
Synthesis and Biological Evaluation of an 18Fluorine-Labeled COX Inhibitor—[18F]Fluorooctyl Fenbufen Amide—For Imaging of Brain Tumors

Abstract
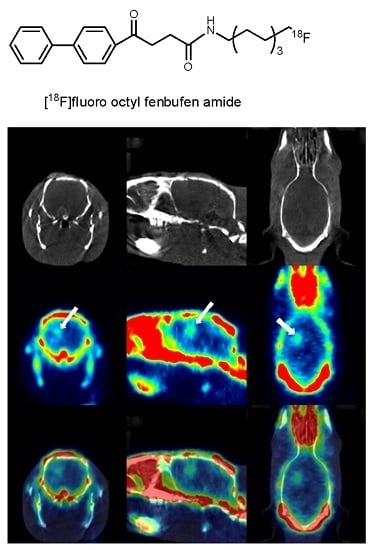
:
1. Introduction
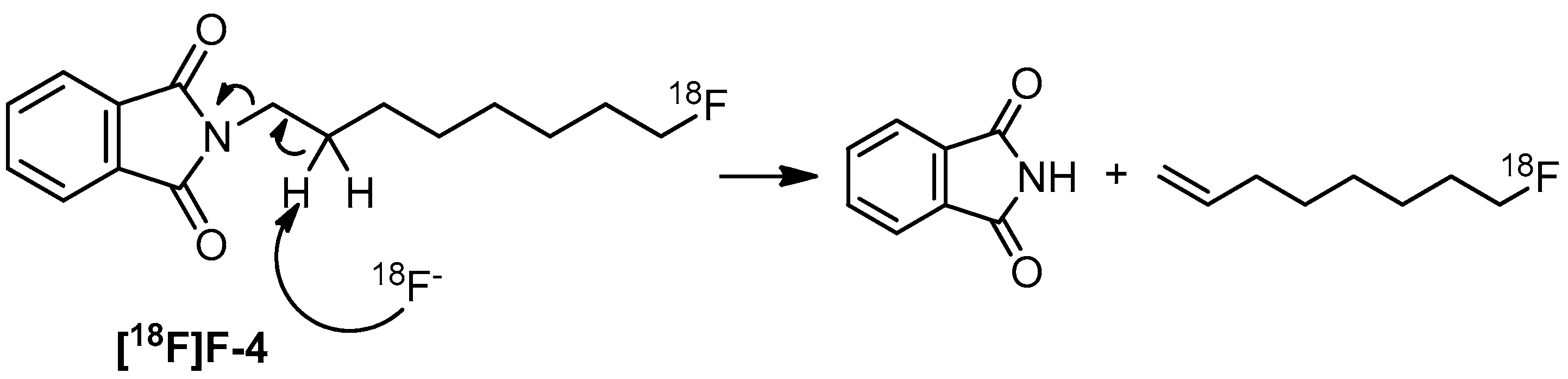
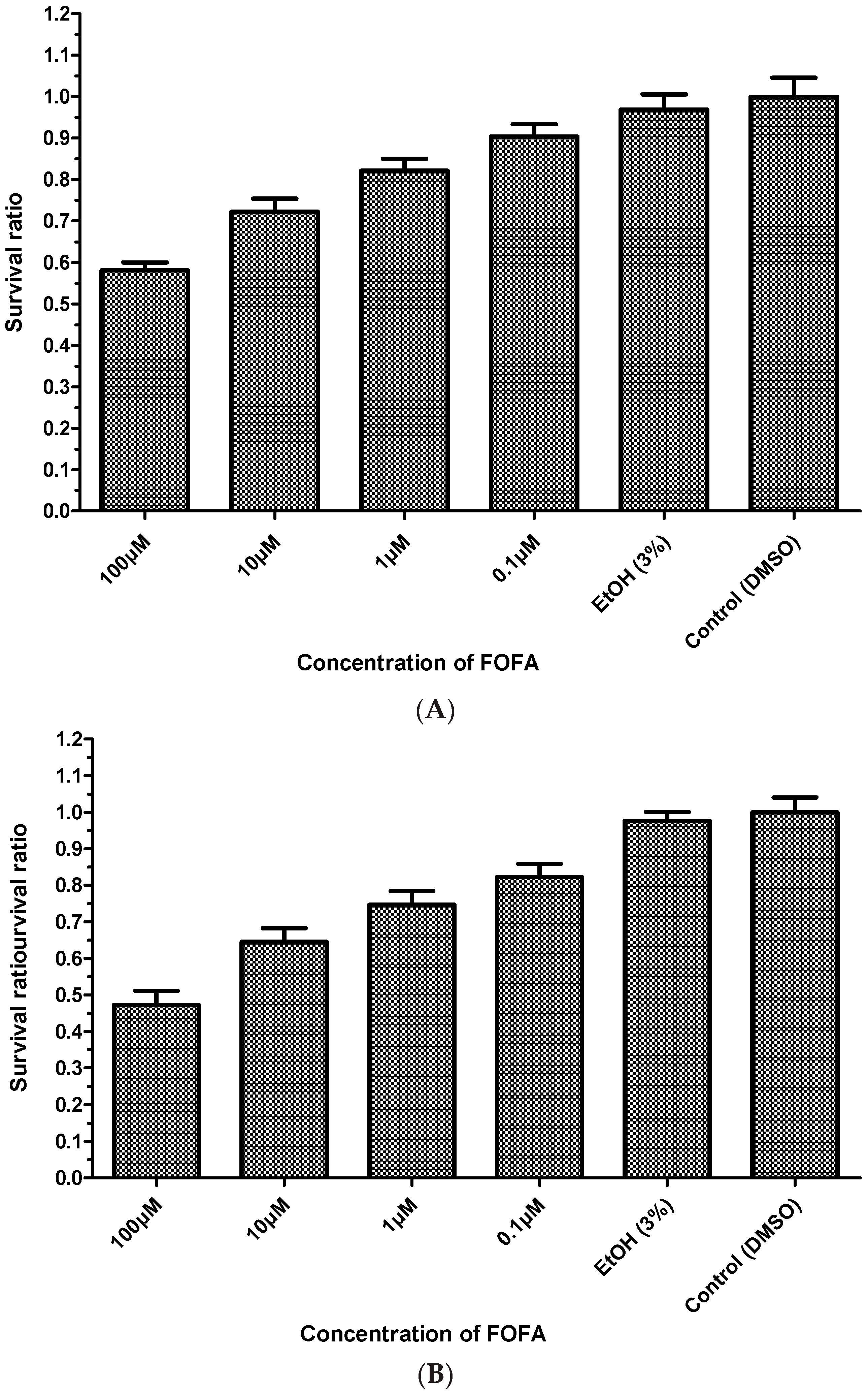
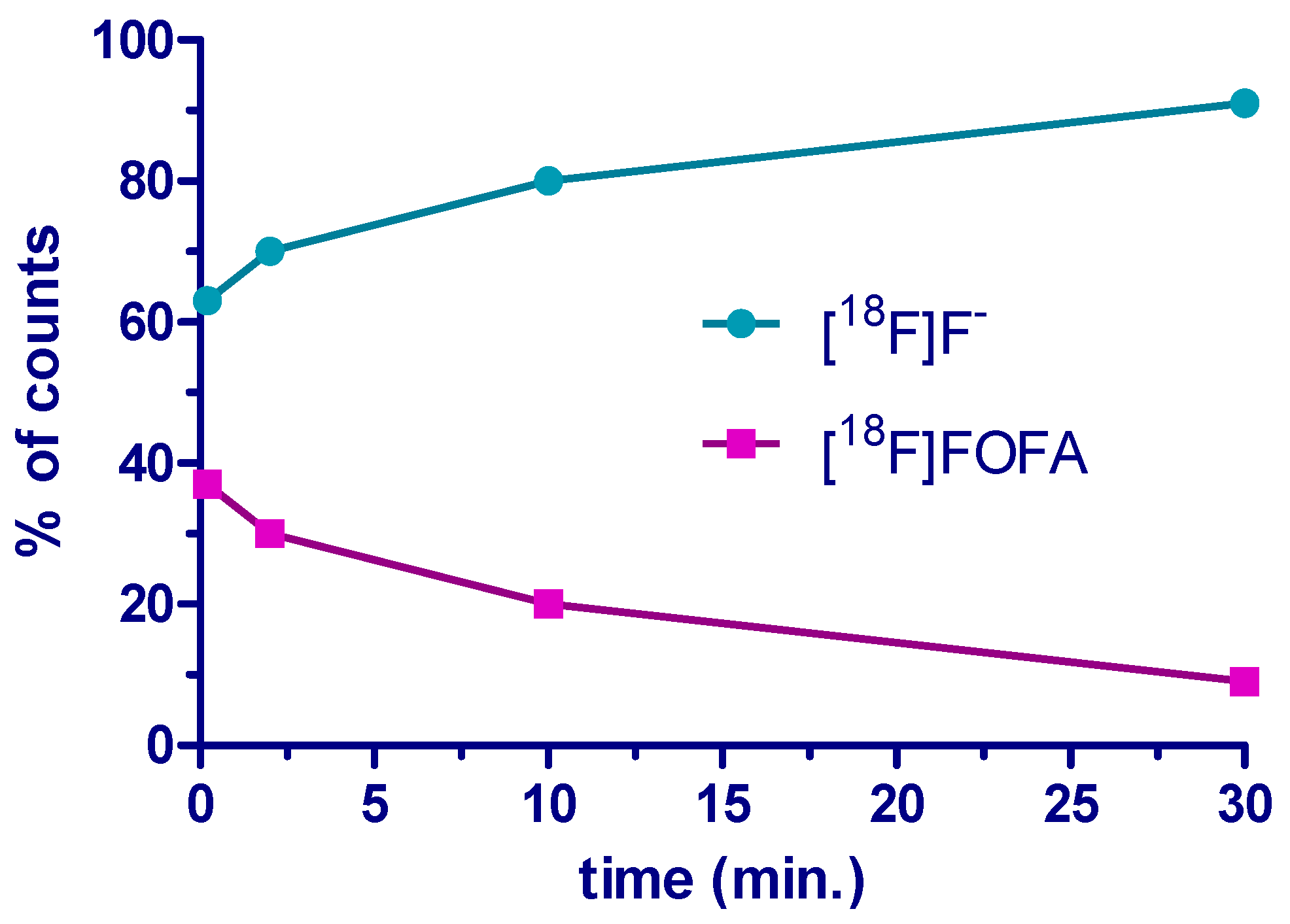
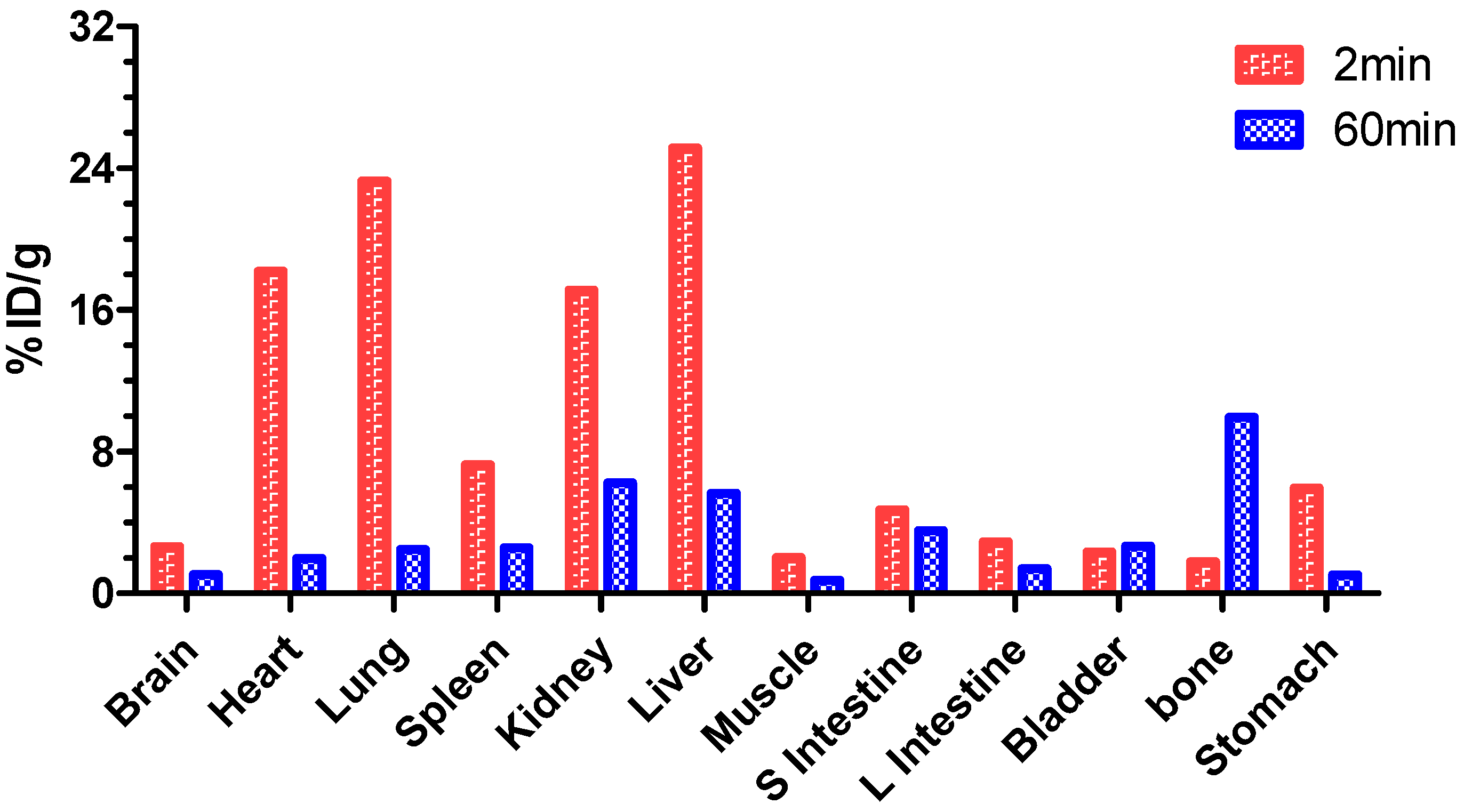
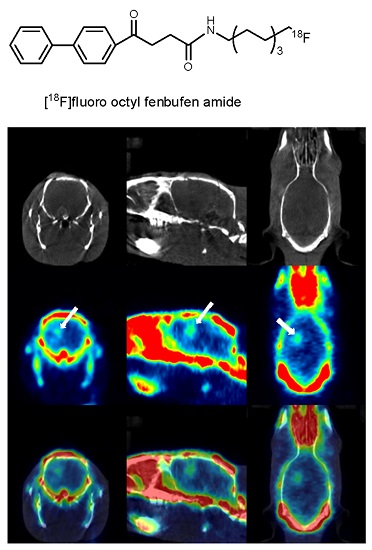
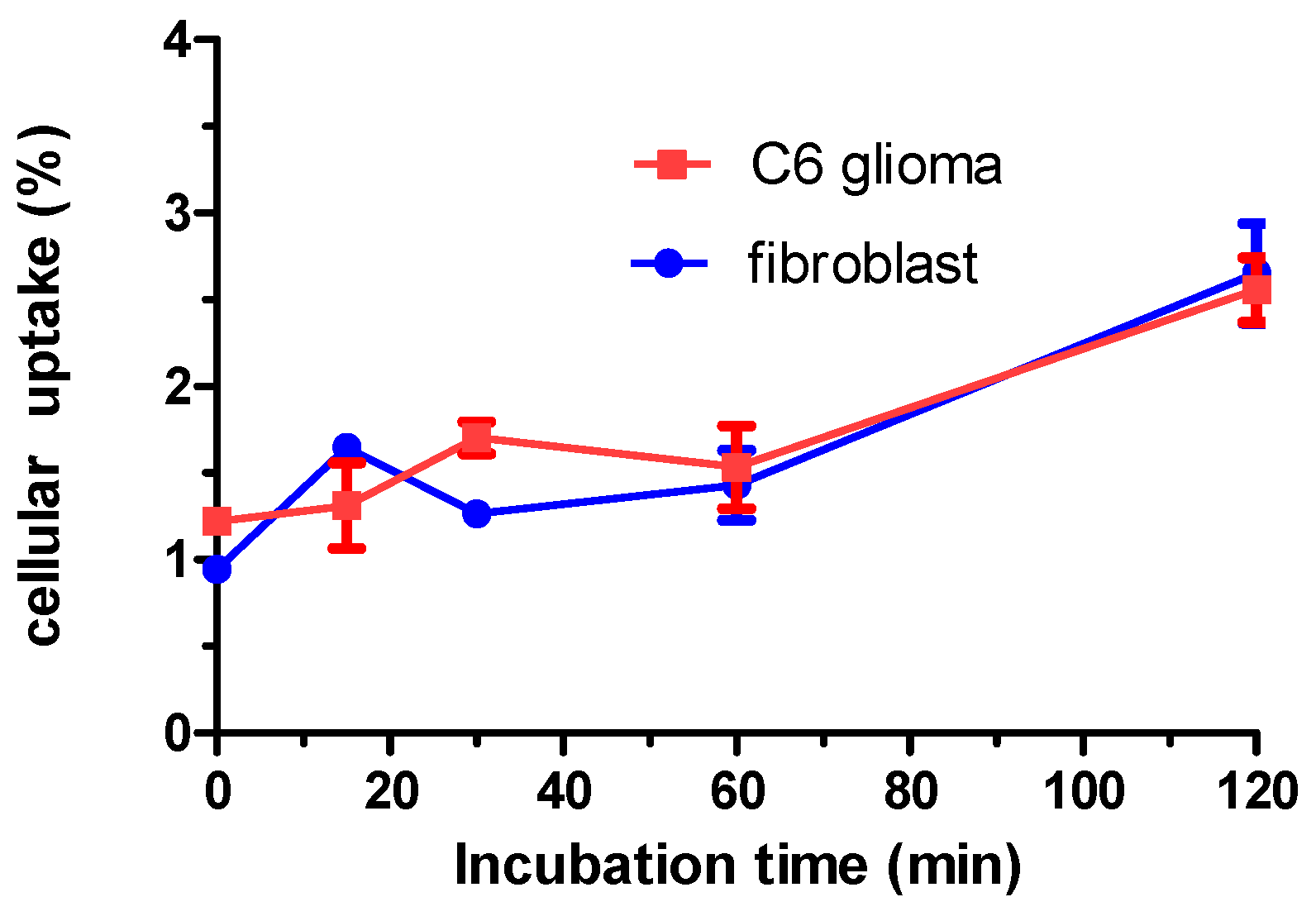
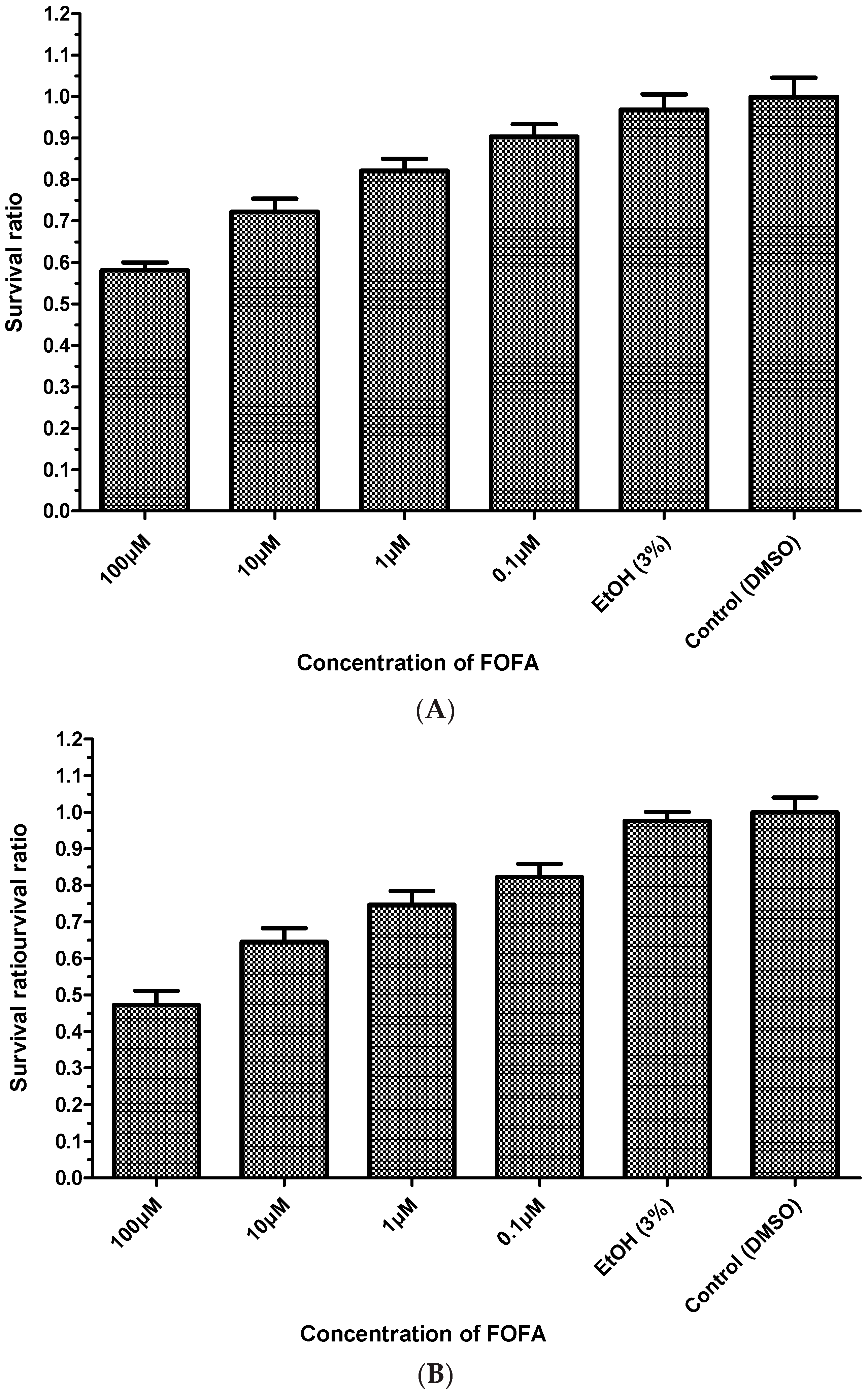
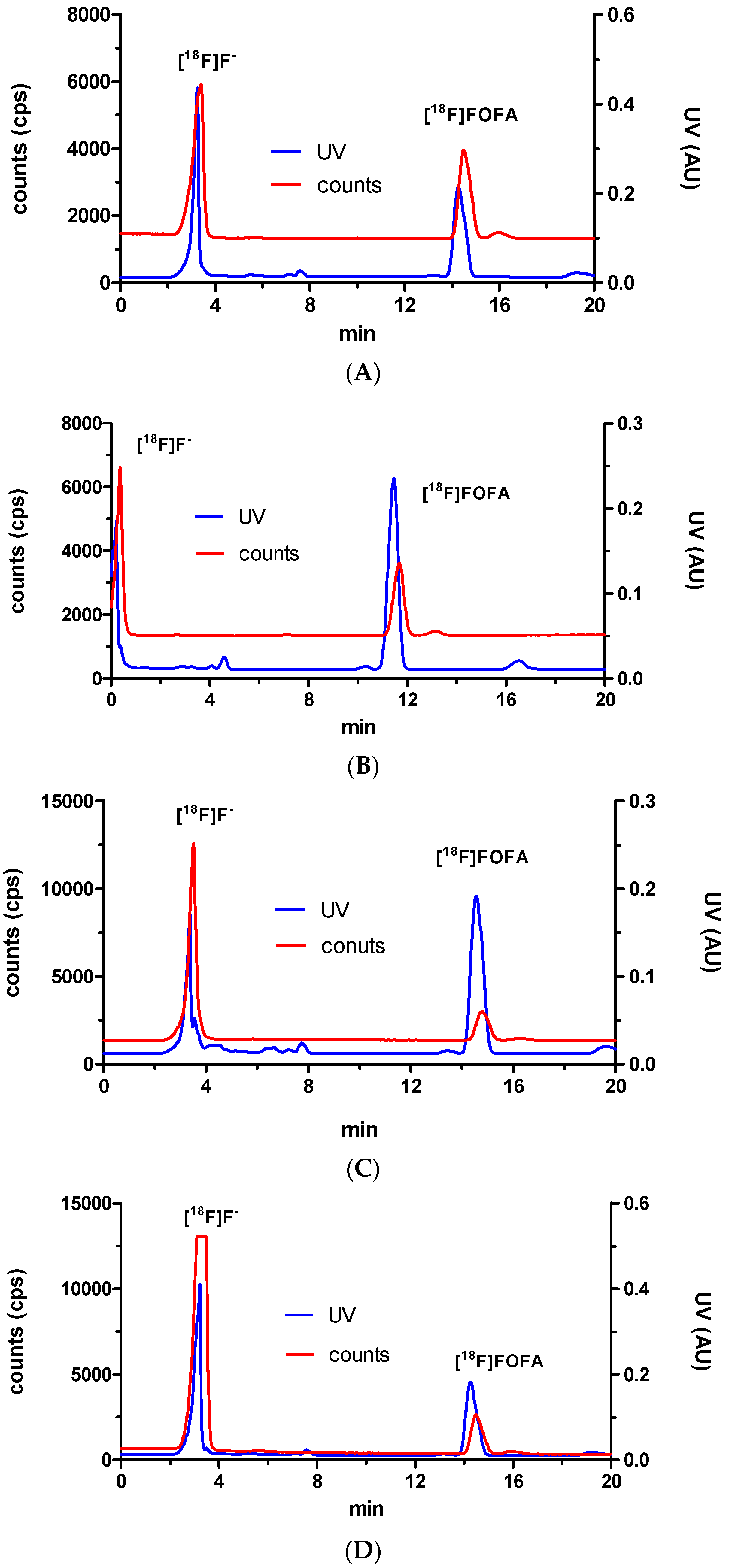
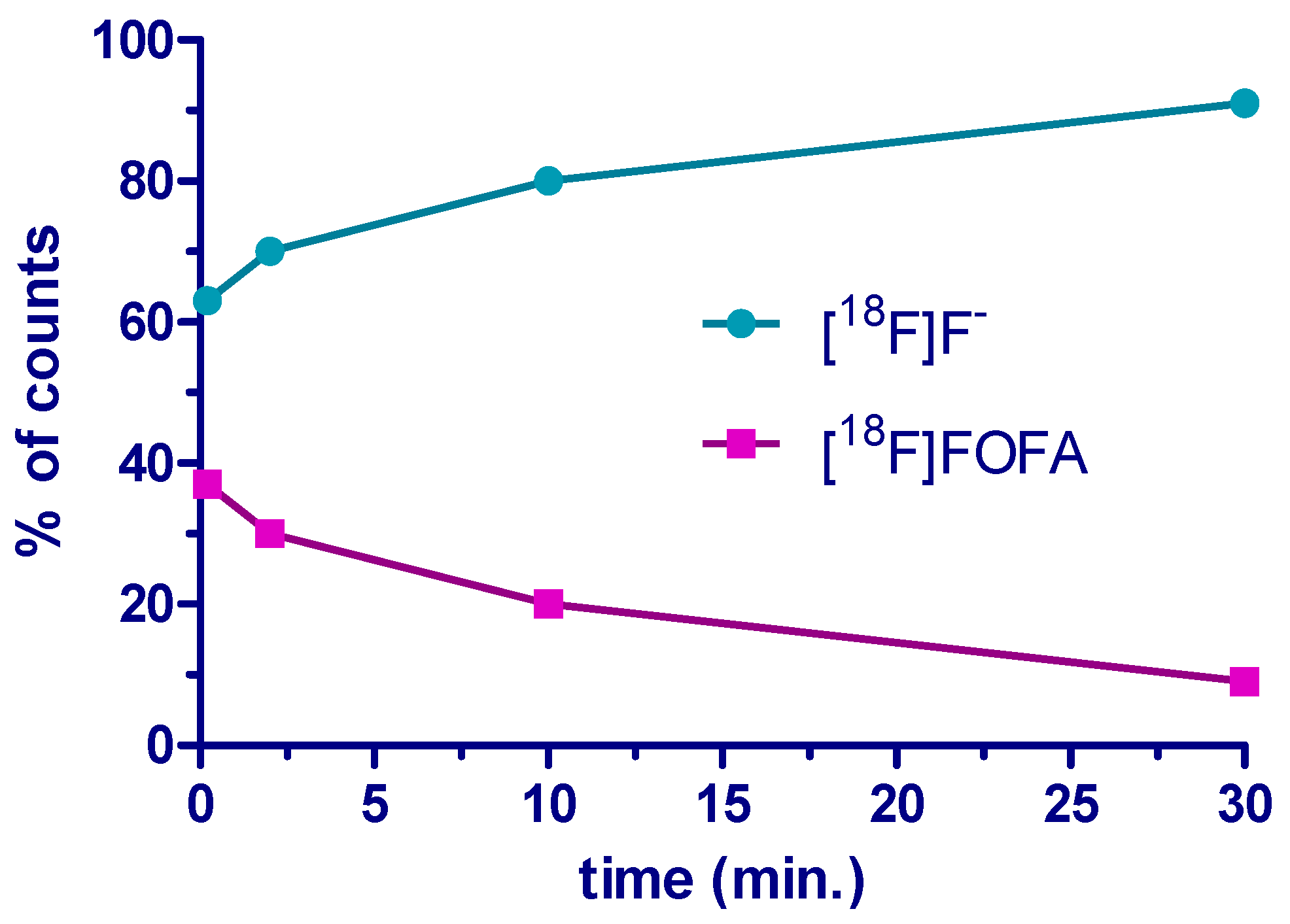
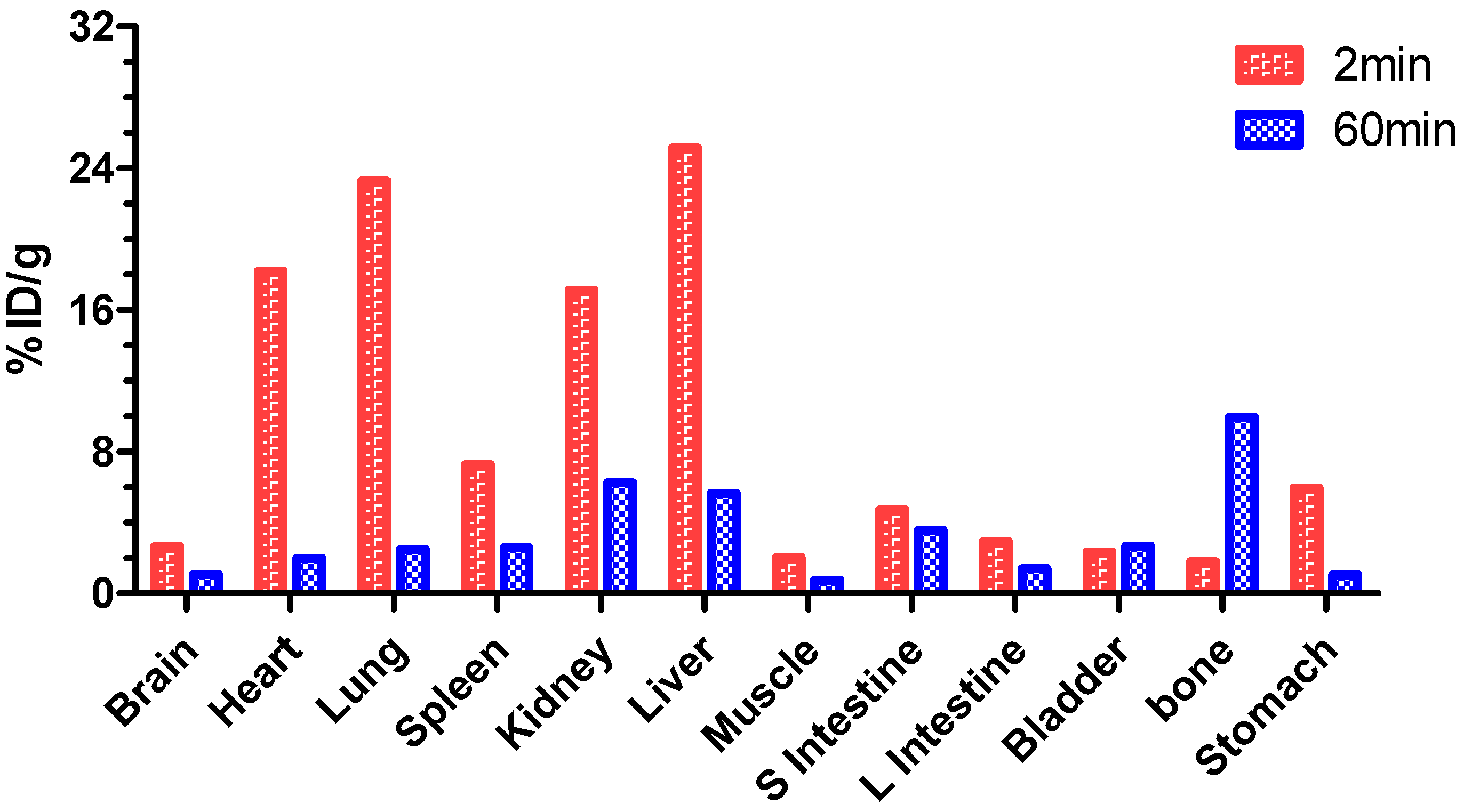
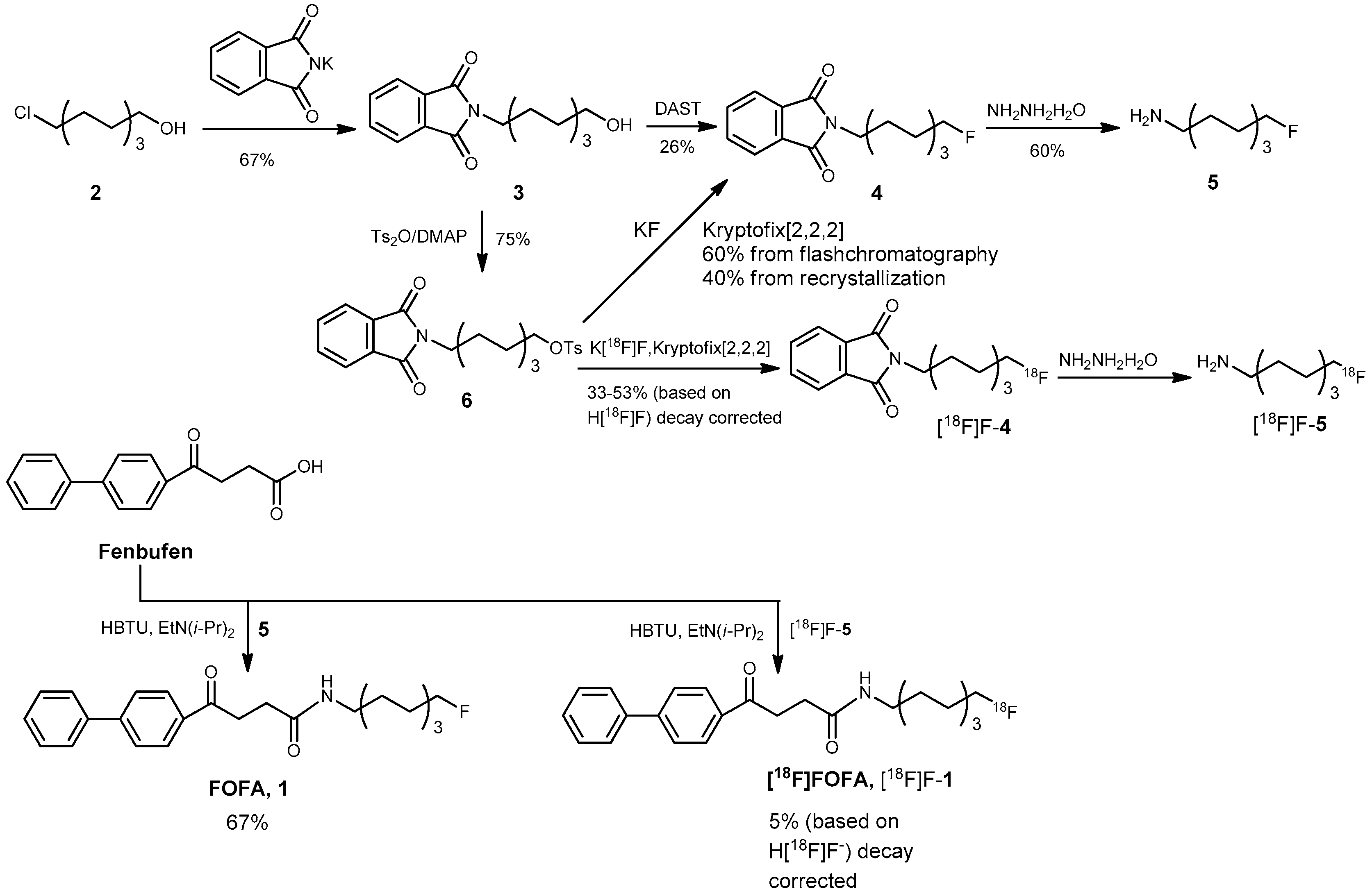
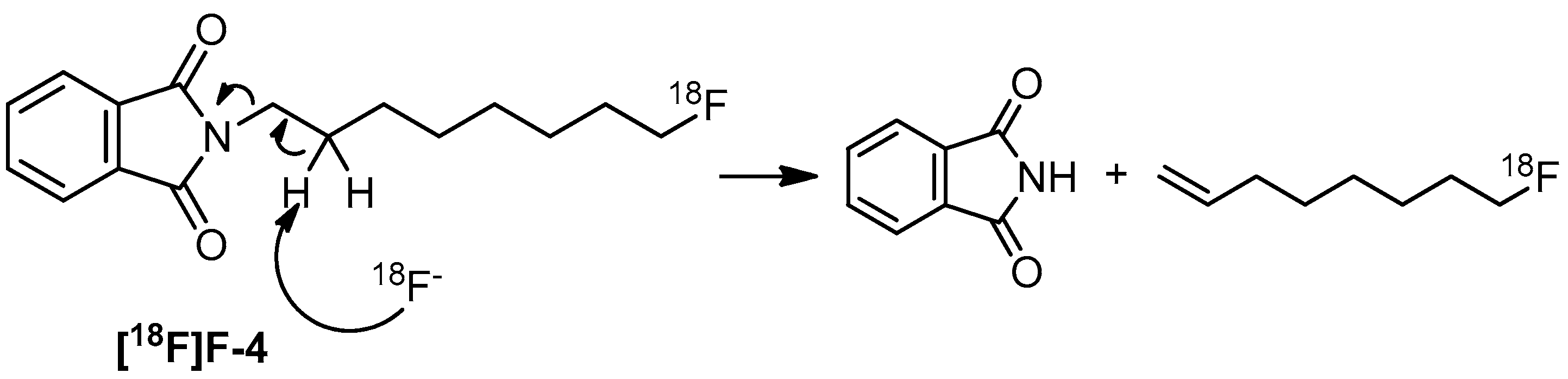
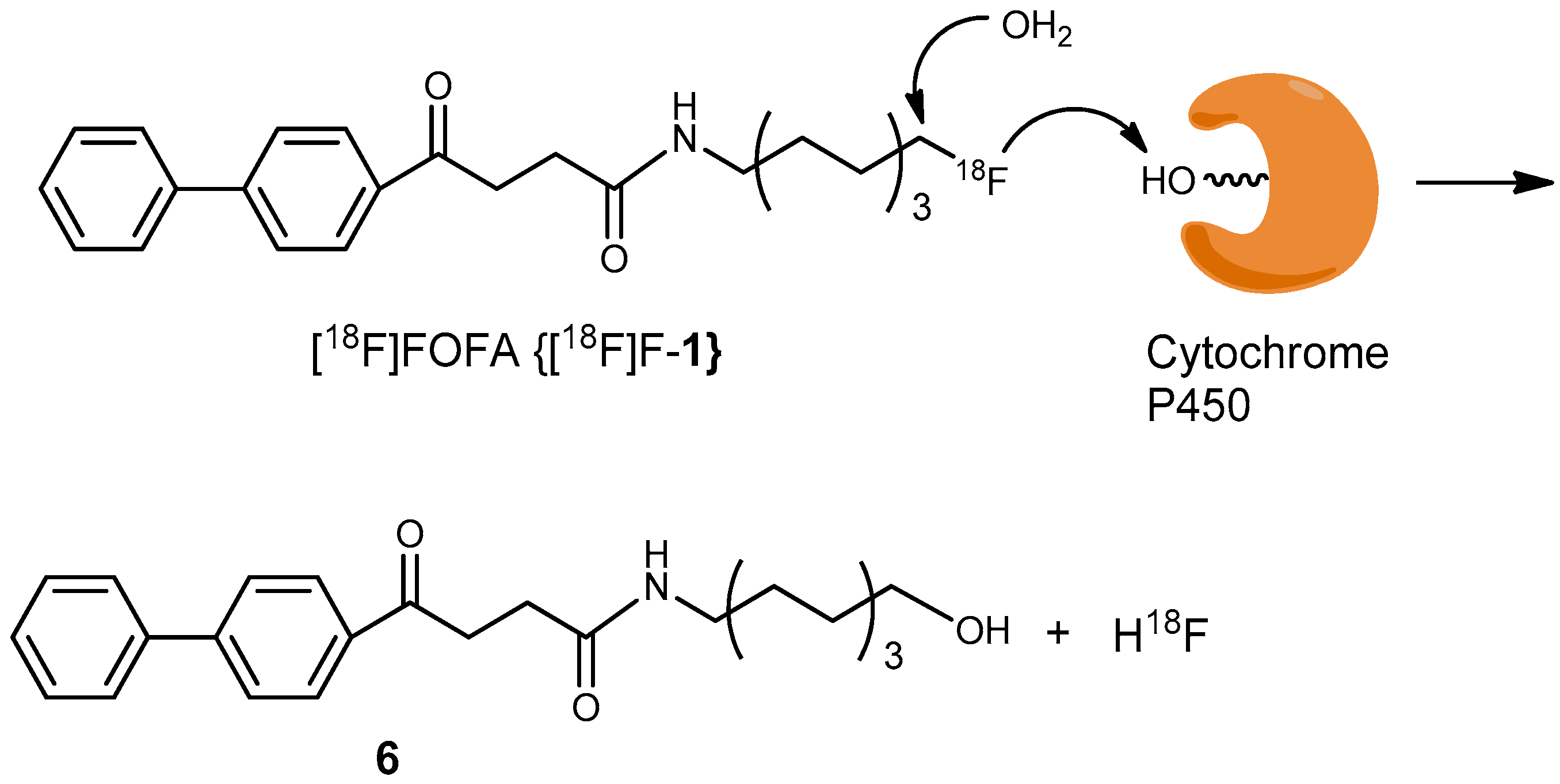
2. Results and Discussion
3. Materials and Methods
3.1. General Information
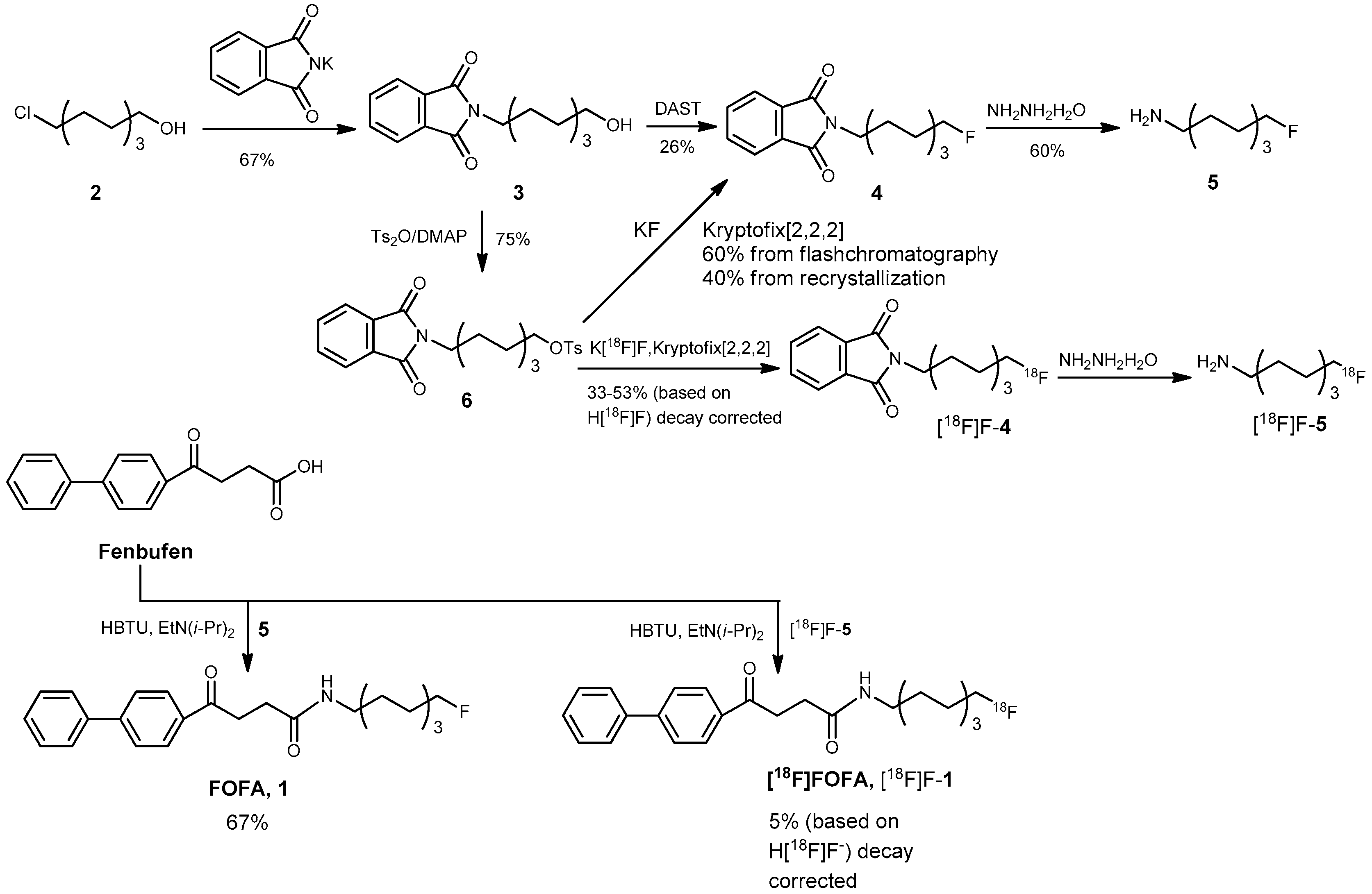
3.2. Chemical Syntheses
2-(8-Hydroxyoctyl)isoindoline-1,3-dione (3)
8-(1,3-Dioxoisoindolin-2-yl)octyl 4-methylbenzenesulfonate (6)
2-(8-Fluorooctyl)isoindoline-1,3-dione (4)
Route 1 via fluorination with diethyl aminosulfur trifluoride (DAST)
Route 2 via fluorination with KF and a phase-transfer catalyst
8-Fluorooctan-1-amine (5)
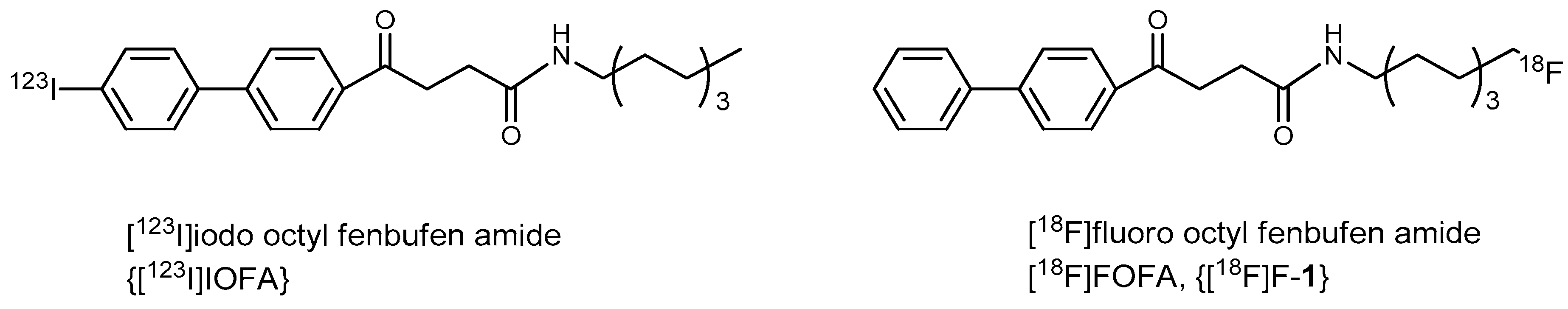
