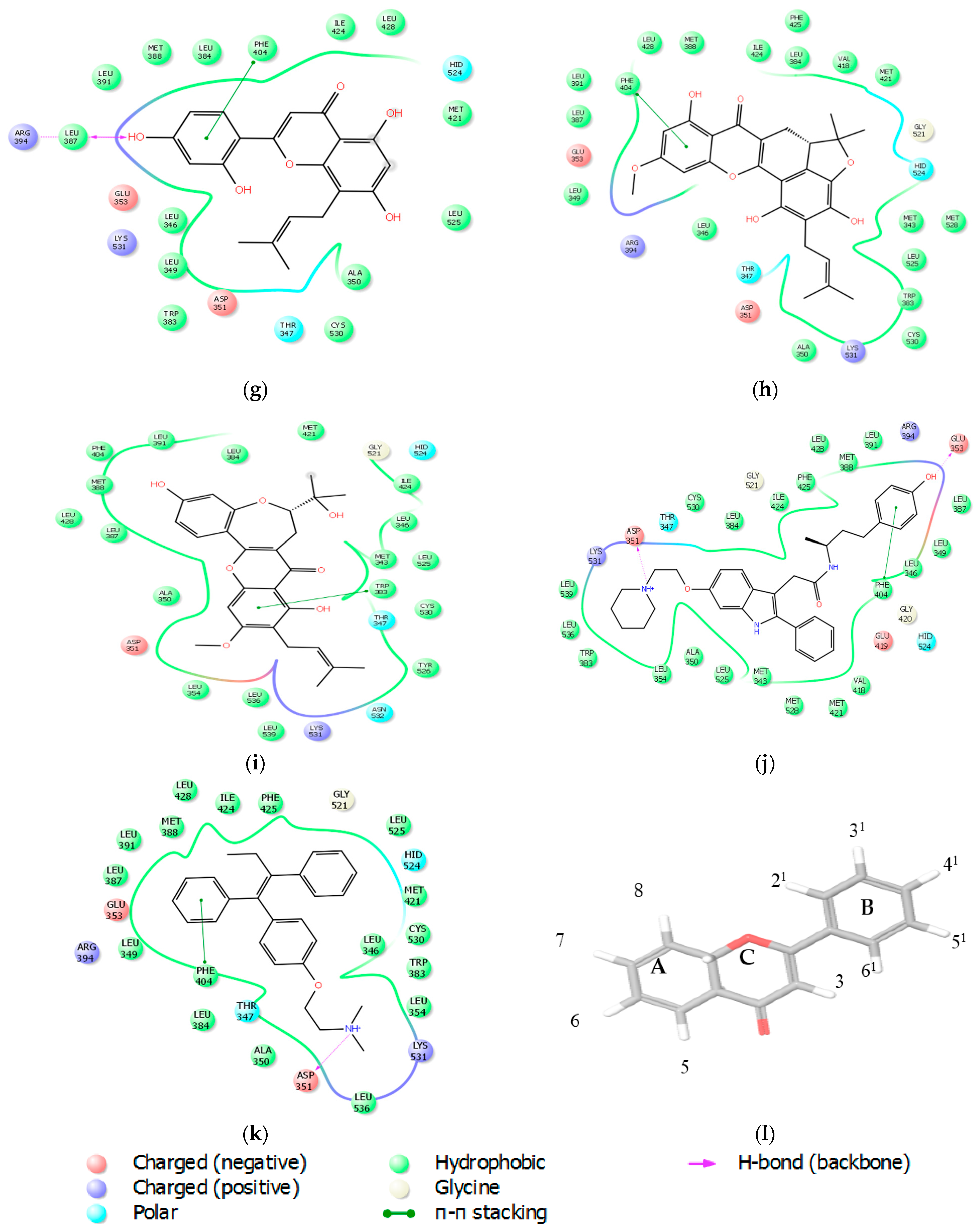
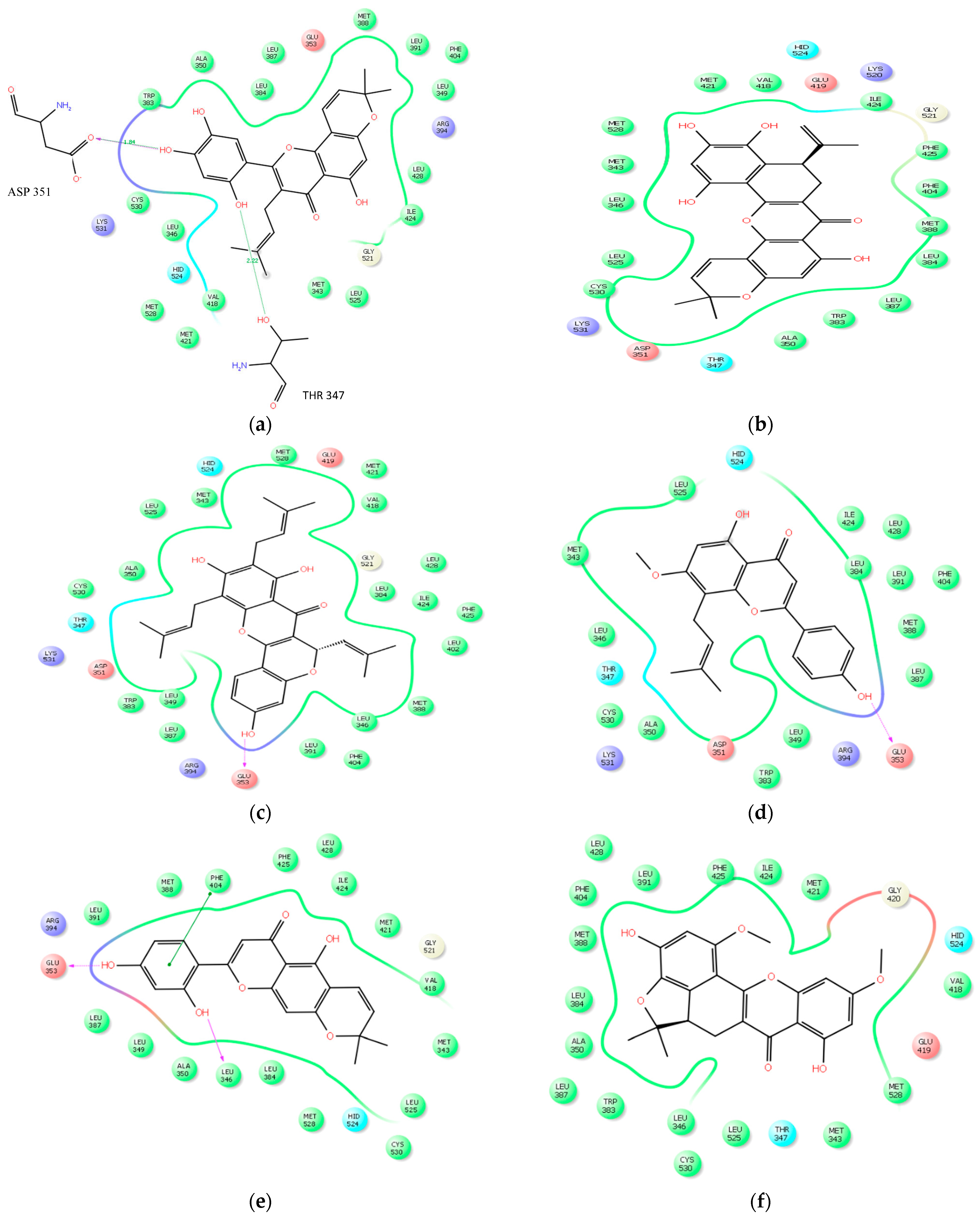
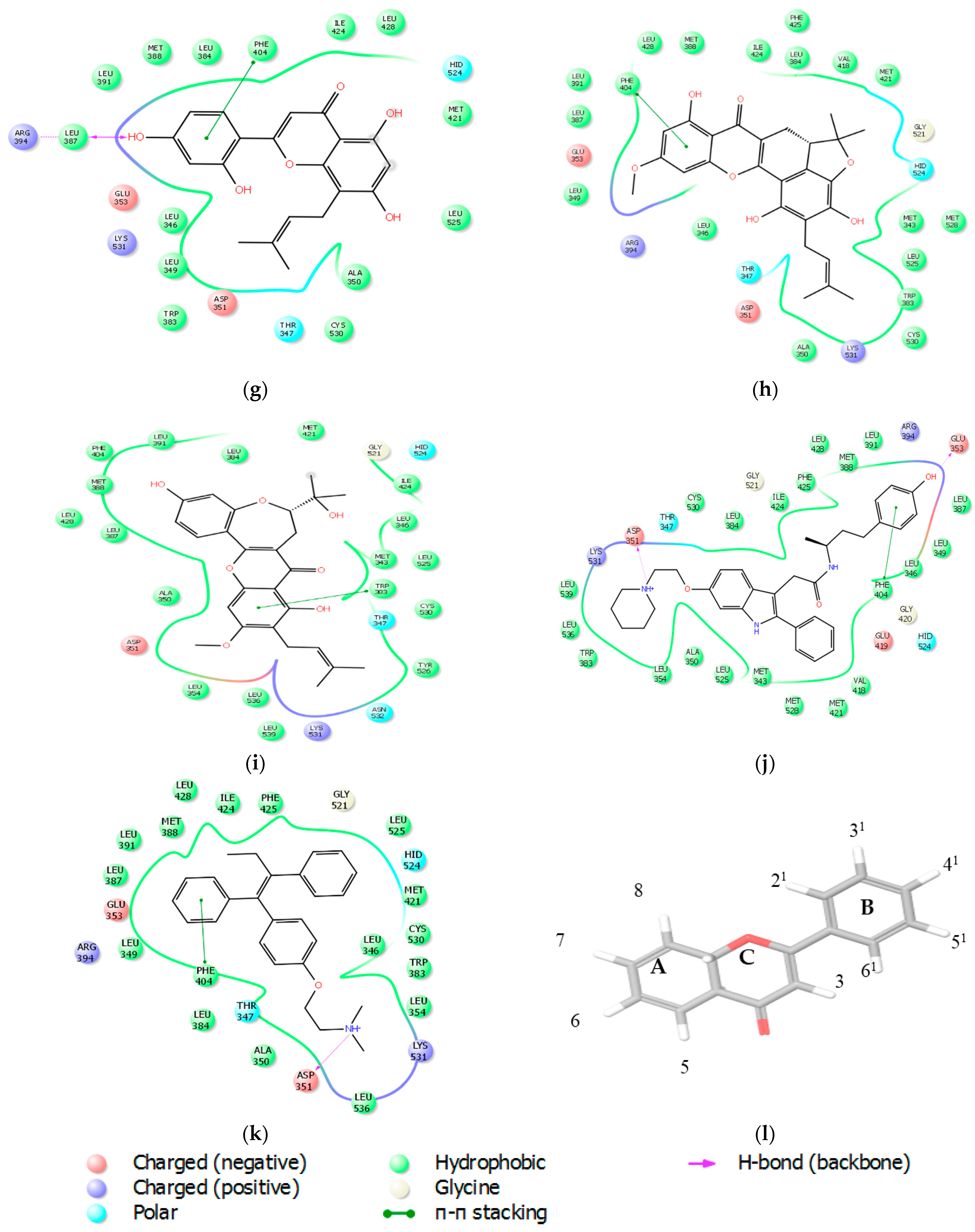
2.2.3. Docking Analysis
2.3. Prime Energy Analysis
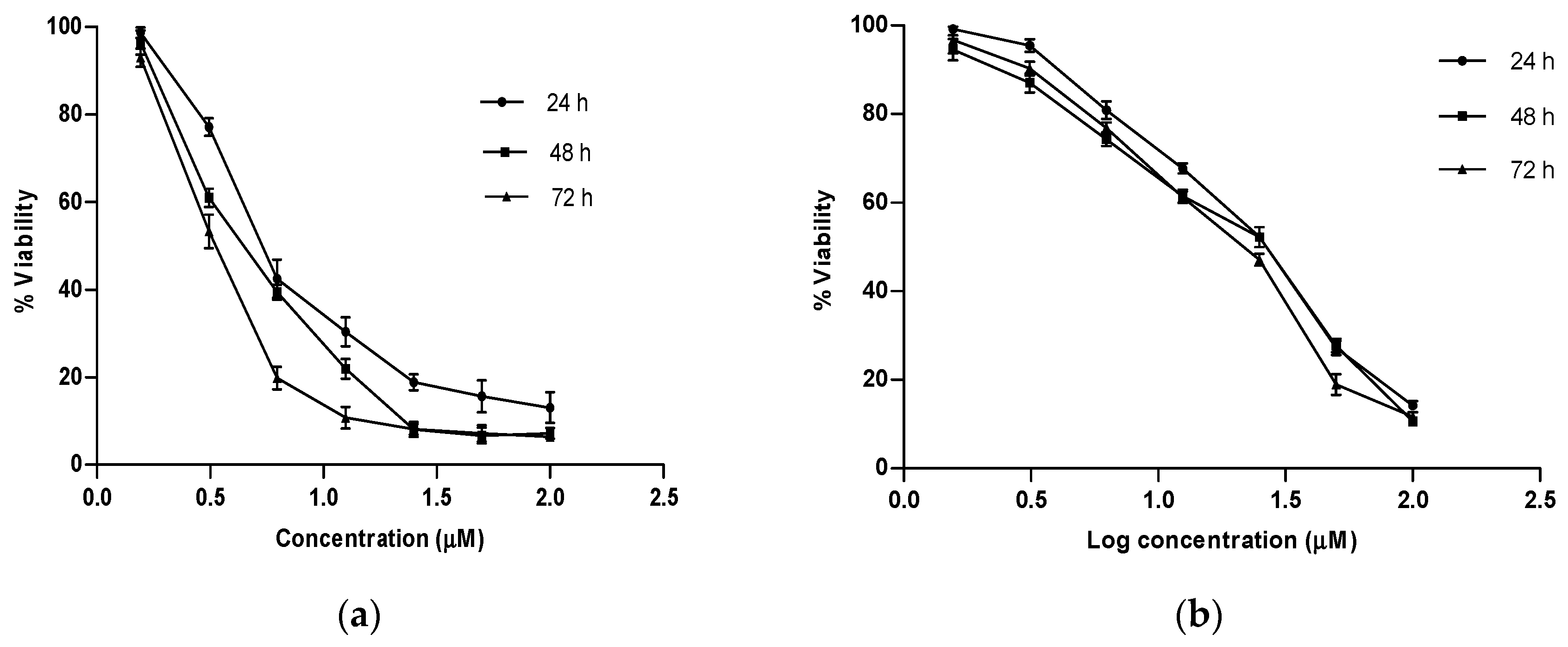
2.4. In Vitro Growth Inhibition Assay
3. Materials and Methods
3.1. Preparation of Ligands
3.2. Determination of ADMET Properties of the Compounds
3.3. Molecular Docking Studies
3.3.1. Identification of Binding Pockets and Validation of Docking Protocol
3.3.2. Preparation of Protein
3.3.3. Molecular Docking Studies
3.4. Prime Energy Analysis
| MM-GBSA_∆G_bind | Ligand binding energy, |
| MM-GBSA_E_complex | Energy of the complex, |
| MM-GBSA_E_protein | Energy of the receptor without the ligand, |
| MM-GBSA_E_ligand | Energy of the unbound ligand, |
| Prime Coulomb energy of the complex | coulomb |
| Prime Van der Waals energy of the complex | vdW |
| Prime Hydrogen Bond of the Complex | Hbond |
3.5. Preparation of Drugs
3.6. Cell Culture
3.7. Growth Inhibitory Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hamilton, K.; Arao, Y.; Korach, K. Estrogen hormone physiology: Reproductive findings from estrogen receptor mutant mice. Reprod. Biol. 2014, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanism of Estrogen Action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [PubMed]
- Burns, K.; Korach, K. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 10, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.E.; Qian, Y.; Liu, G.; Chen, H.; Chen, X. p53, A Target of Estrogen Receptor (ER), Modulates DNA Damage-induced Growth Suppression in ER-positive Breast Cancer Cells. J. Biol. Chem. 2012, 287, 30117–30127. [Google Scholar] [CrossRef] [PubMed]
- Belev, B.; Vrbanec, D. Hormonal resistance in breast- and prostate cancer. Period Biol. 2012, 114, 511–517. [Google Scholar]
