
Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets



Abstract
:
1. Introduction
2. Results
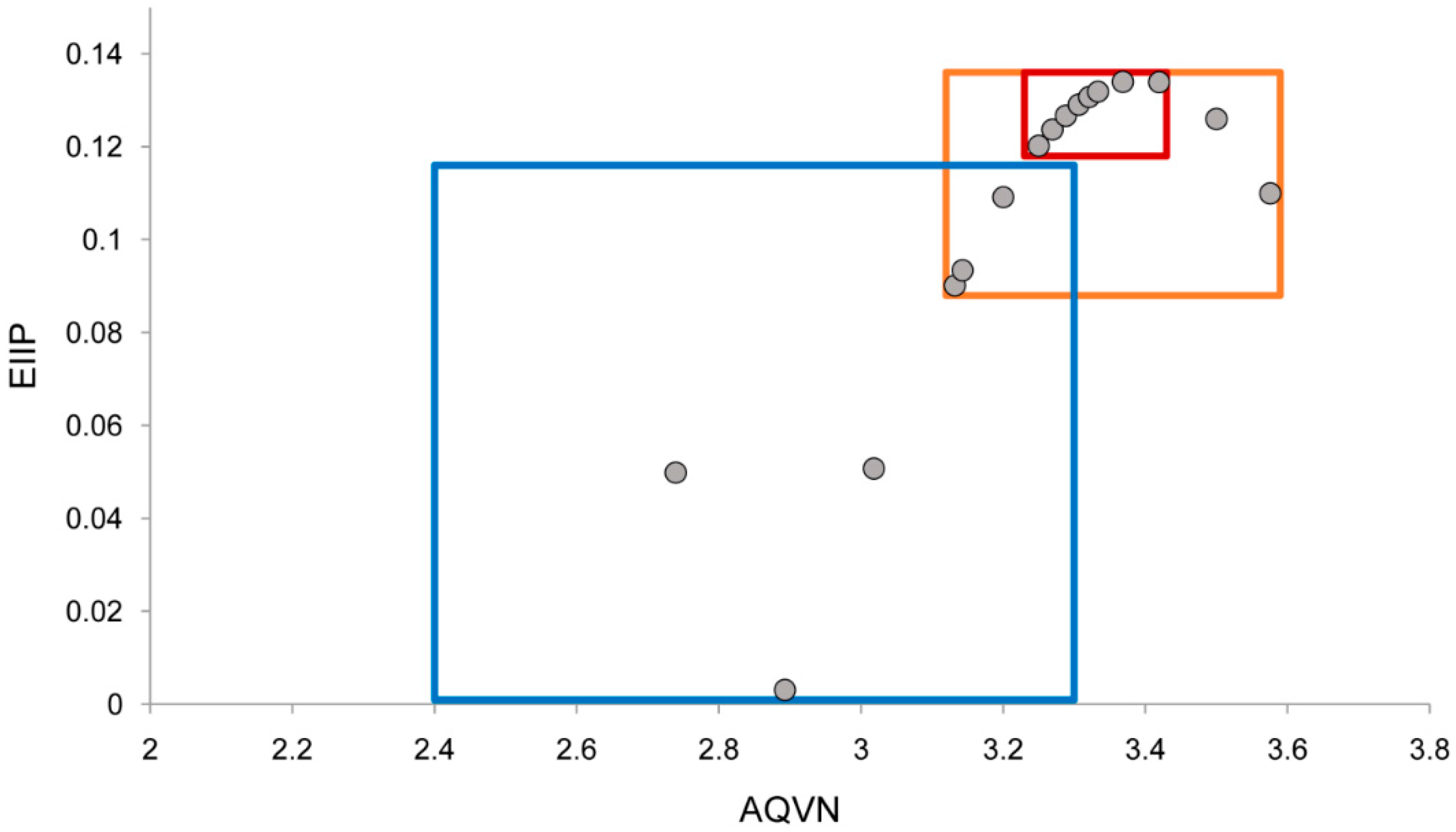
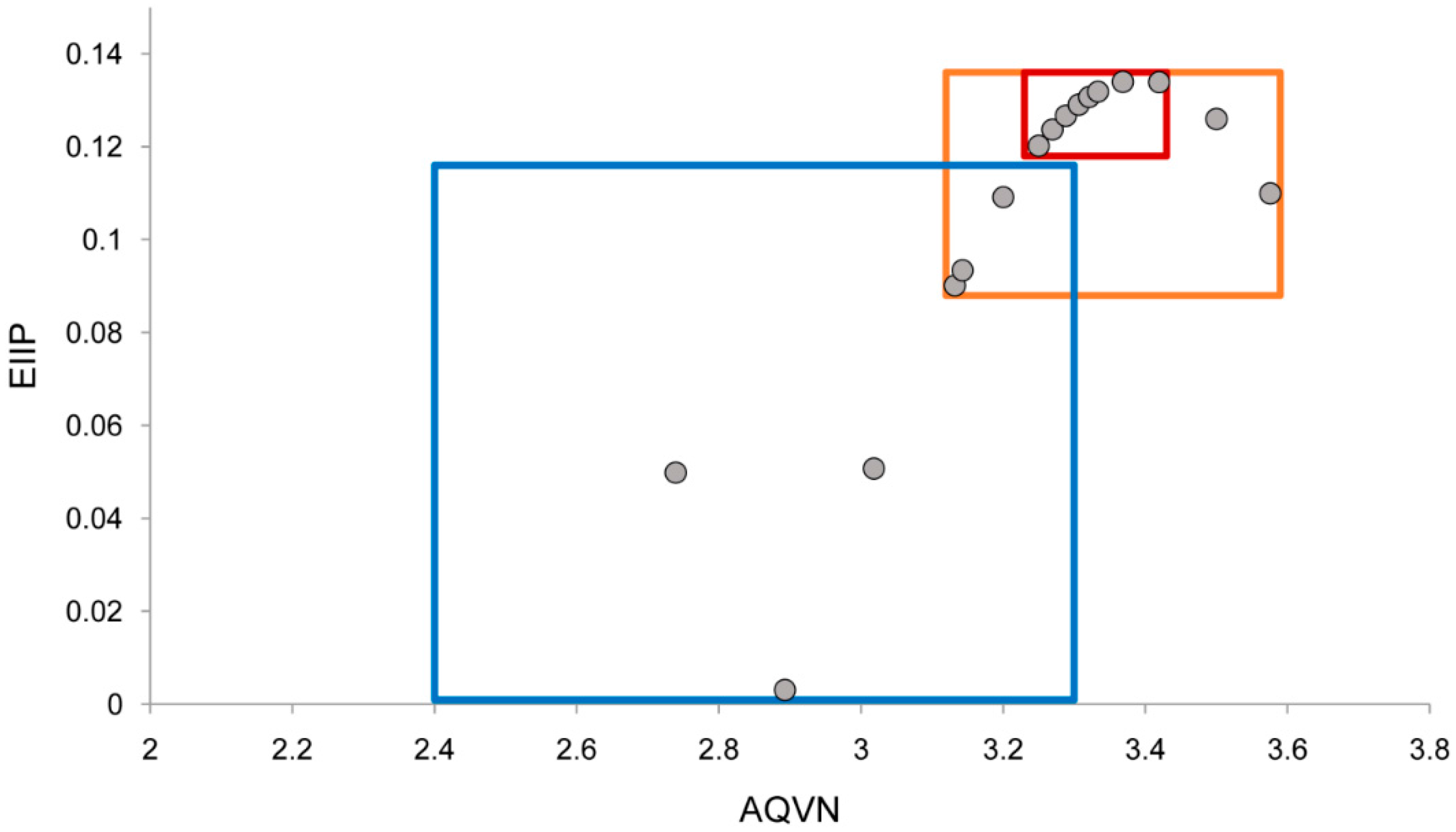
2.1. EIIP/AQVN Filter
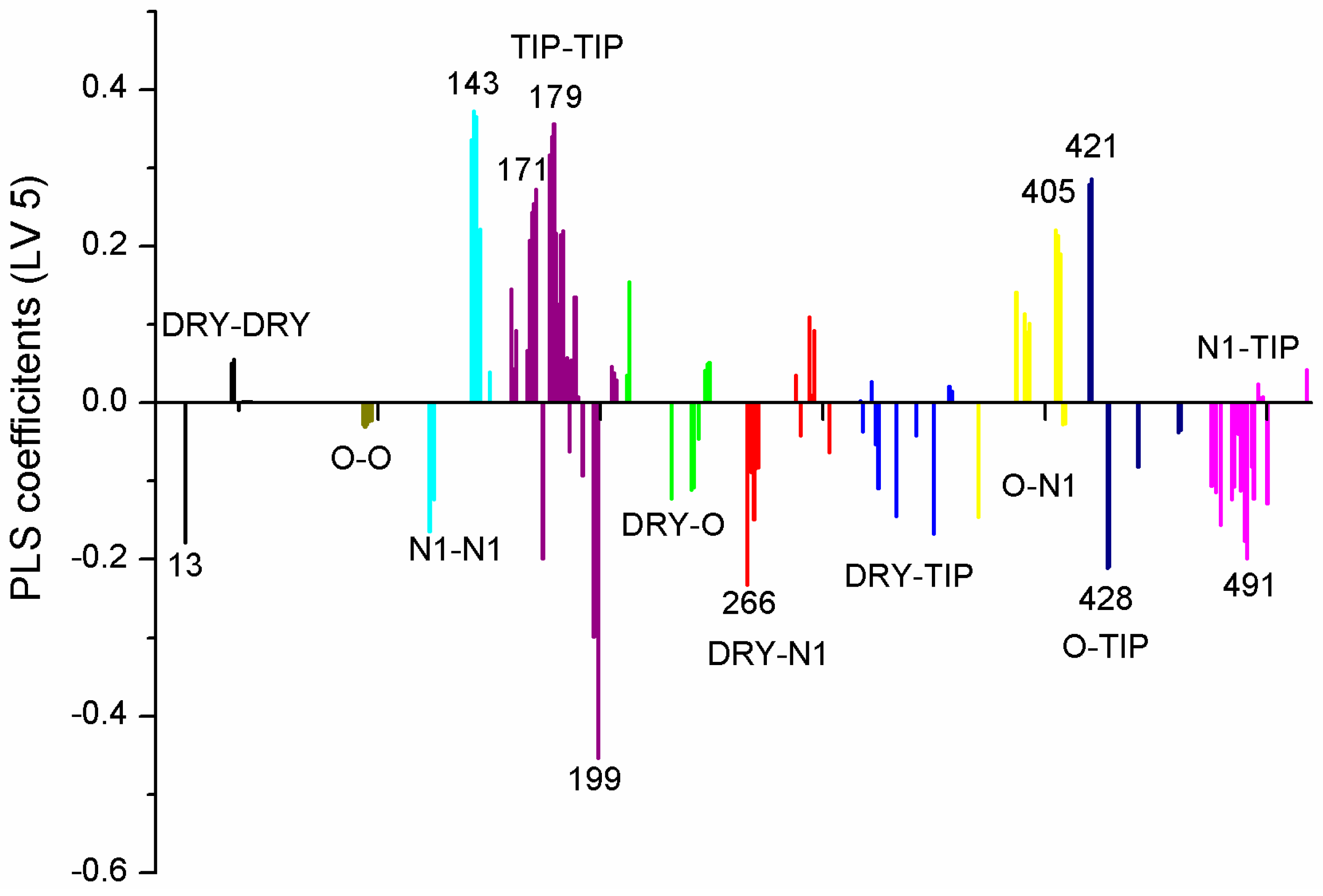
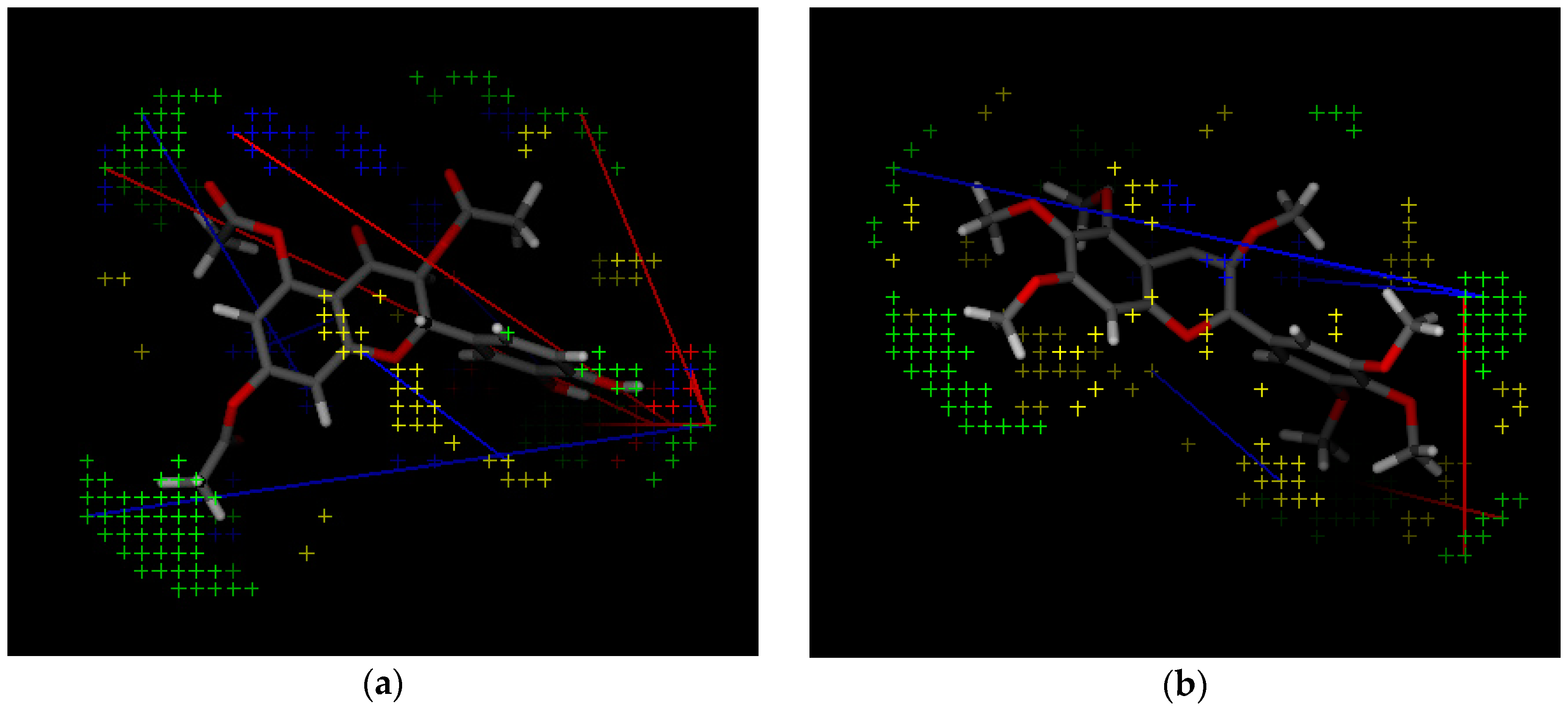
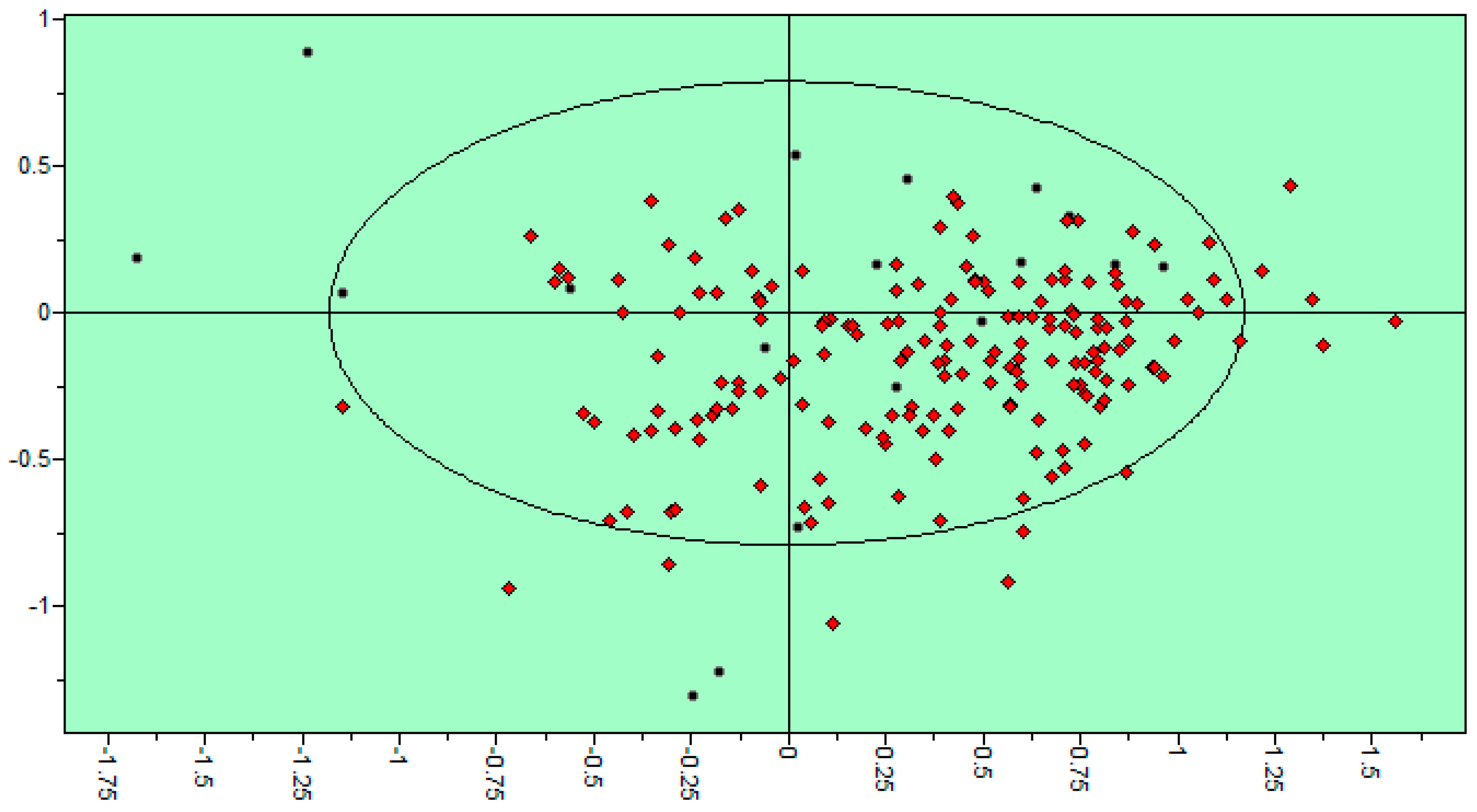
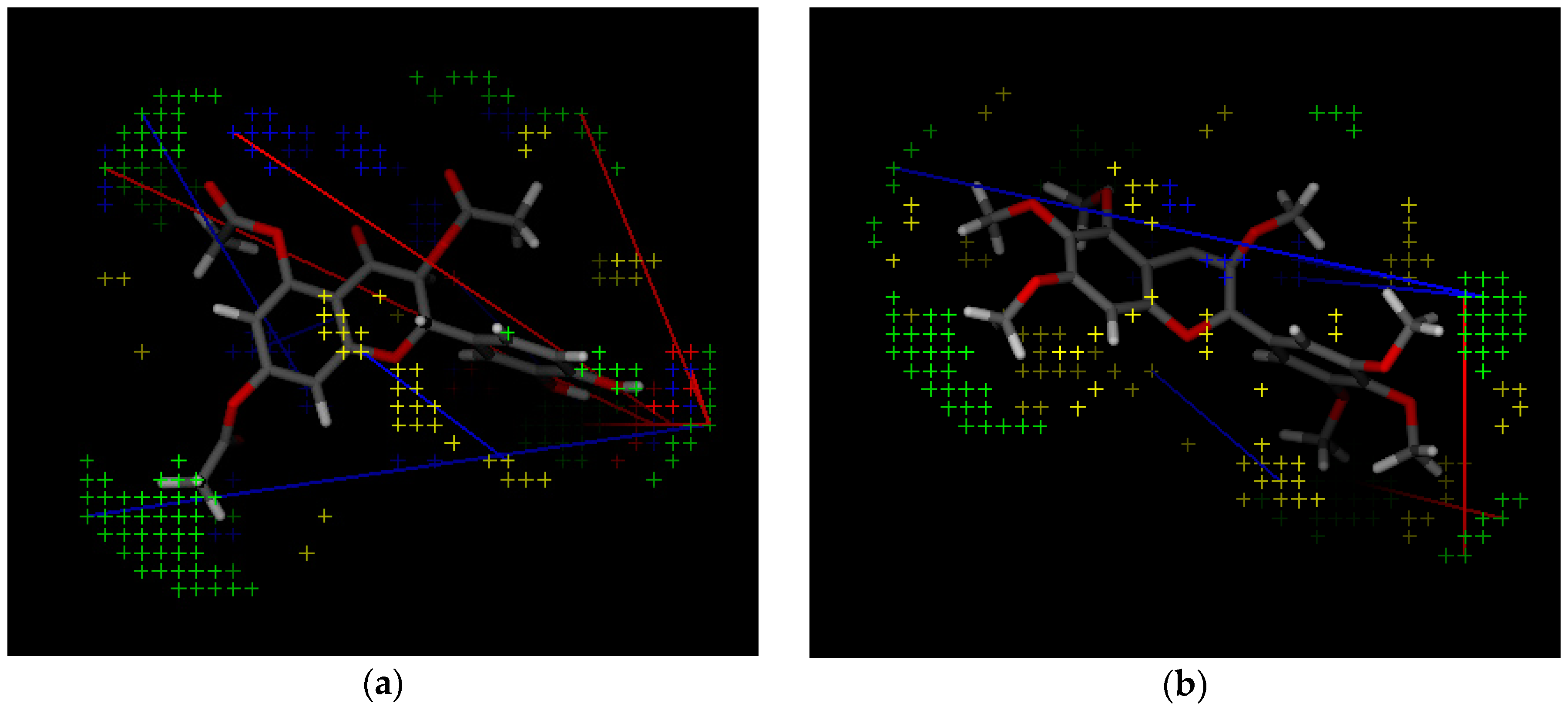
2.2. 3D-QSAR Model Building
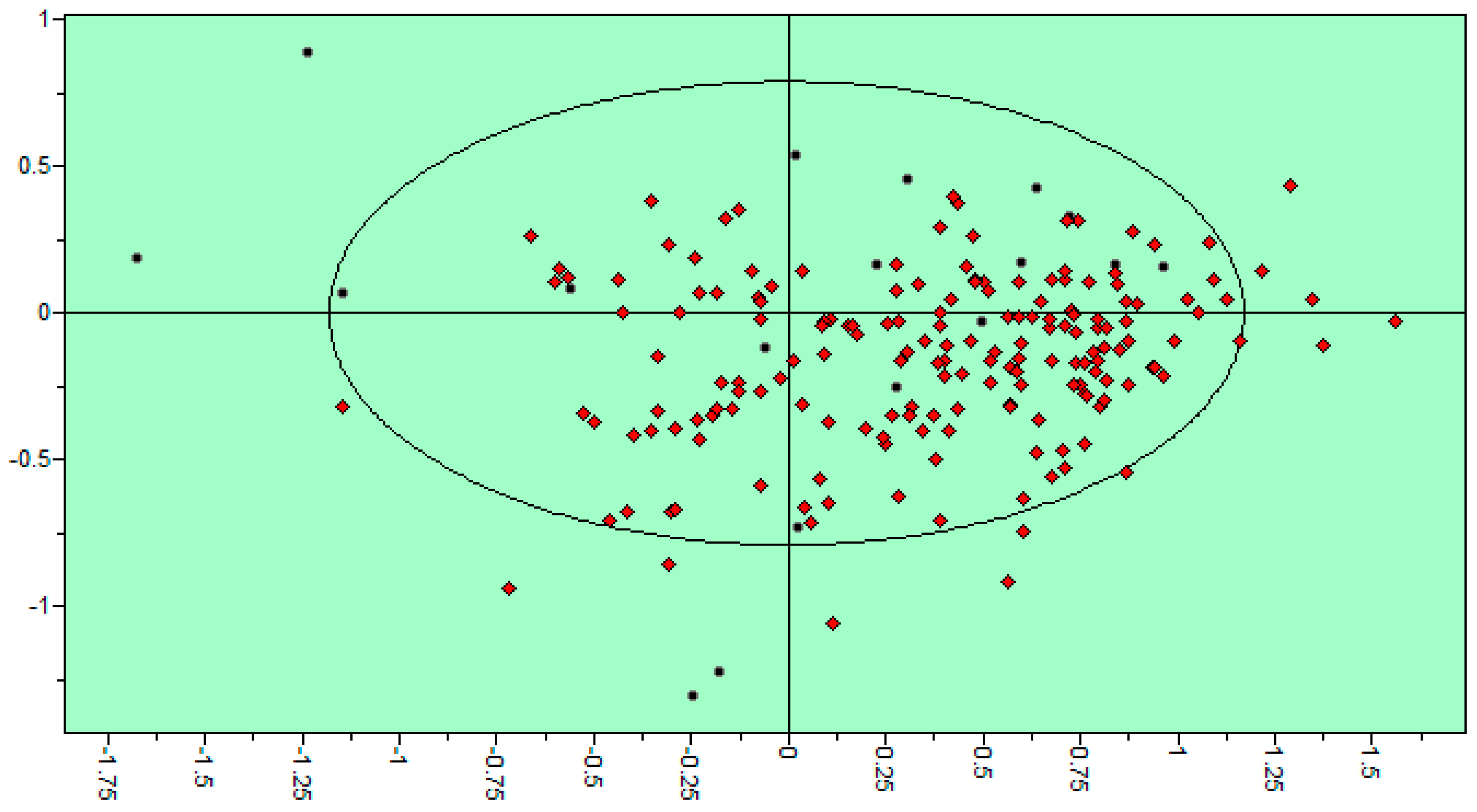
2.3. 3D QSAR Filtering
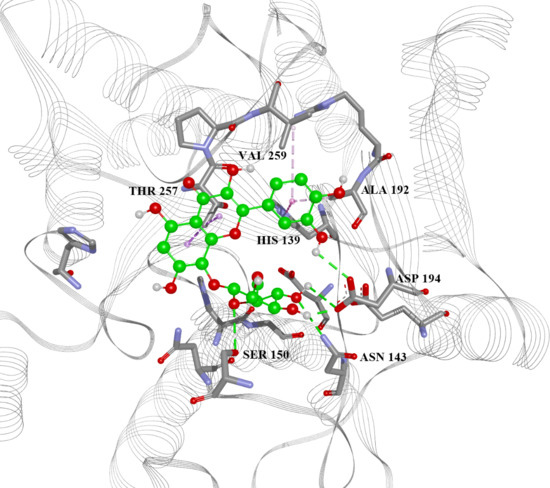
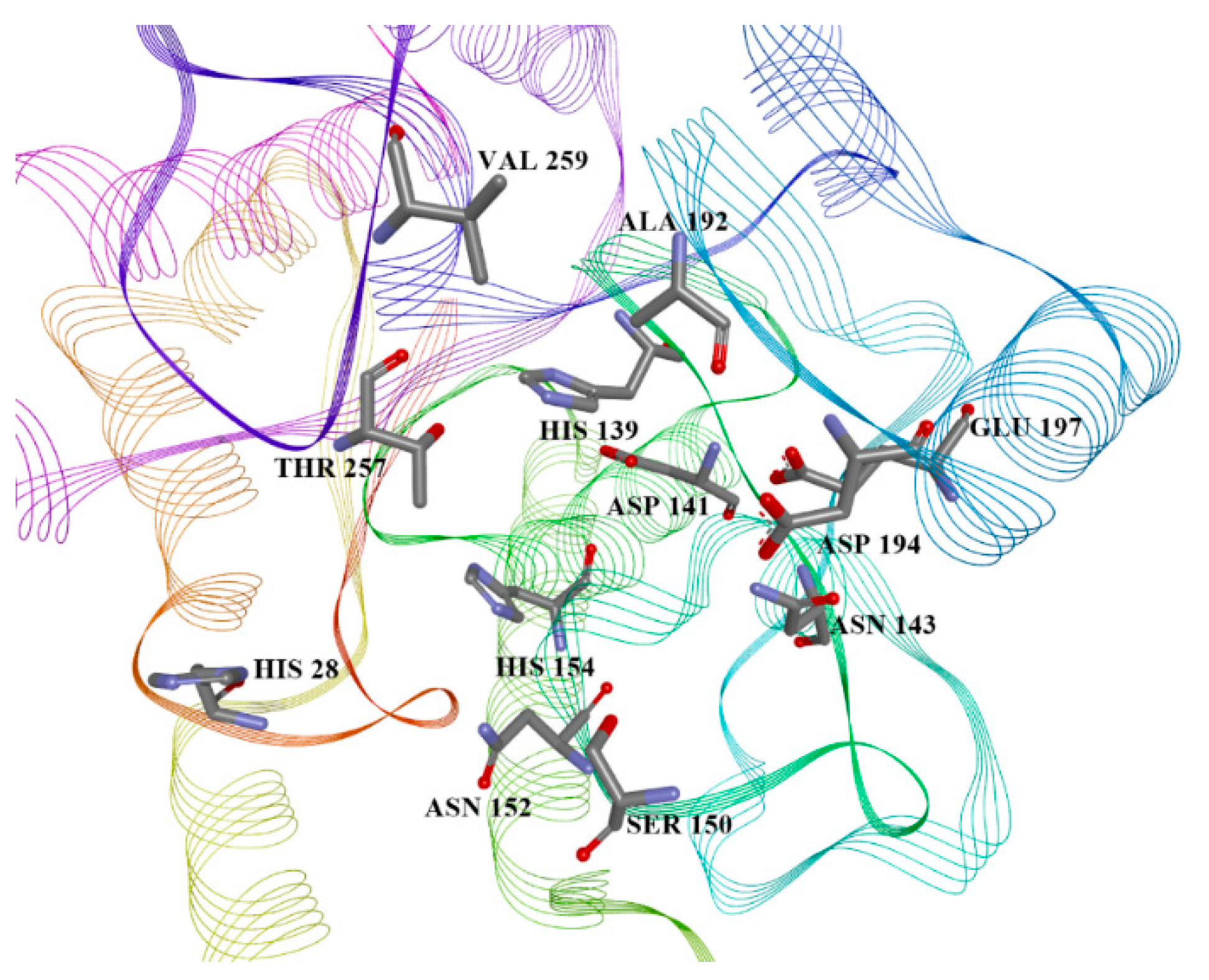
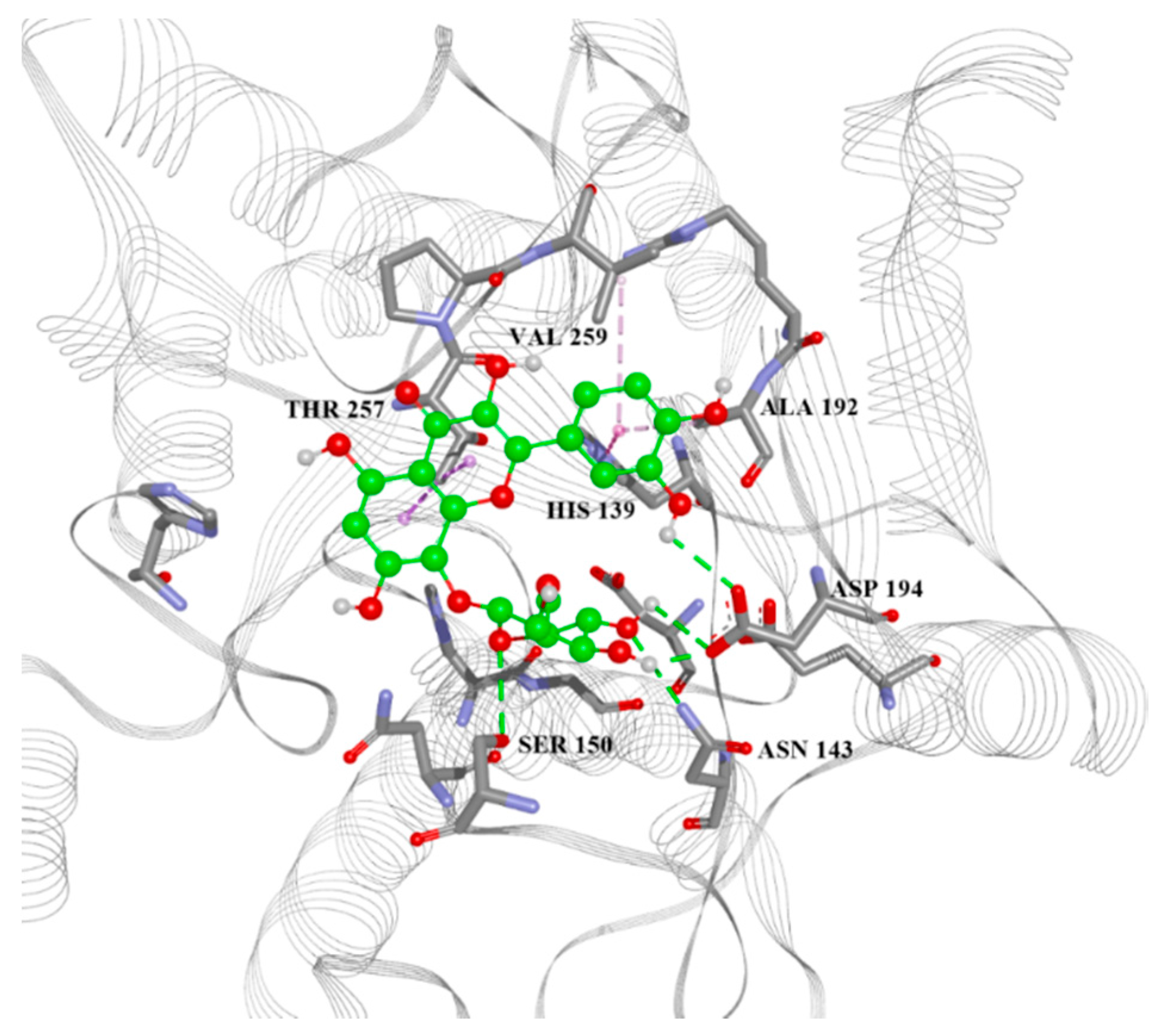
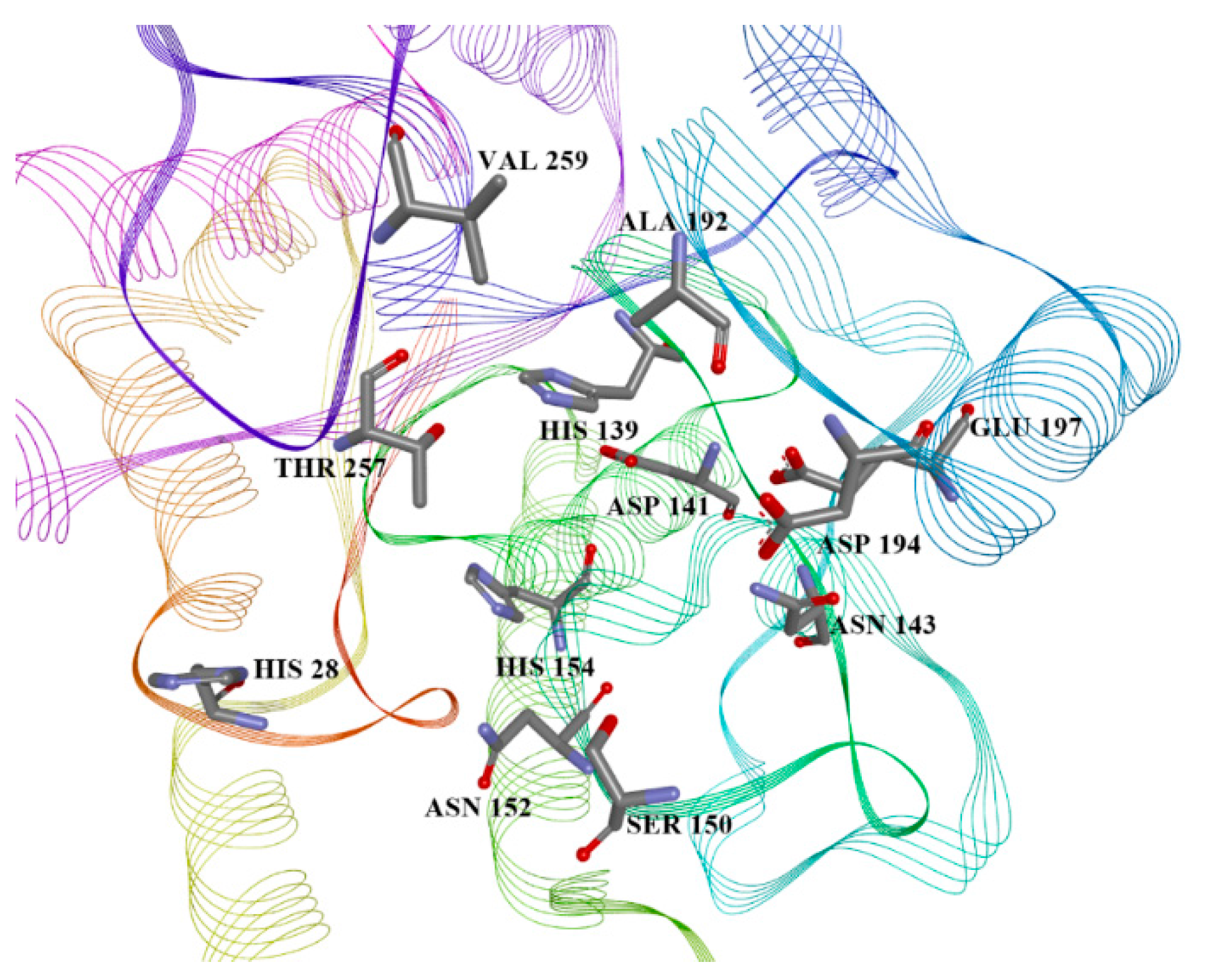
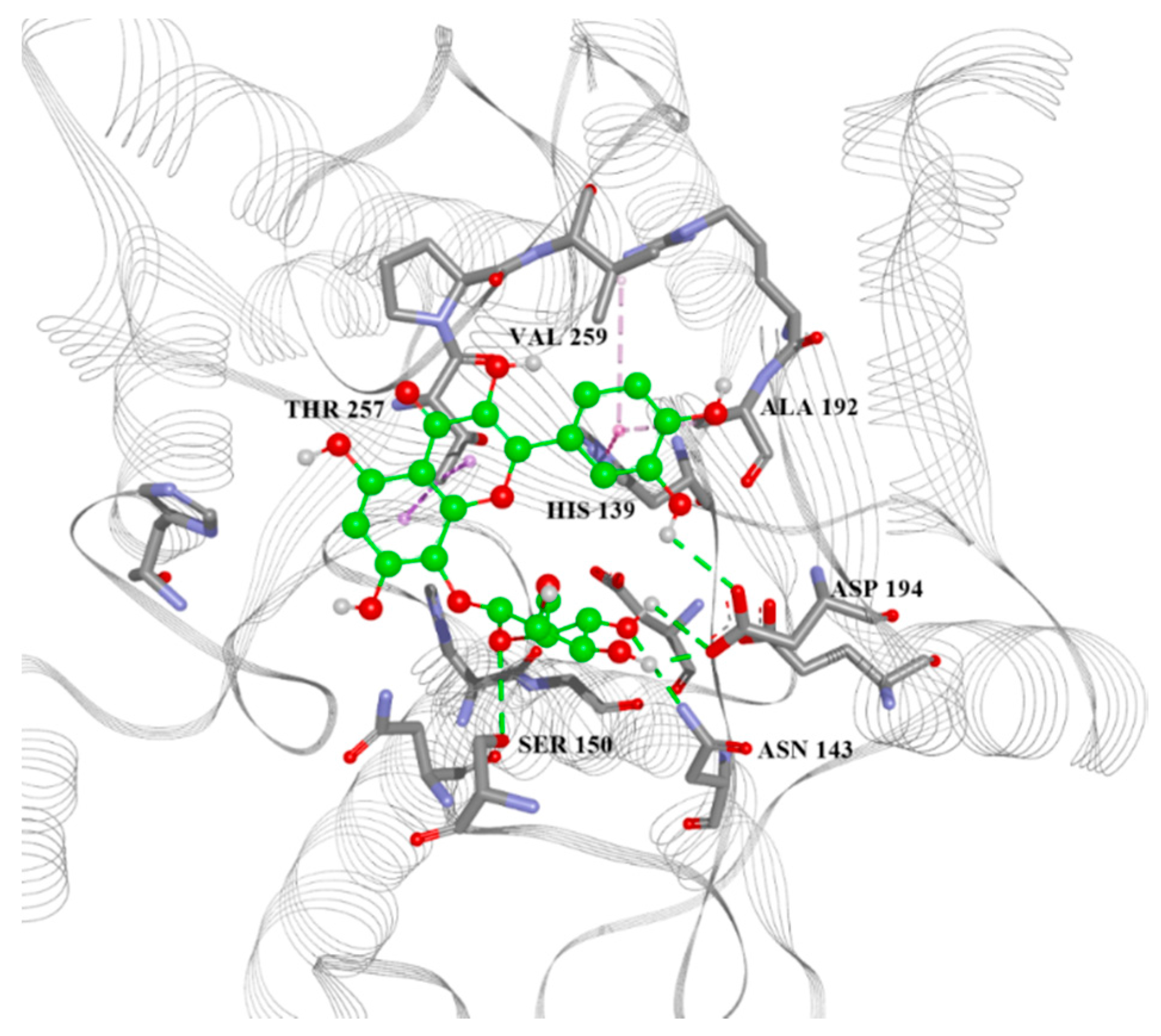
2.4. Arginase Docking
2.5. Anti-Target Interaction Matrix
3. Discussion
4. Materials and Methods
4.1. Ligand Data: Sources, Curation, Processing
4.2. EIIP/AQVN Filter
4.3. 3D QSAR
4.4. Molecular Docking for Human and Leishmania Arginase
4.5. Anti-Target Set
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HIV | Human immunodeficiency virus |
| NTD | Neglected tropical disease |
| PA | polyamines |
| AQVN | average quasi valence number |
| EIIP | electron-ion interaction potential |
| VS | virtual screening |
| PLS | partial least squares |
| Var | Variable |
| CYP | cytochrome |
| SULT | sulfotransferase |
| PXR | pregnane-X-receptor |
| PCA | principal components analysis |
| XP | Glide XP |
| AD4 | AutoDock 4 |
| Vina | AutoDock Vina |
| PDB | Protein DataBank |