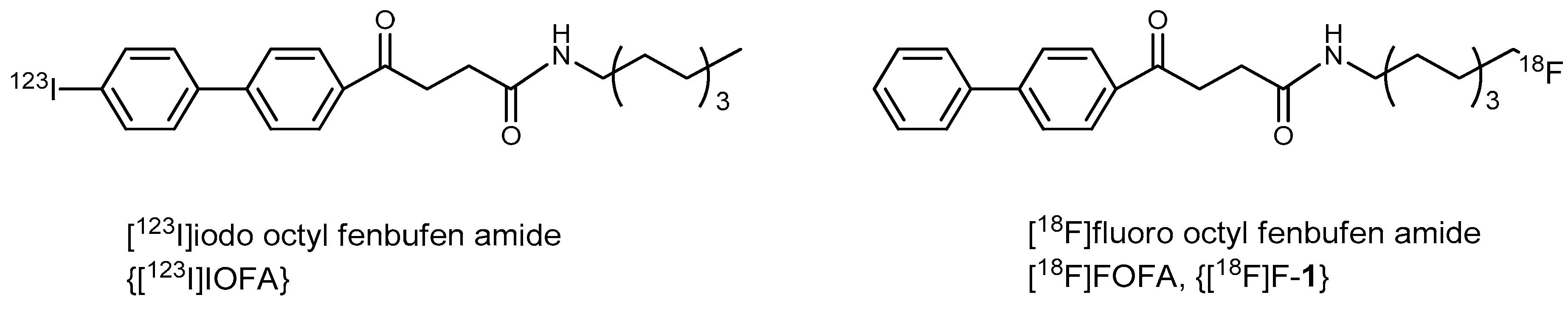
8-Fluorooctyl Fenbufen Amide (FOFA, 1)
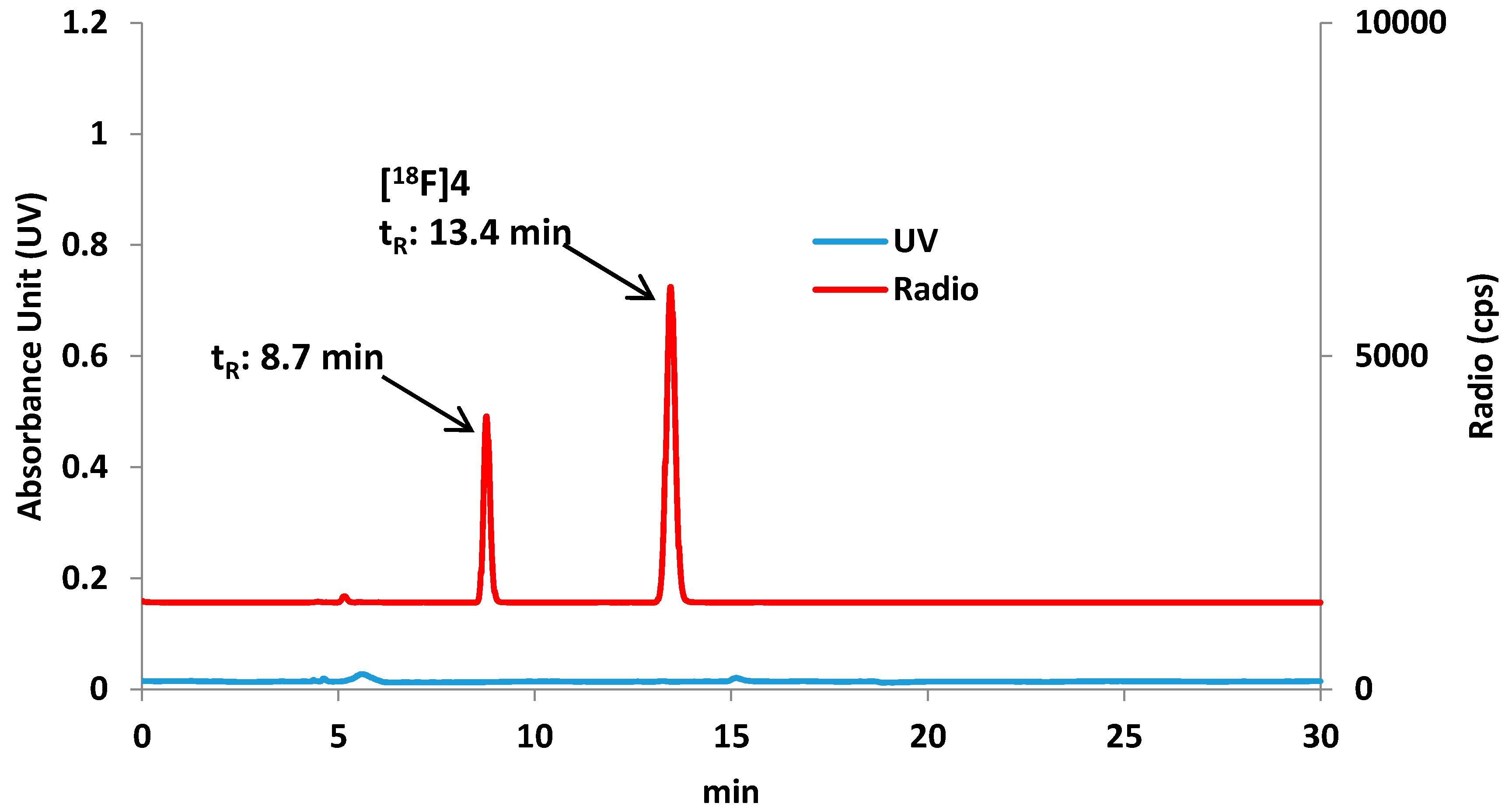
3.3. Radiochemistry
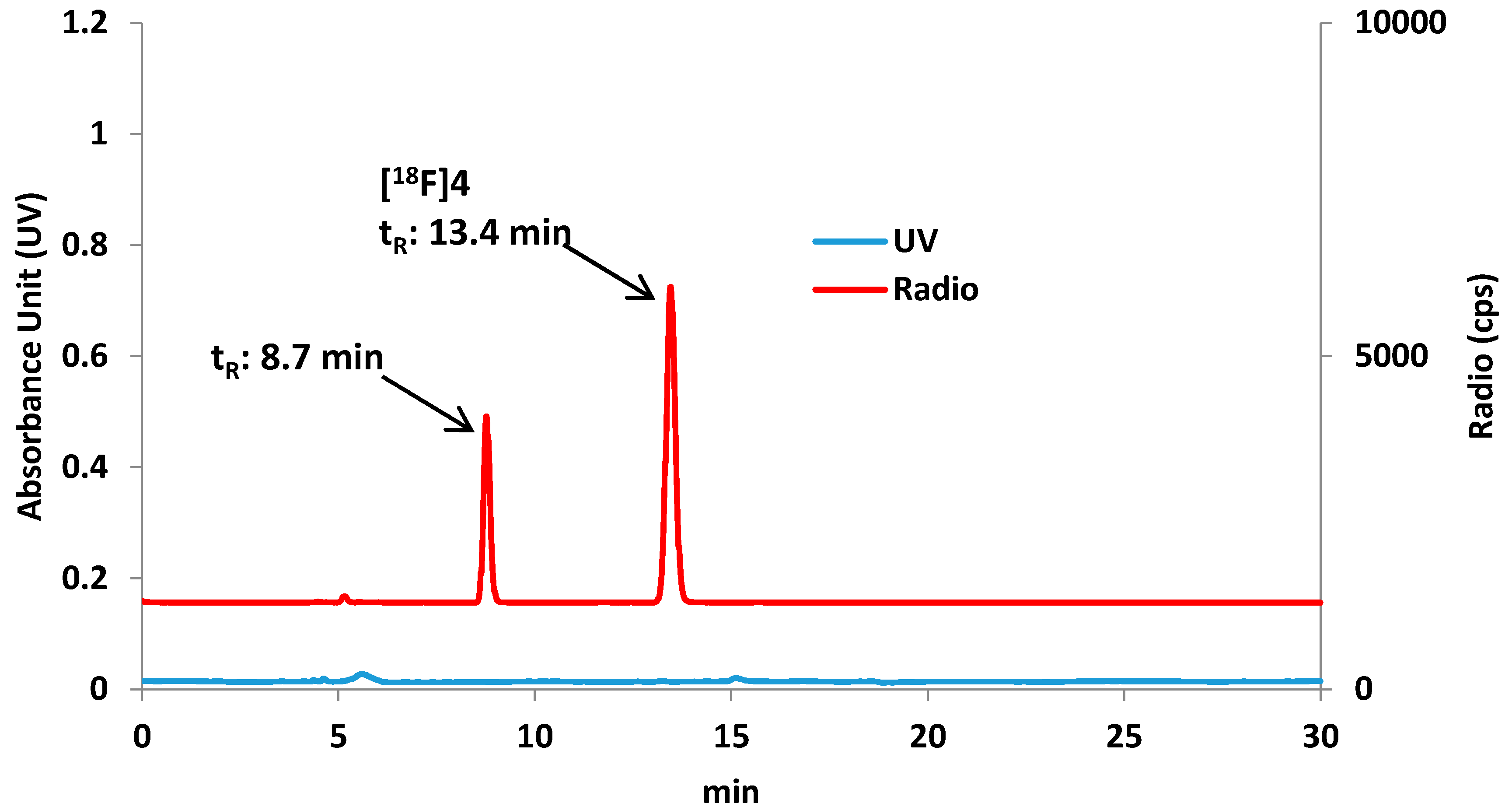
Preparation of 8-[18F]fluorooctyl-1-amine ([18F]F-5)
Preparation of 8-[18F]fluorooctylfenbufen amide ([18F]F-1)
3.4. Cell Culture for C6 Glioma and Fibroblasts
3.5. Animal Model of Cells Implanted in Rat Brains
3.6. MTT Assay (Cell Viability Assay)
3.7. Analysis of the Stability of [18F]FOFA {[18F]F-1} in Plasma
3.8. Biodistribution of [18F]F-1 in Normal Rats
3.9. PET/CT Imaging Study Using [18F]F-1 in an Animal Model
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Phillis, W.; Horrocks, L.A.; Farooqui, A.A. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders Brain Res. Rev. 2006, 52, 201–243. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.F.J. Imaging of cyclooxygenase-2 (COX-2) expression: Potential use in diagnosis and drug evaluation. Curr. Pharm. Des. 2006, 12, 3847–3856. [Google Scholar] [PubMed]
- Patrignani, P.; Tacconelli, S.; Sciulli, M.G.; Capone, M.L. New insights into COX-2 biology and inhibition. Brain Res. Rev. 2005, 48, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Uddin, Md.J.; Elleman, A.V.; Ghebreselasie, K.; Daniel, C.K.; Crews, B.C.; Nance, K.D.; Huda, T.; Marnett, L.J. Design