- Andruska, N.D.; Zheng, X.; Yang, X.; Mao, C.; Cherian, M.M.; Mahapatra, L. Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc. Natl. Acad. Sci. USA 2015, 112, 4737–4742. [Google Scholar] [CrossRef] [PubMed]
- Kochummen, K.M.; Go, R. Tree of Sabah and Sarawak. In Moraceae; Seopadmo, E., Saw, L.G., Eds.; Ampang Press: Kuala Lumpur, Malaysia, 2000; pp. 181–334. [Google Scholar]
- Jagtap, U.; Bapat, V.A. Artocarpus: A review of its traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2010, 129, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yan, G.-R.; Pan, S.-L.; Wang, H.-Y.; Hou, A.-J. New isoprenylated 2-arylbenzofurans and pancreatic lipase inhibitory constituents from Artocarpus nitidus. Chem. Biodivers. 2009, 6, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Boonphong, S.; Baramee, A.; Kittakoop, P. Antitubercular and Antiplasmodial Prenylated Flavones from the Roots of Artocarpus altilis. Chiang Mai J. Sci. 2007, 34, 339–344. [Google Scholar]
- Jayasinghe, U.L.B.; Samarakoon, T.B.; Kumarihamy, B.M.M. Four new prenylated flavonoids and xanthones from the root bark of Artocarpus nobilis. Fitoterapia 2008, 79, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Jamil, S.; King, A.; Jamil, S.; Mariam, S.; Lathiff, A.; Abdullah, S.A. Antimicrobial Flavonoids from Artocarpus anisophyllus Miq and Artocarpus lowii King. J. Teknol. 2014, 71. [Google Scholar] [CrossRef]
- Wongpankam, E.; Chunhacha, P.; Pongrakhananon, V. Artonin E Mediates MCL1 Down-regulation and Sensitizes Lung Cancer Cells to Anoikis. Anticancer Res. 2012, 32, 5343–5351. [Google Scholar] [PubMed]
- Ramli, F.; Rahmani, M.; Sukari, A.; Hashim, N.M. New Flavonoids Derivatives from Artocarpus Elasticus with Antioxidant, Antimicrobial and Cyotoxic Activities. Open Conf. Proc. J. 2013, 4, 2013. [Google Scholar] [CrossRef]
