
Phosphorylation of Akt by SC79 Prevents Iron Accumulation and Ameliorates Early Brain Injury in a Model of Experimental Subarachnoid Hemorrhage

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
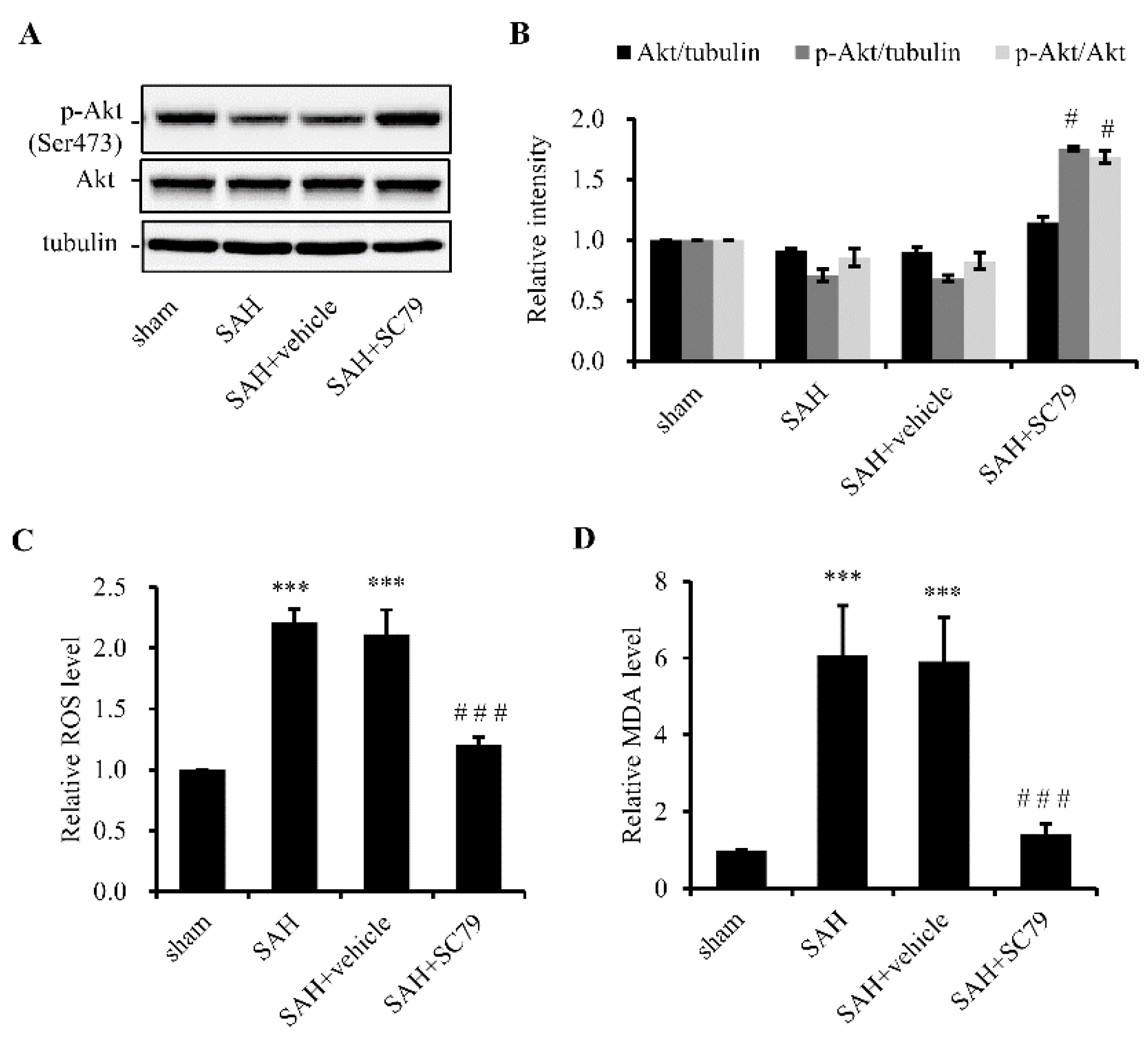
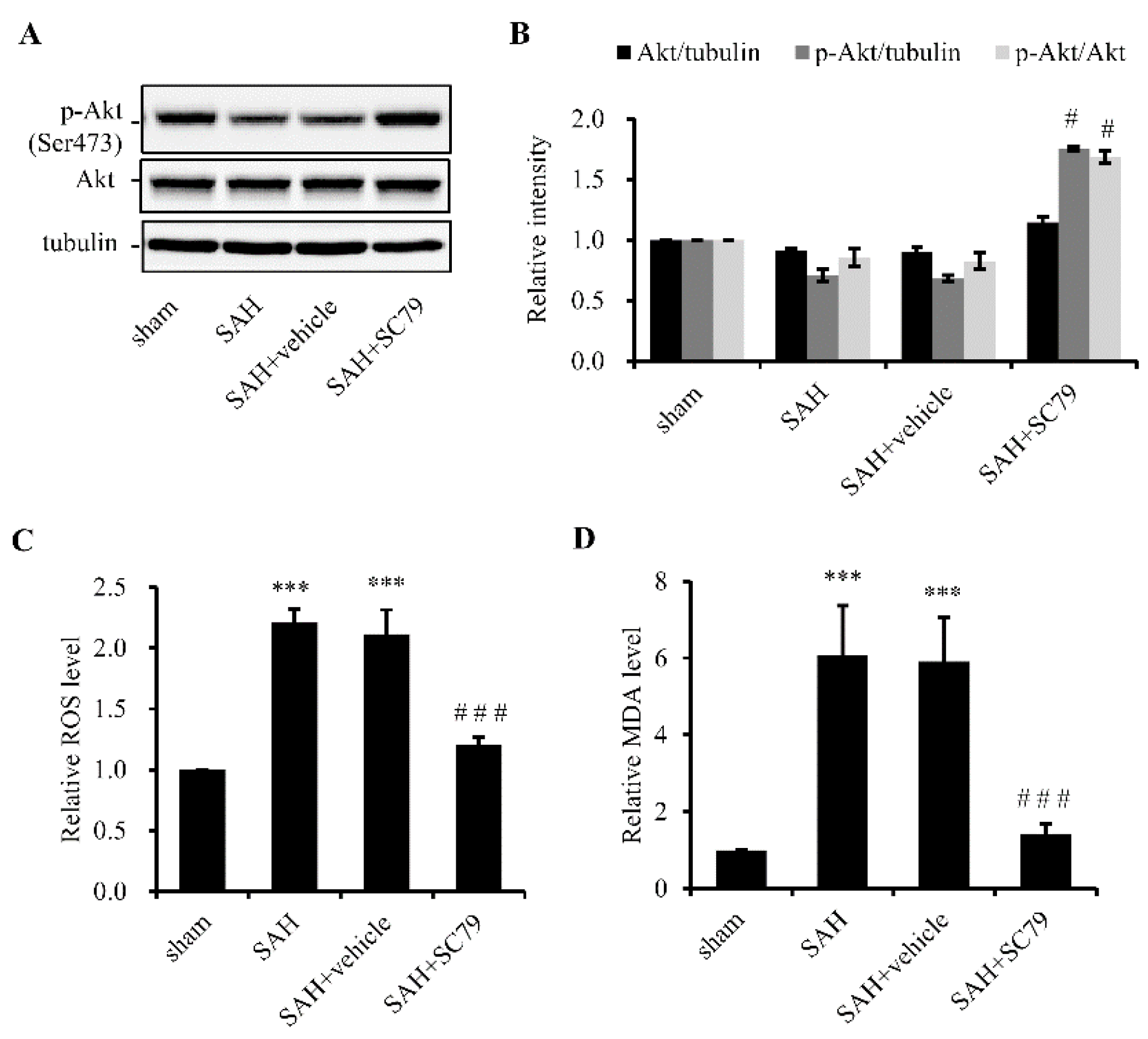
2.1. SC79 Activates Akt and Protects Cells