of fluorine-containing 3,4-diarylfuran-2(5H)-ones as selective COX‑1 inhibitors. ACS Med. Chem. Lett. 2014, 5, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Uddin, Md.J.; Crews, B.C.; Ghebreselasie, K.; Huda, I.; Kingsley, P.J.; Ansari, M.S.; Tantawy, M.N.; Reese, J.; Marnett, L.J. Fluorinated COX-2 Inhibitors as Agents in PET Imaging of Inflammation and Cancer. Cancer Prev. Res. 2011, 4, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J. Natl. Can. Inst. 1998, 90, 1529–1536. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jokobsson, P.-J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-I.; Yang, C.-H.; Huang, C.-W.; Jian, J.-Y.; Huang, Y.-C.; Yu, C.-S. Synthesis and structure-activity relationships of fenbufen amide analogs. Molecules 2010, 15, 8796–8803. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-H.; Chiang, L.-W.; Jeng, K.-C.; Huang, H.-L.; Chen, J.; Lin, W.; Huang, C.-W.; Yu, C.-S. Solution-phase parallel synthesis and screening of anti-tumor activities from fenbufen and ethacrynic acid libraries. Bioorg. Med. Chem. Lett. 2011, 21, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-L.; Yeh, C.-N.; Lee, W.-Y.; Huang, Y.-C.; Chang, K.