- López-Vallejo, F.; Caulfield, T.; Martínez-Mayorga, K.; Giulianotti, M.A.; Nefzi, A.; Houghten, R.A.; Medina-Franco, J.L. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High Throughput Screen. 2011, 6, 475–487. [Google Scholar] [CrossRef]
- Jacobs, M. In silico tools to aid risk assessment of endocrine disrupting chemicals. Toxicology 2004, 205, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Malami, I.; Abdul, A.; Abdullah, R.; Bt Kassim, N.; Waziri, P.; Christopher Etti, I. In silico Discovery of Potential Uridine-Cytidine Kinase 2 Inhibitors from the Rhizome of Alpinia mutica. Molecules 2016, 21, 417. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.F.P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Delaney, J.S. Predicting aqueous solubility from structure. Drug Discov. Today 2005, 4, 289–295. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 12, 2615–2623. [Google Scholar] [CrossRef]
- QikProp, Version 4.6; Schrödinger, LLC: New York, NY, USA, 2015.
- Usansky, H.H.; Sinko, P.J. Estimating Human Drug Oral Absorption Kinetics from Caco-2 Permeability Using an Absorption-Disposition Model: Model Development and Evaluation and Derivation of Analytical Solutions for ka and Fa. J. Pharmacol. Exp. Ther. 2005, 314, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B Elsevier 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Suganya, J.; Radha, M.; Naorem, D.L. In silico Docking Studies of Selected Flavonoids—Natural Healing Agents against Breast Cancer. Asian Pac. J. Cancer Prev. 2014, 15, 8155–8159. [Google Scholar] [CrossRef] [PubMed]