| MM/GBSA | Molecular Mechanics/Generalized Born Solvent Surface Area |
| MIF | Molecular Interaction Fields |
References
- Centers for Disease Control and Prevention. “Parasites: Leishmaniasis”. Available online: http://www.cdc.gov/parasites/leishmaniasis (accessed on 1 February 2016).
- Gradoni, L. Epidemiological surveillance of leishmaniasis in the European Union: Operational and research challenges. Euro Surveill. 2013, 18, 20539. [Google Scholar] [CrossRef] [PubMed]
- WHO. Leishmaniasis: Situation and Trends. World Health Organization, 2015. Available online: http://www.who.int/gho/neglected_diseases/leishmaniasis/en/ (accessed on 1 February 2016).
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. “Leishmaniasis Fact Sheet”. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 1 February 2016).
- Rogers, M.; Kropf, P.; Choi, B.S.; Dillon, R.; Podinovskaia, M.; Bates, P.; Müller, I. Proteophosophoglycans regurgitated by Leishmania-infected sand flies target the l-arginine metabolism of host macrophages to promote parasite survival. PLoS Pathog. 2009, 5, e1000555. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.F.; Floeter-Winter, L.M. Arginase in Leishmania. Subcell. Biochem. 2014, 74, 103–117. [Google Scholar] [PubMed]
