
Organocatalyzed Intramolecular Carbonyl-Ene Reactions

Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure for Intramolecular Carbonyl-Ene Reactions
3.2.1. Synthesis of Compounds 2a/a′
3.2.2. Synthesis of Compounds 5a/a′
3.2.3. Synthesis of Compounds 5b/b′
3.2.4. Synthesis of Compounds 5c/c′
3.2.5. Synthesis of Compounds 5d/d′
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Clarke, M.L.; France, M.B. The carbonyl ene reaction. Tetrahedron 2008, 64, 9003–9031. [Google Scholar] [CrossRef]
- Carlos Dias, L. Chiral Lewis Acid Catalyzed Ene-Reactions. Curr. Org. Chem. 2000, 4, 305–342. [Google Scholar] [CrossRef]
- Snider, B.B. Lewis-acid catalyzed ene reactions. Acc. Chem. Res. 1980, 13, 426–432. [Google Scholar] [CrossRef]
- Pihko, P.M. Activation of Carbonyl Compounds by Double Hydrogen Bonding: An Emerging Tool in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2004, 43, 2062–2064. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Bolm, C.; Rantanen, T.; Schiffers, I.; Zani, L. Protonated Chiral Catalysts: Versatile Tools for Asymmetric Synthesis. Angew. Chem. Int. Ed. 2005, 44, 1758–1763. [Google Scholar] [CrossRef] [PubMed]
- Wittkopp, A.; Schreiner, P.R. Metal-Free, Noncovalent Catalysis of Diels-Alder Reactions by Neutral Hydrogen Bond Donors in Organic Solvents and in Water. Chem. Eur. J. 2003, 9, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.L.; Jones, C.E.; France, M.B. The first organocatalytic carbonyl-ene reaction: Isomerisation-free C-C bond formations catalysed by H-bonding thio-ureas. Beilstein J. Org. Chem. 2007, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueping, M.; Theissmann, T.; Kuenkel, A.; Koenigs, R.M. Highly Enantioselective Organocatalytic Carbonyl-Ene Reaction with Strongly Acidic, Chiral Brønsted Acids as Efficient Catalysts. Angew. Chem. Int. Ed. 2008, 47, 6798–6801. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Leutzsch, M.; Zheng, Y.; Alachraf, M.W.; Thiel, W.; List, B. Confined Acid-Catalyzed Asymmetric Carbonyl–Ene Cyclization. J. Am. Chem. Soc. 2015, 137, 13268–13271. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E.; Thomas, R.C.; Kruse, L.I.; Silberman, L. Rules for ring closure: Ring formation by conjugate addition of oxygen nucleophiles. J. Org. Chem. 1977, 42, 3846–3852. [Google Scholar] [CrossRef]
- Grachan, M.L.; Tudge, M.T.; Jacobsen, E.N. Enantioselective Catalytic Carbonyl–Ene Cyclization Reactions. Angew. Chem. Int. Ed. 2008, 47, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Bahia, P.S.; Kariuki, B.M.; Spencer, N.; Philp, D.; Snaith, J.S. Synthesis of 3,4-Disubstituted Piperidines by Carbonyl Ene and Prins Cyclizations: Switching between Kinetic and Thermodynamic Control with Brønsted and Lewis Acid Catalysts. J. Org. Chem. 2006, 71, 2460–2471. [Google Scholar] [CrossRef] [PubMed]
- Christ, P.; Lindsay, A.G.; Vormittag, S.S.; Neudörfl, J.-M.; Berkessel, A.; O’Donoghue, A.C. pKa Values of Chiral Brønsted Acid Catalysts: Phosphoric Acids/Amides, Sulfonyl/Sulfuryl Imides, and Perfluorinated TADDOLs (TEFDDOLs). Chem. Eur. J. 2011, 17, 8524–8528. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Vennall, G.P.; Davey, P.N.; Newman, C. Scandium trifluoromethaneslfonate, an efficient catalyst for the intermolecular carbonyl-ene reaction and the intramolecular cyclisation of citronellal. Tetrahedron Lett. 1998, 39, 1997–2000. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kawashima, K. A Highly Stereoselective Preparation of l -Isopulegol. Synthesis 1978, 1978, 147–148. [Google Scholar] [CrossRef]
- Kočovský, P.; Ahmed, G.; Šrogl, J.; Malkov, A.V.; Steele, J. New Lewis-Acidic Molybdenum(II) and Tungsten(II) Catalysts for Intramolecular Carbonyl Ene and Prins Reactions. Reversal of the Stereoselectivity of Cyclization of Citronellal. J. Org. Chem. 1999, 64, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Cheon, C.-H.; Yamamoto, H. Synthesis of N,N-Bis(nonaflyl) Squaric Acid Diamide and its Application to Organic Reactions. Bull. Korean Chem. Soc. 2010, 31, 539–540. [Google Scholar] [CrossRef]
- At 24 h, the reaction described in entry 4 had only reached 85% conversion.