- Henrich, S.; Salo-ahen, O.M.H.; Huang, B.; Rippmann, F.F.; Cruciani, G.; Wade, R.C. Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Mol. Recognit. 2010, 23, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Celik, L.; Lund, J.; Schiøtt, B. Exploring Interactions of Endocrine-Disrupting Compounds with Different Conformations of the Human Estrogen Receptor α Ligand Binding Domain: A Molecular Docking Study. Chem. Res. Toxicol. 2008, 21, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Josh, M.K.S.; Pradeep, S.; Adarsh, V.K.; Amma, K.S.V.; Devi, R.S.; Balachandran, S.; Sreejith, M.N.; Jaleel, U.C.A.; Benjamin, S. In silico evidences for the binding of phthalates onto human estrogen receptor α, β subtypes and human estrogen-related receptor γ. Mol. Simul. 2014, 40, 408–417. [Google Scholar] [CrossRef]
- Desai, N.; Mahto, M.K.; Alekhya, B.; Naveen, C.R.; Bhaskar, P.M. Comparative docking studies of estrogen receptor inhibitors and their binding interaction analysis. Int. J. Pharm. Sci. Rev. Res. 2012, 16, 91–95. [Google Scholar]
- Dykstra, K.D.; Guo, L.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Hayes, E.C.; DaSilva, C.A.; Pai, L.Y.; Mosley, R.T.; Kraker, B.; et al. Estrogen receptor ligands. Part 16: 2-Aryl indoles as highly subtype selective ligands for ER? Bioorg. Med. Chem. Lett. 2007, 17, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.R.; Ueda, N.; Hada, T.; Sackeyfio, A.C.; Yamamoto, S.; Hano, Y.; Aida, M.; Nomura, T. A prenylflavone, artonin E, as arachidonate 5-lipoxygenase inhibitor. Biochem. Pharmacol. 1991, 41, 115–118. [Google Scholar] [CrossRef]
- Jiang, Q.; Zheng, S.; Wang, G. Development of new estrogen receptor-targeting therapeutic agents for tamoxifen-resistant breast cancer. Future Med. Chem. 2013, 9. [Google Scholar] [CrossRef]
- Prime, Version 4.2; Schrodinger, LLC: New York, NY, USA, 2015.