- Colotti, G.; Ilari, A. Polyamine metabolism in Leishmania: From arginine to trypanothione. Amino Acids 2011, 40, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Reguera, R.M.; Balaña-Fouce, R.; Showalter, M.; Hickerson, S.; Beverley, S.M. Leishmania major lacking arginase (ARG) are auxotrophic for polyamines but retain infectivity to susceptible BALB/c mice. Mol. Biochem. Parasitol. 2009, 165, 48–56. [Google Scholar] [CrossRef] [PubMed]
- da Silva, E.R.; Castilho, T.M.; Pioker, F.C.; Tomich de Paula Silva, C.H.; Floeter-Winter, L.M. Genomic organisation and transcription characterisation of the gene encoding Leishmania (Leishmania) amazonensis arginase and its protein structure prediction. Int. J. Parasitol. 2002, 32, 727–737. [Google Scholar] [CrossRef]
- Chawla, B.; Madhubala, R. Drug targets in Leishmania. J. Parasit. Dis. 2010, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Machado-Silva, A.; Guimarães, P.P.; Tavares, C.A.; Sinisterra, R.D. New perspectives for leishmaniasis chemotherapy over current anti-leishmanial drugs: A patent landscape. Expert Opin. Ther. Pat. 2015, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, R.; Chen, Y.P. Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discov. Today 2015, 20, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Fabricant, D.S.; Farnsworth, N.R. The value of plants used in traditional medicine for drug discovery. Environ. Health Perspect. 2001, 109, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Espinosa, O.A.; Montenegro, H.; Cubilla, L.; Capson, T.L.; Ortega-Barría, E.; Romero, L.I. Hydrosoluble formazan XTT: Its application to natural products drug discovery for Leishmania. J. Microbiol. Methods 2003, 55, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 2014, 114, 10369–10428. [Google Scholar] [CrossRef] [PubMed]