-W.; Lin, K.-J.; Tien, S.-F.; Su, W.-C.; Yang, C.-H.; Chen, J.-T.; et al. [123I]Iodooctyl fenbufen amide as a SPECT tracer for imaging tumors that overexpress COX enzymes. Biomaterials 2013, 34, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.Y.; Chang, J.Y.; Yen, Y. Increasing incidence of intrahepatic cholangiocarcinoma and its relationship to chronic viral hepatitis. J. Natl. Compr. Cancer Netw. 2009, 7, 423–427. [Google Scholar]
- Yeh, C.N.; Maitra, A.; Lee, K.F.; Jan, Y.Y.; Chen, M.F. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: An animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis 2004, 25, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.N.; Chiang, K.C.; Juang, H.H.; Pang, J.H.S.; Yu, C.S.; Lin, K.J.; Yeh, T.S.; Jan, Y.Y. Reappraisal of the therapeutic role of celecoxib in cholangiocarcinoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Yeh, C.N.; Weng, W.H.; Lenka, G.; Tsao, L.C.; Chiang, K.C.; Pang, S.T.; Chen, T.W.; Jan, Y.Y.; Chen, M.F. cDNA microarray profiling of rat cholangiocarcinoma induced by thioacetamide. Mol. Med. Rep. 2013, 8, 350–360. [Google Scholar] [PubMed]
- Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18F]fluoride ion. Eur. J. Org. Chem. 2008, 2853–2873. [Google Scholar] [CrossRef]