- Abu, N.; Akhtar, M.N.; Ho, W.Y.; Yeap, S.K.; Alitheen, N.B. 3-Bromo-1-hydroxy-9,10-anthraquinone (BHAQ) inhibits growth and migration of the human breast cancer cell lines MCF-7 and MDA-MB231. Molecules 2013, 18, 10367–10377. [Google Scholar] [CrossRef] [PubMed]
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef] [PubMed]
- LigPrep, Version 3.6; Schrödinger, LLC: New York, NY, USA, 2015.
- Ekins, S.; Waller, C.; Swaan, P.; Cruciai, G.; Wrigton, S.; Wikel, J. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods 2000, 44, 251–272. [Google Scholar] [CrossRef]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. Computed atlas of surface topography of proteins. Nucleic Acids Nucleic Acid Res. 2003, 13, 3352–3355. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. 2001, 28, 6474–6487. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Ramli, F.; Karimian, H.; Dehghan, F.; Nordin, N.; Mohd Ali, H.; Mohan, S.; Mohd Hashim, N. Artonin E Induces Apoptosis via Mitochondrial Dysregulation in SKOV-3 Ovarian Cancer Cells. PLoS ONE 2016, 11, e0151466. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of Artonin E are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | MW | SASA | Donor HB | Accepted HB | QPlogPo/w | QPlogS | QPPCaco | QPlogBB | # Metab | % Human-Oral Absorption | Lipinskis Rule of Five |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Artonin E | 436.5 | 734.1 | 3 | 5.3 | 3.9 | −6.4 | 124.6 | −1.9 | 7 | 88 | 0 |
| Artobiloxanthone | 434.4 | 689.9 | 3 | 5.3 | 3.6 | −5.8 | 156.3 | −1.6 | 7 | 87 | 0 |
| Cycloartocarpesin | 352.3 | 608.4 | 2 | 4.5 | 2.9 | −5.1 | 193.3 | −1.4 | 3 | 85 | 0 |
| Artoelastin | 488.6 | 821.3 | 2 | 4.5 | 6.2 | −8.1 | 704.4 | −1.2 | 12 | 100 | 1 |
| Artonin Y | 354.4 | 580.3 | 3 | 4.5 | 2.4 | −4.0 | 75.4 | −1.8 | 7 | 75 | 0 |
| Artonin U | 352.4 | 618.9 | 1 | 3.8 | 3.9 | −5.4 | 380.3 | −1.2 | 6 | 96 | 0 |