- Girard-Thernier, C.; Pham, T.N.; Demougeot, C. The Promise of Plant-Derived Substances as Inhibitors of Arginase. Mini Rev. Med. Chem. 2015, 15, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Ishiki, H.; Mendonça Júnior, F.J.; Da Silva, M.S.; Scotti, M.T. In-silico analyses of natural products on leishmania enzyme targets. Mini Rev. Med. Chem. 2015, 15, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Cruz Ede, M.; da Silva, E.R.; Maquiaveli Cdo, C.; Alves, E.S.; Lucon, J.F., Jr.; dos Reis, M.B.; de Toledo, C.E.; Cruz, F.G.; Vannier-Santos, M.A. Leishmanicidal activity of Cecropia pachystachya flavonoids: Arginase inhibition and altered mitochondrial DNA arrangement. Phytochemistry 2013, 89, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Manjolin, L.C.; dos Reis, M.B.; Maquiaveli Cdo, C.; Santos-Filho, O.A.; da Silva, E.R. Dietary flavonoids fisetin, luteolin and their derived compounds inhibit arginase, a central enzyme in Leishmania (Leishmania) amazonensis infection. Food Chem. 2013, 141, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.J.; Klabunde, T. Antitargets. Prediction and Prevention of Drug Side Effects. In Series: Methods and Principles in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2008; pp. xix–xxv. [Google Scholar]