- Reeves, J.T.; Song, J.J.; Tan, Z.; Lee, H.; Yee, N.K.; Senanayake, C.H. Trifluoromethyl Ketones from Enolizable Carboxylic Acids via Enediolate Trifluoroacetylation/Decarboxylation. J. Org. Chem. 2008, 73, 9476–9478. [Google Scholar] [CrossRef] [PubMed]
- Tomooka, K.; Suzuki, M.; Shimada, M.; Ni, R.; Uehara, K. Stereoselective Multimodal Transformations of Planar Chiral 9-Membered Diallylic Amides. Org. Lett. 2011, 13, 4926–4929. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Nachtsheim, B.J.; Koenigs, R.M.; Ieawsuwan, W. Synthesis and Structural Aspects of N-Triflylphosphoramides and Their Calcium Salts—Highly Acidic and Effective Brønsted Acids. Chem. Eur. J. 2010, 16, 13116–13126. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Kesler, B.S.; Moon, Y.C. Inter- and intramolecular [4 + 2] cycloadditions of nitroalkenes with olefins. 2-Nitrostyrenes. J. Org. Chem. 1992, 57, 4912–4924. [Google Scholar] [CrossRef]
- Gandon, L.A.; Russell, A.G.; Güveli, T.; Brodwolf, A.E.; Kariuki, B.M.; Spencer, N.; Snaith, J.S. Synthesis of 2,4-Disubstituted Piperidines via Radical Cyclization: Unexpected Enhancement in Diastereoselectivity with Tris(trimethylsilyl)silane. J. Org. Chem. 2006, 71, 5198–5207. [Google Scholar] [CrossRef] [PubMed]
- We attempted to determine the stereochemistry of the carbinol using both NOESY NMR and derivatization. However, cross peaks between the hydroxyl proton and other protons in the molecule were not observed. Acetylation of the alcohols in products 2a/a’ resulted in an inseparable mixture of oils, while efforts to install the corresponding benzoyl groups were unsuccessful.
- Kropp, P.J.; Breton, G.W.; Craig, S.L.; Crawford, S.D.; Durland, W.F.; Jones, J.E.; Raleigh, J.S. Surface-Mediated Reactions. 6. Effects of Silica Gel and Alumina on Acid-Catalyzed Reactions. J. Org. Chem. 1995, 60, 4146–4152. [Google Scholar] [CrossRef]
- Demole, E.; Enggist, P. A Chemical Study ofVirginia Tobacco Flavour (Nicotiana tabacum L.) II. Isolation and synthesis ofcis-2-isopropenyl-8-methyl-1,2,3,4-tetrahydro-1-naphthalenol and 3-isopropenyl-5-methyl-1,2-dihydronaphthalene. Helv. Chim. Acta 1978, 61, 1335–1341. [Google Scholar] [CrossRef]