| Artonin L | 396.4 | 635.0 | 1 | 5.3 | 3.5 | −5.3 | 461.1 | −1.0 | 6 | 95 | 0 |
| Artonin T | 450.5 | 718.9 | 2 | 5.3 | 4.5 | −6.3 | 451.0 | −1.2 | 9 | 100 | 0 |
| Artonin S | 452.5 | 728.0 | 2 | 5.3 | 4.6 | −6.4 | 333.7 | −1.4 | 8 | 100 | 0 |
| Tamoxifen | 371.5 | 730.6 | 0 | 2.6 | 6.6 | −5.9 | 2203.2 | 0.4 | 3 | 100 | 1 |
| Native Ligand | 547.9 | 1021.8 | 4 | 10.1 | 4.3 | −4.9 | 20.7 | −0.6 | 4 | 62.9 | 1 |
| Ligands | Glide Score | THR 347 | ASP 351 | GLU 353 | ARG 394 | ||||
|---|---|---|---|---|---|---|---|---|---|
| VDW | Coloumb | VDW | Coloumb | VDW | Coloumb | VDW | Coloumb | ||
| Native ligand | −16.81 | −2.46 | −3.23 | −1.99 | −50.43 | 0.63 | −23.30 | −0.24 | 7.52 |
| Tamoxifen | −13.93 | −2.35 | −2.36 | −1.47 | −40.68 | −0.85 | −17.79 | −0.26 | 13.57 |
| Artonin E | −12.72 | −2.35 | −1.76 | −1.03 | −12.11 | −1.42 | −2.87 | −0.60 | 1.21 |
| Cycloartocarpesin | −11.72 | −0.41 | −0.23 | −0.10 | −1.09 | 1.19 | −13.83 | 0.15 | −1.67 |
| Artonin U | −11.03 | −3.21 | 0.27 | −0.74 | −1.51 | −0.11 | −11.60 | −0.35 | −1.43 |
| Artoelastin | −10.90 | −3.35 | −0.11 | −1.16 | −1.32 | −1.05 | −10.05 | −0.45 | −1.58 |
| Artonin L | −10.70 | −1.11 | 0.36 | −0.15 | −0.34 | −0.24 | −1.45 | −0.06 | 0.79 |
| Artobiloxanthone | −10.50 | −1.21 | −1.26 | 0.69 | −1.20 | −0.22 | 2.33 | −0.04 | −1.63 |
| Artonin Y | −10.50 | −2.89 | −0.77 | −0.57 | −1.93 | −1.50 | −1.21 | −0.39 | 0.66 |
| Artonin T | −9.10 | −1.47 | −0.26 | −1.19 | −1.45 | −0.79 | −1.17 | −0.00 | −0.01 |
| Artonin S | −9.10 | −2.80 | −0.61 | −2.99 | 1.04 | −0.37 | −3.18 | −0.09 | 2.49 |
| IUPAC Names | (kcal/mol) | Coulomb | vdW | Prime MMGBSA Complex Energy | Prime MMGBSA Ligand Energy | Prime MMGBSA Receptor Energy | H Bond |
|---|---|---|---|---|---|---|---|