against Oxidative Stress after SAH
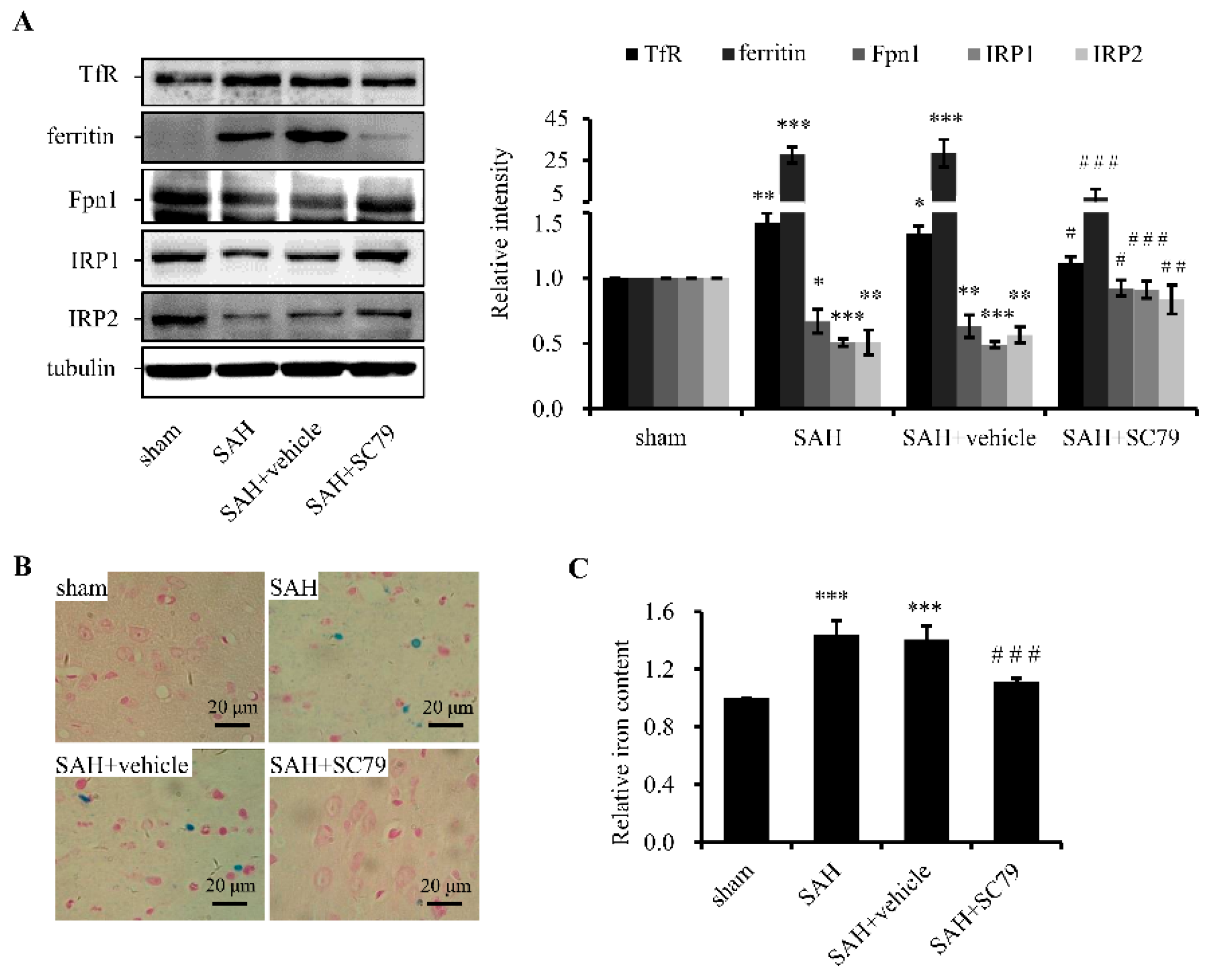
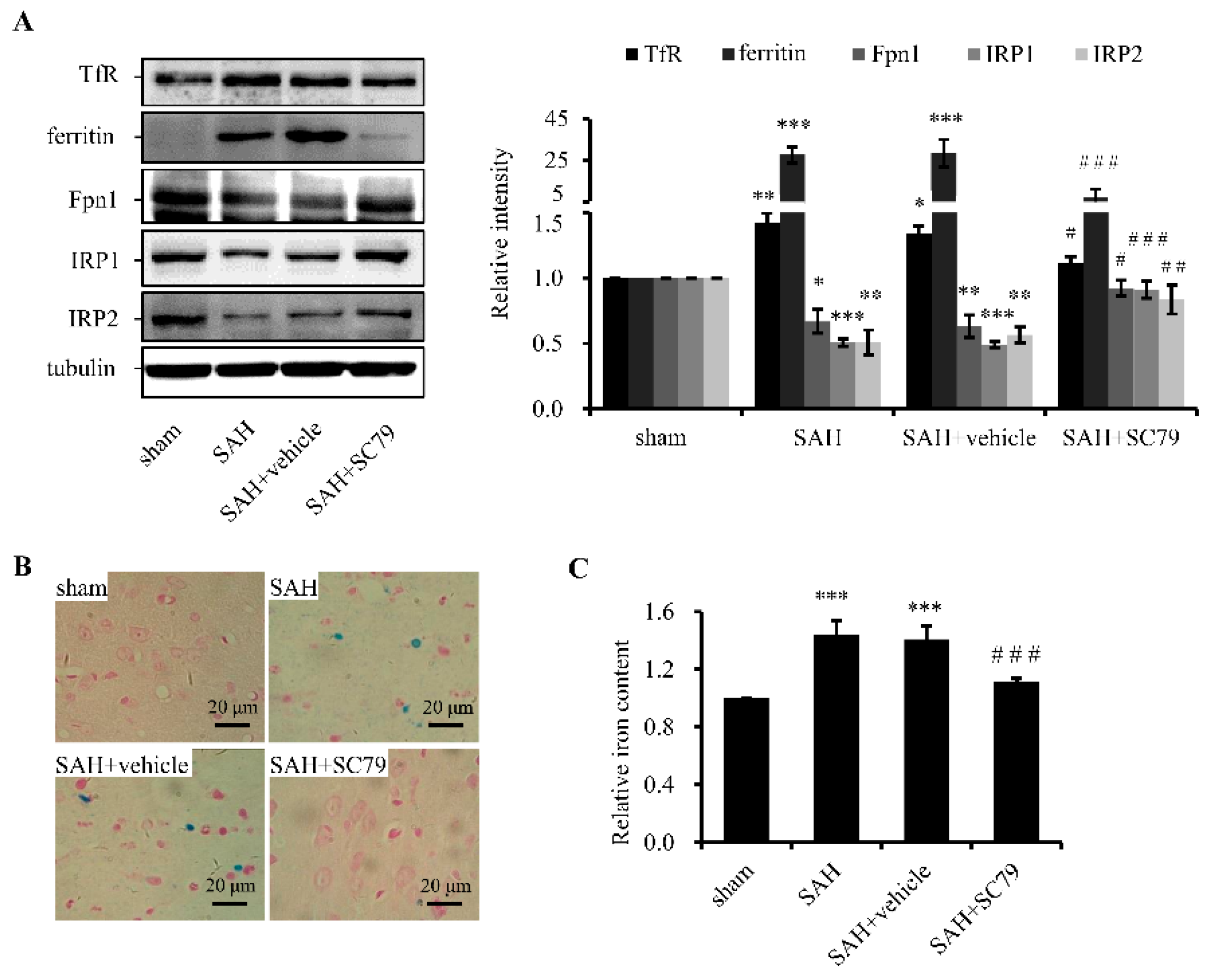