- García-Sosa, A.T.; Sild, S.; Takkis, K.; Maran, U. Combined approach using ligand efficiency, cross-docking, and antitarget hits for wild-type and drug-resistant Y181C HIV-1 reverse transcriptase. J. Chem. Inf. Model. 2011, 51, 2595–2611. [Google Scholar]
- García-Sosa, A.T.; Maran, U. Improving the use of ranking in virtual screening against HIV-1 integrase with triangular numbers and including ligand profiling with Antitargets. J. Chem. Inf. Model. 2014, 54, 3172–3185. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Mouscadet, J.F.; Veljkovic, N.; Glisic, S.; Debyser, Z. Simple criterion for selection of flavonoid compounds with anti-HIV activity. Bioorg. Med. Chem. Lett. 2007, 17, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, N.; Glisic, S.; Perovic, V.; Veljkovic, V. The role of long-range intermolecular interactions in discovery of new drugs. Exp. Opin. Drug Disc. 2011, 6, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Loiseau, P.M.; Figadere, B.; Glisic, S.; Veljkovic, N.; Perovic, V.R.; Cavanaugh, D.P.; Branch, D.R. Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection. F1000Res. 2015, 4. [Google Scholar] [CrossRef]
- Veljkovic, N.; Glisic, S.; Prljic, J.; Perovic, V.; Veljkovic, V. Simple and general criterion for “in silico” screening of candidate HIV drugs. Curr. Pharm. Biotechnol. 2013, 14, 561–569. [Google Scholar] [CrossRef] [PubMed]
- EMBL-EBI. ChEMBL. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 12 December 2015).