| Artonin E 5-hydroxy-8,8-dimethyl-3-(3-methylbut-2-enyl)-2-(2,4,5-trihydroxyphenyl)pyrano[2,3-h]chromen-4-one | −47.68 | −22.59 | −33.53 | −9989.98 | −119.08 | −9823.22 | −1.76 |
| Cycloartocarpesin 8-(2,4-dihydroxyphenyl)-5-hydroxy-2,2-dimethylpyrano[3,2-g]chromen-6-one | −51.28 | −18.48 | −38.52 | −10,022.60 | −148.09 | −9823.22 | −2.13 |
| Artonin U 5-Hydroxy-2-(4-hydroxyphenyl)-7-methoxy-8-(3-methyl-2-buten-1-yl)-4H-chromen-4-one | −60.35 | −16.22 | −52.39 | −9987.84 | −104.26 | −9823.22 | −1.99 |
| Artoelastin 3,8,10-trihydroxy-9,11-bis(3-methylbut-2-enyl)-6-(2-methylprop-1-enyl)-6H-chromeno[4,3-b]chromen-7-one | −35.29 | −13.48 | −30.50 | −10,017.30 | −158.81 | −9823.22 | −1.65 |
| Artonin L 3,8-Dihydroxy-1,10-dimethoxy-5,5-dimethyl-5a,6-dihydro-5H,7H-[1]benzofuro[3,4-bc]xanthen-7-one | −32.69 | −5.91 | −26.01 | −9949.88 | −93.968 | −9823.22 | −0.05 |
| Artobiloxanthone 6,10,11,13-Tetrahydroxy-9-isopropenyl-3,3-dimethyl-8,9-dihydro-3H,7H-benzo[c]pyrano[3,2-h]xanthen-7-one | −11.32 | −5.45 | −16.28 | −9930.81 | −96.27 | −9823.22 | −0.15 |
| Artonin Y 2-(2,4-Dihydroxyphenyl)-5,7-dihydroxy-8-(3-methyl-2-buten-1-yl)-4H-chromen-4-one | −44.70 | −9.09 | −47.89 | −10,023.60 | −155.67 | −9823.22 | −1.20 |
| Artonin T 1,3,8-Trihydroxy-10-methoxy-5,5-dimethyl-2-(3-methyl-2-buten-1-yl)-5a,6-dihydro-5H,7H-[1]benzofuro[3,4-bc]xanthen-7-one | −27.36 | −3.95 | −46.21 | −9953.46 | −102.87 | −9823.22 | −0.24 |
| ARTONIN S 3,9-dihydroxy-6-(2-hydroxypropan-2-yl)-11-methoxy-10-(3-methylbut-2-enyl)-6,7-dihydrochromeno[3,2-d][1]benzoxepin-8-one | −25.70 | 3.67 | −28.01 | −9957.42 | −108.50 | −9823.22 | −0.33 |
| Ligands | No of Bonds | Residues | Distance |
|---|---|---|---|
| Artonin E | 4 | THR 347, ASP 351, LYS 531, CYS 530 | 2.22, 1.84, 3.04, 3.22 |
| Cycloartocarpesin | 3 | ARG 394, GLU 353, LEU 346 | 1.94, 3.18, 2.70 |
| Artonin U | 3 | GLU 353, ARG 394 GLY 521 | 1.81, 3.41, 3.44 |