- Lasne, M.C.; Perrio, C.; Rouden, J.; Barre, L.; Roeda, D.; Dolle, F.; Crouzel, C. Chemistry of beta(+)-emitting compounds based on fluorine-18. Top. Curr. Chem. 2002, 222, 201–258. [Google Scholar]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Zhang, X.; Xiong, Z.; Chen, X. Comparative in vitro and in vivo evaluation of two 64Cu-labeled bombesin analogs in a mouse model of human prostate adenocarcinoma. Nucl. Med. Biol. 2006, 33, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M.A. Philosophy for CNS Radiotracer Design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.W.; Djulbegovic, B.; Soares, H.P.; Siegel, B.A.; Lowe, V.J.; Lyman, G.H.; Coleman, R.E.; Wahl, R.; Paschold, J.C.; Avril, N.; et al. Recommendations on the Use of 18F-FDG PET in Oncology. J. Nucl. Med. 2008, 49, 480–508. [Google Scholar] [CrossRef] [PubMed]
- Dunet, V.; Rossier, C.; Buck, A.; Stupp, R.; Prior, J.O. Performance of 18F-Fluoro-Ethyl-Tyrosine (18F-FET) PET for the Differential Diagnosis of Primary Brain Tumor: A Systematic Review and Metaanalysis. J. Nucl. Med. 2012, 53, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Kniess, T.; Pietz, J. Radiolabeled COX-2 Inhibitors for Non-Invasive Visualization of COX-2 Expression and Activity—A Critical Update. Molecules 2013, 18, 6311–6355. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Oberdorfer, F. Synthesis of 4-O-methyl-protected 5-(2-hydroxyethy)-2’-deoxyuridine derivatives and their nucleophilic fluorination to 5-(2-fluoroethyl)-2’-deoxyuridine. Synthesis 1999, 2057–2064. [Google Scholar] [CrossRef]
- Huang, H.-L.; Huang, Y.-C.; Lee, W.-L.; Yeh, C.-N.; Lin, K.-J.; Yu, C.-S. 18F-Glutathione conjugate as a PET tracer for imaging tumors that overexpress L-PGDS enzyme. PLoS ONE 2014, 9, e104118. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Huang, H.-L.; Yeh, C.-N.; Lin, K.-J.; Yu, C.-S. Investigation of brain tumors using 18F-fluorobutyl ethacrynic amide and its metabolite with positron emission tomography. Oncotarg. Ther. 2015, 8, 1877–1885. [Google Scholar]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Di Car, F.J. Human cytochrome p450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors drug. Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Armstrong, A.S.; Gunn, K. Clinical sevoflurane metabolism and dispodition. II. The role of cytochrome P450 2E1 in fluoride and hexafluoroisopropanol formation. Anesthesiology 1995, 82, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.I.; Ganellin, C.R.; Piegentili, A.; Dunn, P.M.; Jenkinson, D.H. Synthesis and pharmacological testing of polyamino quinolones as blockers of the apamin-sensitive Ca2+-activated K+ channel (SKCa). Bioorg. Med. Chem. 2007, 15, 5457–5479. [Google Scholar] [CrossRef] [PubMed]
- Bartholomä, M.D.; Vortherms, A.R.; Hillier, S.; Ploier, B.; Joyal, J.; Babich, J.; Doyle, R.P.; Zubieta, J. Synthesis, cytotoxicity, and insight into the mode of action of Re(CO)3 thymidine complexes. Chem. Med. Chem. 2010, 5, 1513–1529. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Z.; Zhu, L.; Tan, X.; Li, C. Silver-catalyzed radical fluorination of alkylboronates in azueous solution. J. Am. Chem. Soc. 2014, 136, 16439–16443. [Google Scholar] [CrossRef] [PubMed]
- Pattison, F.L.M.; Howell, W.C.; White, R.W. Toxic fluorine compounds. VI w-Fluoro alkylamine. J. Am. Chem. Soc. 1956, 78, 13487–13489. [Google Scholar]
- Windhorst, A.D.; Bechger, L.; Visser, G.W.M.; Menge, W.P.M.B.; Leurs, R.; Timmerman, H.; Herscheid, J.D.M. Approaches towards the synthesis of fluoro(cyclo)alkylamines. J. Fluo. Chem. 1996, 80, 35–40. [Google Scholar] [CrossRef]