- Mihaleva, V.V.; te Beek, T.A.; van Zimmeren, F.; Moco, S.; Laatikainen, R.; Niemitz, M.; Korhonen, S.P.; van Driel, M.A.; Vervoort, J. MetIDB: A publicly accessible database of predicted and experimental 1H NMR spectra of flavonoids. Anal. Chem. 2013, 85, 8700–8707. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Veljkovic, N.; Este, J.; Huther, A.; Dietrich, U. Application of the EIIP/ISM bioinformatics concept in development of new drugs. Curr. Med. Chem. 2007, 14, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Peraza-Sánchez, S.R.; Cen-Pacheco, F.; Noh-Chimal, A.; May-Pat, F.; Simá-Polanco, P.; Dumonteil, E.; García-Miss, M.R.; Mut-Martín, M. Leishmanicidal evaluation of extracts from native plants of the Yucatan peninsula. Fitoterapia 2007, 78, 315–318. [Google Scholar]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V. A Theoretical Approach to Preselection of Carcinogens and Chemical Carcinogenesis; Gordon & Breach: New York, NY, USA, 1980. [Google Scholar]
- Veljkovic, V.; Slavic, I. Simple general-model pseudopotential. Phys. Rev. Lett. 1972, 29, 105–107. [Google Scholar] [CrossRef]
- Duran, A.; Zamora, I.; Pastor, M. Suitability of GRIND-Based Principal Properties for the Description of Molecular Similarity and Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2009, 49, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Comesaña, G.; Pastor, M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in molecular interaction fields. J. Chem. Inf. Model. 2008, 48, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.; McLay, I.; Pickett, S.; Clementi, S. Grid-Independent descriptors (GRIND): A novel class of alignment-independent three-dimensional molecular descriptors. Med. Chem. 2000, 43, 3233–3243. [Google Scholar] [CrossRef]
- Bohn, M.F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. Research Collaboratory for Structural Bioinformatics. Available online: http://www.pdb.org/pdb/home/home.do (accessed on 1 February 2014).
- PSI|The Protein Model Portal. Available online: http://www.proteinmodelportal.org/ (accessed on 10 March 2016).
- Trott, O.; Olson, A.J. Autodock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Virtual Screening Workflow; Schrödinger, LLC: New York, NY, USA, 2015.
- Prime MM/GBSA; Schrödinger, LLC: New York, NY, USA, 2015.
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- García-Sosa, A.T. Hydration properties of ligands and drugs in protein binding sites: Tightly-bound, bridging water molecules and their effects and consequences on molecular design strategies. J. Chem. Inf. Model. 2013, 53, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Mancera, R.L. Free energy calculations of mutations involving a tightly bound water molecule and ligand substitutions in a ligand-protein complex. Mol. Inf. 2010, 29, 589–600. [Google Scholar]