| Artoelastin | 3 | GLU 353, ARG 394, GLY 521 | 1.94, 2.31, 3.41 |
| Artonin L | 2 | CYS 530, MET 528, | 3.37, 2.08 |
| Artobiloxanthone | 2 | LEU 525, MET 343 | 3.37, 3.29 |
| Artonin Y | 2 | LEU387, ARG394 | 2.02, 2.25 |
| Artonin T | 1 | ARG 394 | 3.50 |
| Artonin S | 1 | THR 347 | 2.23 |
| Native ligand | 3 | GLU 353, LYS 351,ARG 394 | 1.80, 2.08, 2.11 |
| Tamoxifen | 2 | LYS 351, CYS 530 | 1.92, 3.51 |
| Compounds | 24 h | 48 h | 72 h | |||
|---|---|---|---|---|---|---|
| IC50 (µM) | 95% Confidence Interval | IC50 (µM) | 95% Confidence Interval | IC50 (µM) | 95% Confidence Interval | |
| Artonin E | 6.9 | 5.5–8.6 | 5.1 | 4.5–5.8 | 3.8 | 3.4–4.1 |
| Tamoxifen | 24.1 | 22.6–25.7 | 20.6 | 18.6–22.9 | 18.9 | 17.5–20.4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etti, I.; Abdullah, R.; Hashim, N.M.; Kadir, A.; Abdul, A.B.; Etti, C.; Malami, I.; Waziri, P.; How, C.W. Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer. Molecules 2016, 21, 839. https://doi.org/10.3390/molecules21070839
Etti I, Abdullah R, Hashim NM, Kadir A, Abdul AB, Etti C, Malami I, Waziri P, How CW. Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer. Molecules. 2016; 21(7):839. https://doi.org/10.3390/molecules21070839
Chicago/Turabian StyleEtti, Imaobong, Rasedee Abdullah, Najihah Mohd Hashim, Arifah Kadir, Ahmad Bustamam Abdul, Christopher Etti, Ibrahim Malami, Peter Waziri, and Chee Wun How. 2016. "Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer" Molecules 21, no. 7: 839. https://doi.org/10.3390/molecules21070839
APA StyleEtti, I., Abdullah, R., Hashim, N. M., Kadir, A., Abdul, A. B., Etti, C., Malami, I., Waziri, P., & How, C. W. (2016). Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer. Molecules, 21(7), 839. https://doi.org/10.3390/molecules21070839