- Tang, D.; McKinley, E.T.; Hight, M.R.; Uddin, M.I.; Harp, J.M.; Fu, A.; Nickels, M.L.; Buck, J.R.; Manning, H.C. Synthesis and Structure-Activity Relationships of 5,6,7-Substituted Pyrazolopyrimidines: Discovery of a Novel TSPO PET Ligand for Cancer Imaging. J. Med. Chem. 2013, 56, 3429–3433. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent Combination | RCY | Experiment No. |
|---|---|---|---|
| 1 | TsCl/pyridine | 33% ± 4% | 4 |
| 2 | TsCl/DMAP | 39% ± 4% | 3 |
| 3 | Ts2O/DMAP | 53% ± 1% | 4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-C.; Chang, Y.-C.; Yeh, C.-N.; Yu, C.-S. Synthesis and Biological Evaluation of an 18Fluorine-Labeled COX Inhibitor—[18F]Fluorooctyl Fenbufen Amide—For Imaging of Brain Tumors. Molecules 2016, 21, 387. https://doi.org/10.3390/molecules21030387
Huang Y-C, Chang Y-C, Yeh C-N, Yu C-S. Synthesis and Biological Evaluation of an 18Fluorine-Labeled COX Inhibitor—[18F]Fluorooctyl Fenbufen Amide—For Imaging of Brain Tumors. Molecules. 2016; 21(3):387. https://doi.org/10.3390/molecules21030387
Chicago/Turabian StyleHuang, Ying-Cheng, Yu-Chia Chang, Chun-Nan Yeh, and Chung-Shan Yu. 2016. "Synthesis and Biological Evaluation of an 18Fluorine-Labeled COX Inhibitor—[18F]Fluorooctyl Fenbufen Amide—For Imaging of Brain Tumors" Molecules 21, no. 3: 387. https://doi.org/10.3390/molecules21030387