- García-Sosa, A.T.; Mancera, R.L.; Dean, P.M. WaterScore: A novel method for distinguishing between bound and displaceable water molecules in the crystal structure of the binding site of protein-ligand complexes. J. Mol. Model. 2003, 9, 172–182. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Firth-Clark, S.; Mancera, R.L. Including tightly-bound water molecules in de novo drug design. Exemplification through the in silico generation of poly (ADP-ribose) polymerase ligands. J. Chem. Inf. Model. 2005, 45, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.G.; García-Sosa, A.T.; Alberts, I.L.; Todorov, N.P.; Mancera, R.L. The effect of tightly bound water molecules on the structural interpretation of ligand-derived pharmacophore models. J. Comput. Aid. Mol. Des. 2004, 18, 89–100. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
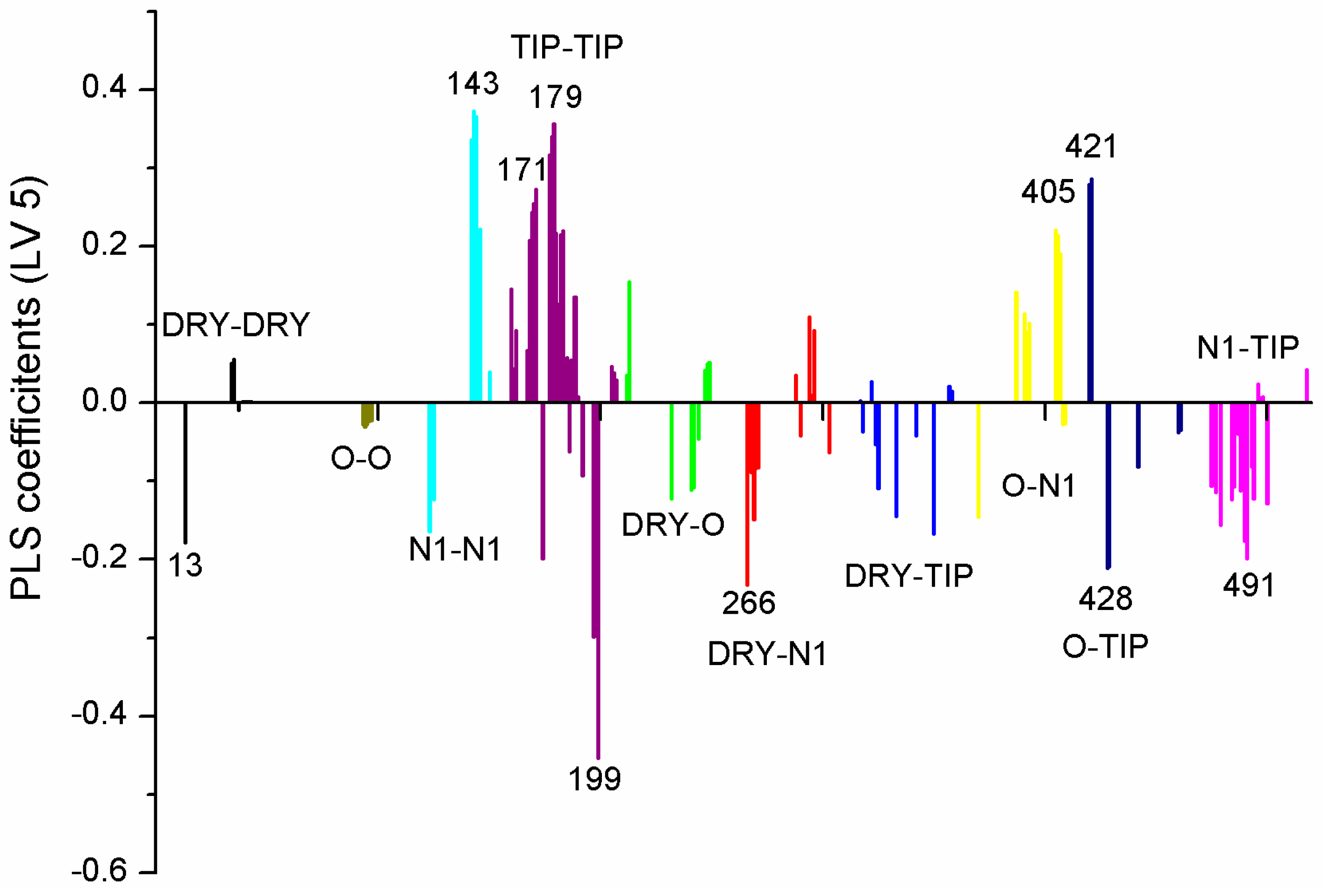
| Variable Number | Type | Distance | Latent Variable (LV5) Values | Regions (see Figure 3) | |
|---|---|---|---|---|---|
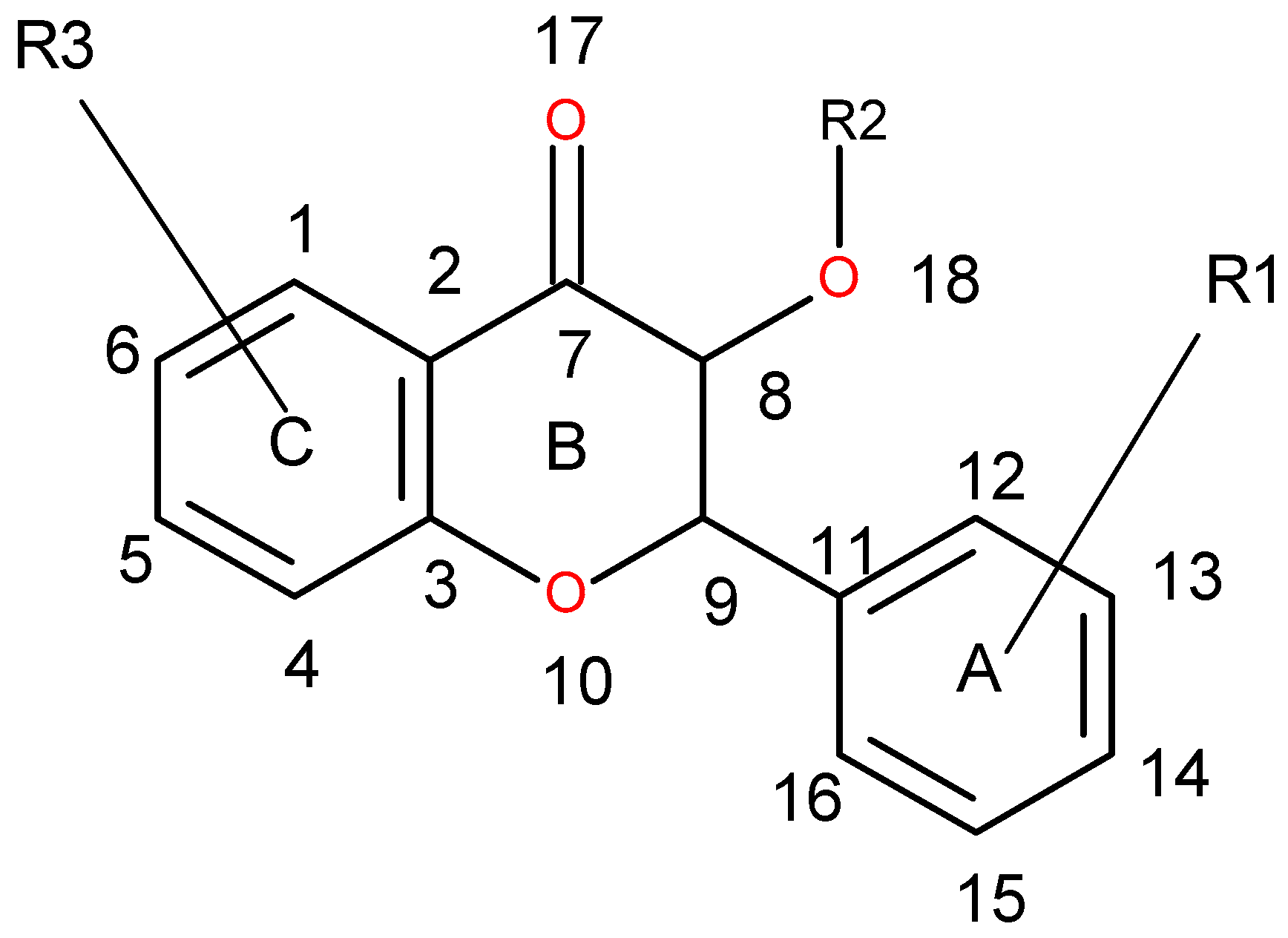
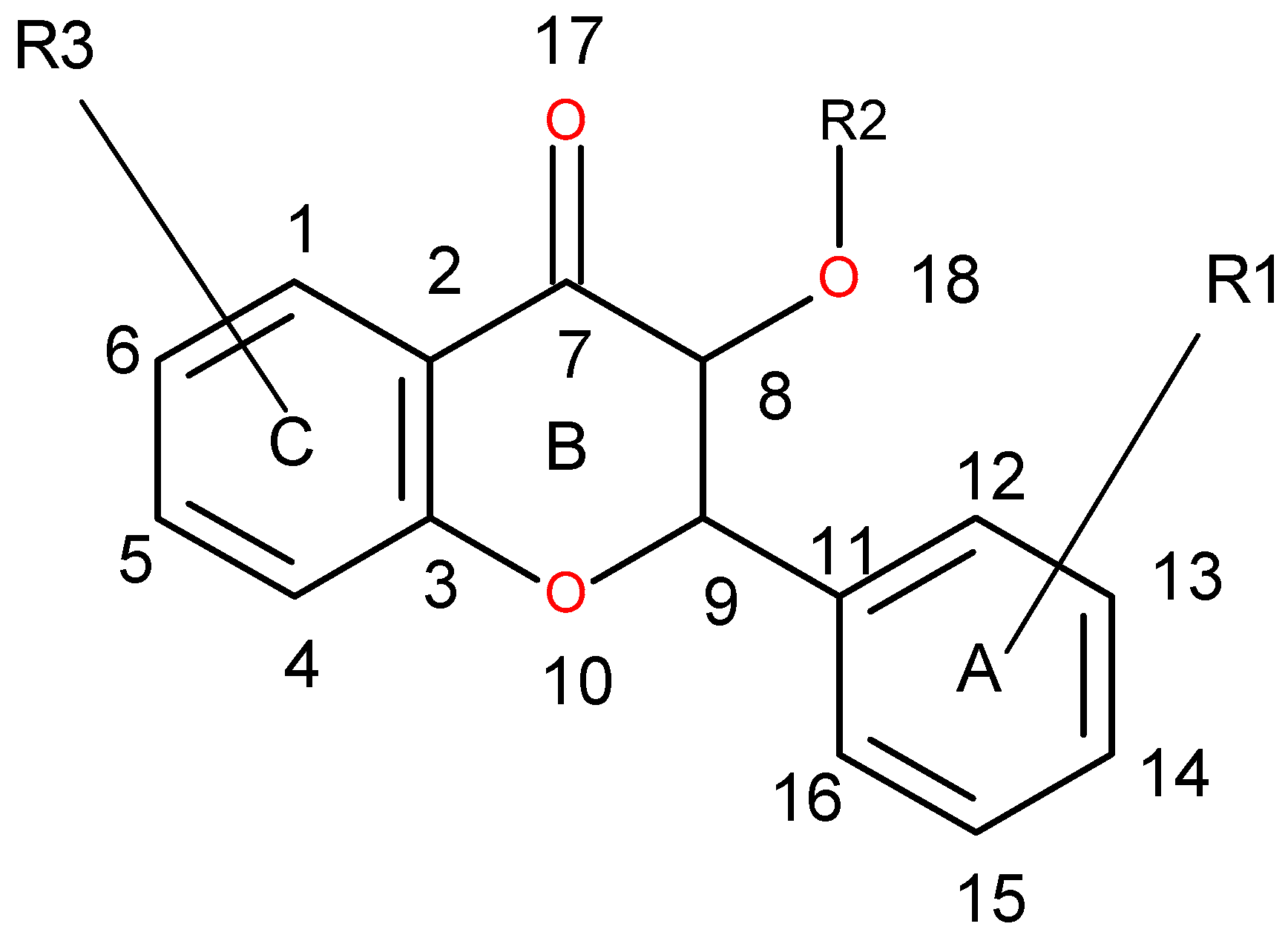
| 1 | 13 | DRY-DRY | 5.20–5.60 | −0.1793 | Distance between two aromatic rings A and C. |
| 2 | 266 | DRY-N1 | 2.40–2.80 | −0.2333 | C3 atom from B ring and carbonyl O10 or carbonyl O17 atom. |
| 3 | 405 | O-N1 | 16.40–16.80 | 0.2214 | Carbonyl oxygen of OAc at position 1, ring C, and OH oxygen atom, position 14, ring A, or C5 ring C-CH2OH oxygen in glucose residue R2 , present only in CHEMBL 3109443, 361362 and 250450. |
| 4 | 199 | TIP-TIP | 17.20–17.60 | −0.4547 | C6-C15 methyl groups. |
| 5 | 428 | O-TIP | 4.80–5.20 | −0.2121 | Ring A, distance between CH3 group C14 and nearest OH group C15. |
| 6 | 143 | N1-N1 | 15.60–16.00 | 0.3729 | Distance between oxygens on A and C rings C14–C5. |
| 7 | 421 | O-TIP | 2.00–2.40 | 0.2867 | Distance between H atom and O curvature on the same OH group on ring A C2 position. |
| 8 | 491 | N1-TIP | 9.20–9.60 | −0.1997 | Distance between link O atom, ring B or hexose ring and CH3 group curvature on ring A O10–C14. |
| 9 | 171 | TIP-TIP | 6.00–6.40 | 0.2737 | Distance between two curvature probes over two neighbor OH groups in ring A C13–C14. |
| 10 | 179 | TIP-TIP | 9.20–9.60 | 0.3568 | Distance between two curvature probes over two neighbor OH groups in ring C C6–C4. |
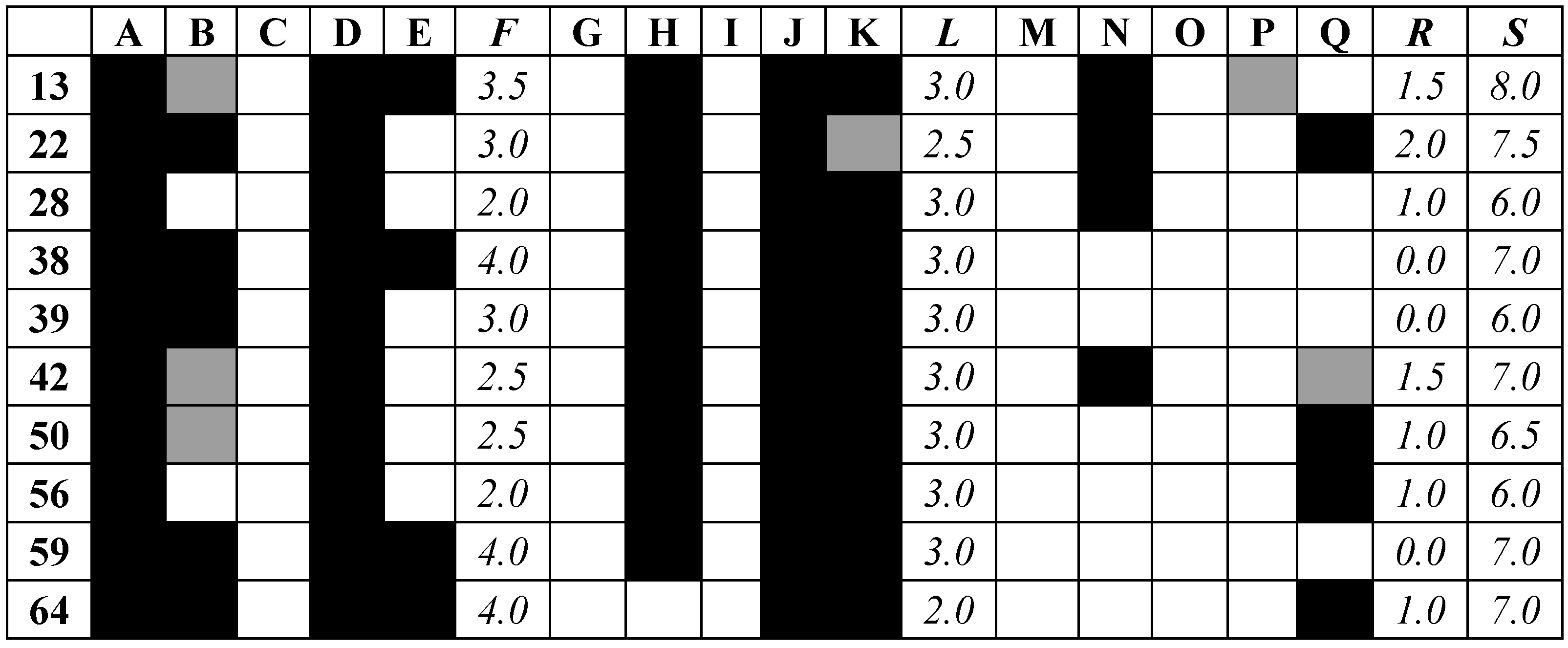