- Shirakawa, E.; Imazaki, Y.; Hayashi, T. Cobalt-catalyzed Coupling of Alkenyl Triflates with Aryl and Alkenyl Grignard Reagents. Chem. Lett. 2008, 37, 654–655. [Google Scholar] [CrossRef]
- Snider, B.B.; Karras, M.; Price, R.T.; Rodini, D.J. Alkylaluminum halide induced cyclization of unsaturated carbonyl compounds. J. Org. Chem. 1982, 47, 4538–4545. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the Compounds 2 and 5b–d are available from the authors.
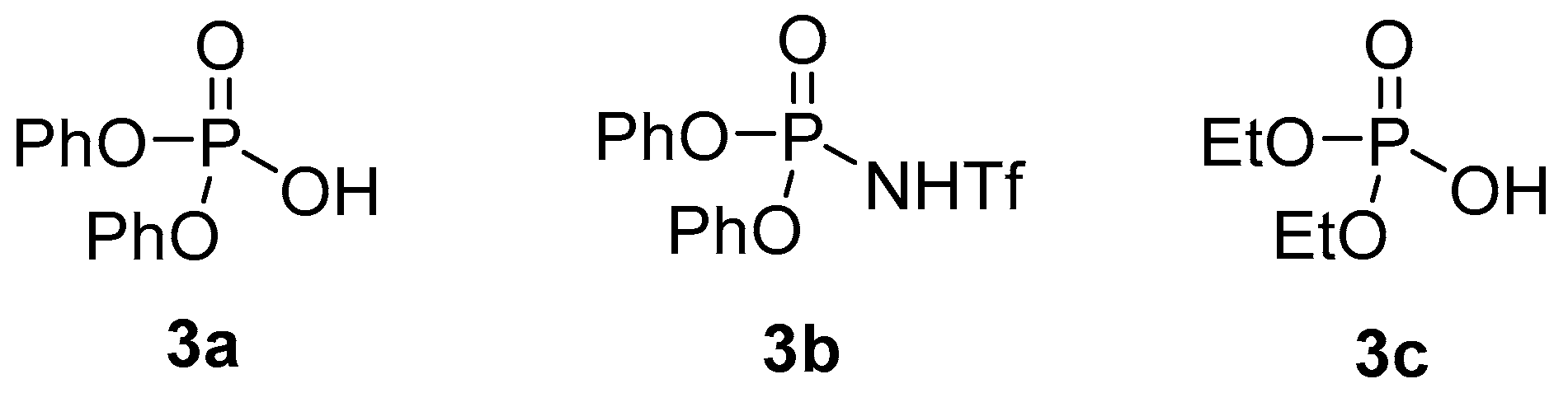


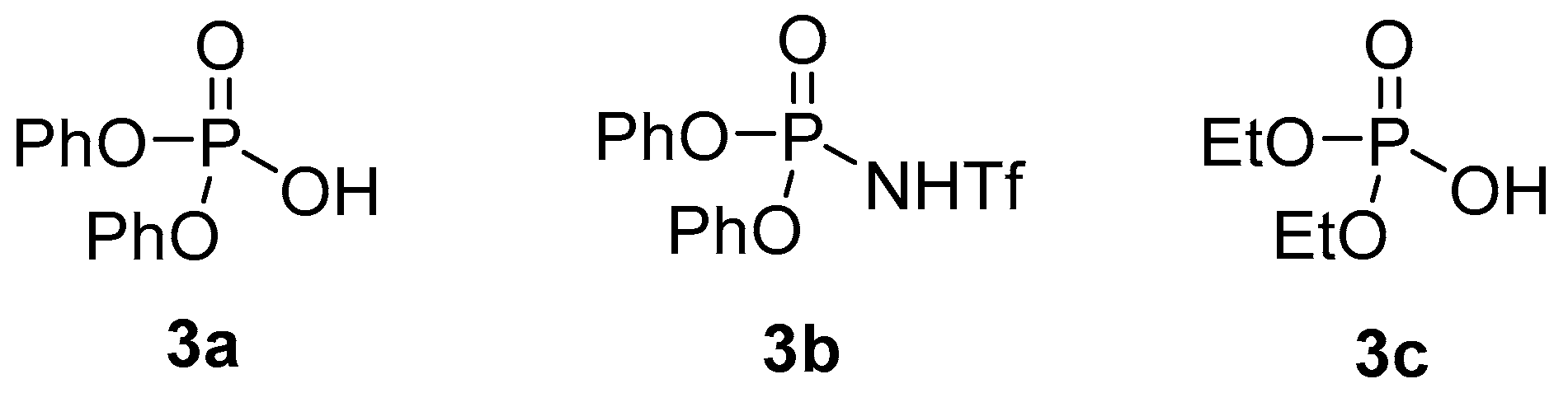
| Entry 1 | Catalyst | Equiv. of Catalyst | [1] (M) | t (h) | Yield 2 2 (%) | d.r. (2a:2a′) 3 |
|---|---|---|---|---|---|---|
| 1 | H3PO4 | 1 | 2 | 24 | 0 | - |
| 2 | HCl | 1 | 2 | 24 | 13 | - |
| 3 | 3a | 0.5 | 2 | 24 | 25 | 2:1 |
| 4 | 3b | 0.5 | 2 | 24 | 84 | 2:1 |
| 5 | 3b | 0.2 | 2 | 24 | 86 | 2:1 |
| 6 | 3b | 0.1 | 2 | 24 | 69 | 2.1:1 |
| 7 | 3b | 0.1 | 0.5 | 48 | 75 | 2.1:1 |
| 8 | 3b | 0.5 | 2 | 7 | 79 | 2.2:1 |

{kind=link}
| Entry 1 | Catalyst | Equiv. of Catalyst | [4a] (M) | t (h) | Yield 5a 2 (%) | d.r. (5a:5a′) 3 |
|---|---|---|---|---|---|---|
| 1 | 3b | 1 | 0.1 | 24 | trace | - |
| 2 | 3c | 0.5 | 0.1 | 24 | 14 | 1:1 |
| 3 | 3a | 0.5 | 0.1 | 22 | 61 | 2.4:1 |
| 4 | 3a | 0.06 | 0.6 | 24 | 65 | 1.5:1 |
| 5 | 3a | 0.1 | 2 | 18 | 31 | 2:1 |
| Entry 1 | Aldehyde | Product | Yield (%) (d.r.) 2,3 |
|---|---|---|---|
| 1 |  |  | 89 (2.7:1) |
| 2 4 |  |  | 44 (3.3:1) |
| 3 |  |  | 37 (>10:1) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlmann, H.A.; McKinney, A.J.; Santos, M.P.; Davis, L.O. Organocatalyzed Intramolecular Carbonyl-Ene Reactions. Molecules 2016, 21, 713. https://doi.org/10.3390/molecules21060713
Dahlmann HA, McKinney AJ, Santos MP, Davis LO. Organocatalyzed Intramolecular Carbonyl-Ene Reactions. Molecules. 2016; 21(6):713. https://doi.org/10.3390/molecules21060713
Chicago/Turabian StyleDahlmann, Heidi A., Amanda J. McKinney, Maria P. Santos, and Lindsey O. Davis. 2016. "Organocatalyzed Intramolecular Carbonyl-Ene Reactions" Molecules 21, no. 6: 713. https://doi.org/10.3390/molecules21060713