| ID | Predicted L.A. pIC50 | L.A. Vina, Glide, and Autodock 4 Scores, MM/GBSA Energy (kcal/mol) | List of Aminoacid Residues Included in Interactions | H.A. Vina, Glide, and Autodock 4 Scores, MM/GBSA Energy (kcal/mol) |
|---|---|---|---|---|
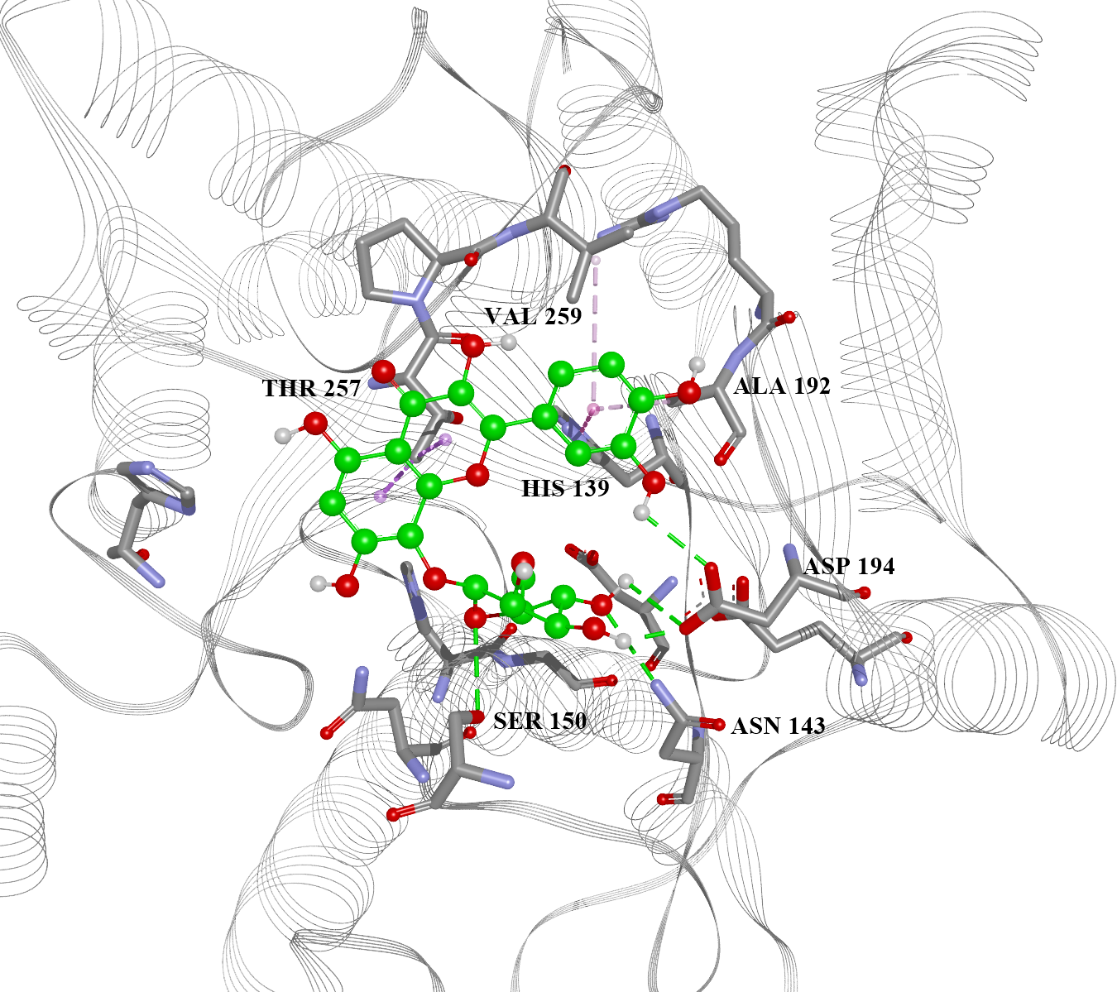
| 13 | 5.777 | −7.6, −5.53, −4.88, −30.80 | His28(H), Thr257(A), Asn143(H), Asp141(H), His139(A), Ala192(A) | −6.4, −4.40, −4.77, −32.18 |
| 22 | 5.667 | −6.8, N/A, −4.51, N/A | Asn143(H), Asp141(H), His139(A), Val259(A) | −6.5, −4.83, −3.82, −36.08 |
| 28 | 5.167 | −6.6, −5.16, −4.75, −32.15 | Asn143(H), His139(A), Thr257(A) | −6.0, −4.07, −3.98, −31.69 |
| 38 | 5.054 | −7.0, −5.14, −4.01, −35.12 | Asp141(H), Asn152(H), Thr257(A), Asp194(H) | −6.9, −5.35, −4.60, −38.47 |
| 39 | 5.465 | −6.8, −5.23, −4.59, −39.11 | Ser150(H), Asn143(H), Asp194(H), Ala192(A), Thr257(A) | −6.5, −4.09, −5.19, −24.99 |
| 42 | 5.738 | −6.6, −5.61, −5.08, −36.63 | His139(A), Asn152(H), Thr257(A) | −6.8, −3.84, −4.01, −32.88 |
| 50 | 5.122 | −6.3, −5.04, −4.32, −49.23 | Ser150(H), Asn152(H), His139(A), Val259(A), Ala192(A) | −7.2, −3.96, −4.35, −18.30 |
| 56 | 5.283 | −6.6, −5.64, −4.32, −35.87 | Asn152(H), His154(A), His139(A), Asp141(H), Val259(A) | −6.0, −5.38, −4.15, −33.92 |
| 59 | 5.204 | −7, −4.88, −5.59, −32.81 | Asn143(H), Glu197(H), Thr257(A), His139(A) | −6.7, −5.61, −4.44, −32.44 |
| 64 | 5.673 | −6.7, −4.04, −4.39, −40.30 | Ser150(H), His139(A), Ala192(A), Val259(A) | −6.0, N/A, −3.89, N/A |
| Number of Latent Variables | SSX | SSXacc | SDEC | SDEP | R2 | R2acc | Q2acc |
|---|---|---|---|---|---|---|---|
| 1 | 28.31 | 28.31 | 0.45 | 0.61 | 0.64 | 0.64 | 0.34 |
| 2 | 12.94 | 41.25 | 0.13 | 0.33 | 0.33 | 0.97 | 0.81 |
| 3 | 7.95 | 49.20 | 0.08 | 0.25 | 0.02 | 0.99 | 0.89 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glisic, S.; Sencanski, M.; Perovic, V.; Stevanovic, S.; García-Sosa, A.T. Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets. Molecules 2016, 21, 589. https://doi.org/10.3390/molecules21050589
Glisic S, Sencanski M, Perovic V, Stevanovic S, García-Sosa AT. Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets. Molecules. 2016; 21(5):589. https://doi.org/10.3390/molecules21050589
Chicago/Turabian StyleGlisic, Sanja, Milan Sencanski, Vladimir Perovic, Strahinja Stevanovic, and Alfonso T. García-Sosa. 2016. "Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets" Molecules 21, no. 5: 589. https://doi.org/10.3390/molecules21050589
APA StyleGlisic, S., Sencanski, M., Perovic, V., Stevanovic, S., & García-Sosa, A. T. (2016). Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets. Molecules, 21(5), 589. https://doi.org/10.3390/molecules21050589