2.2. SC79 Administration Mitigates SAH-Induced Iron Accumulation
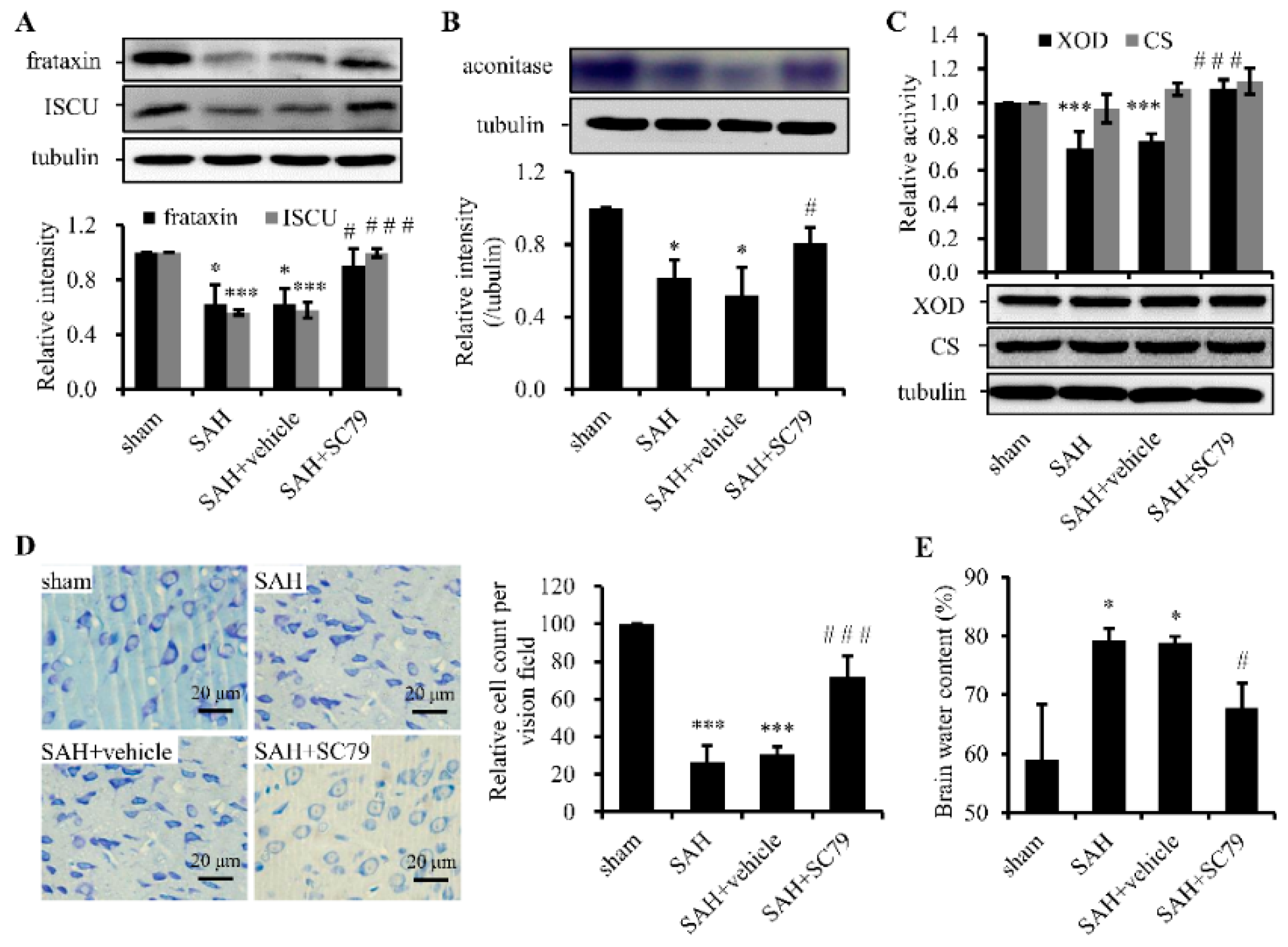
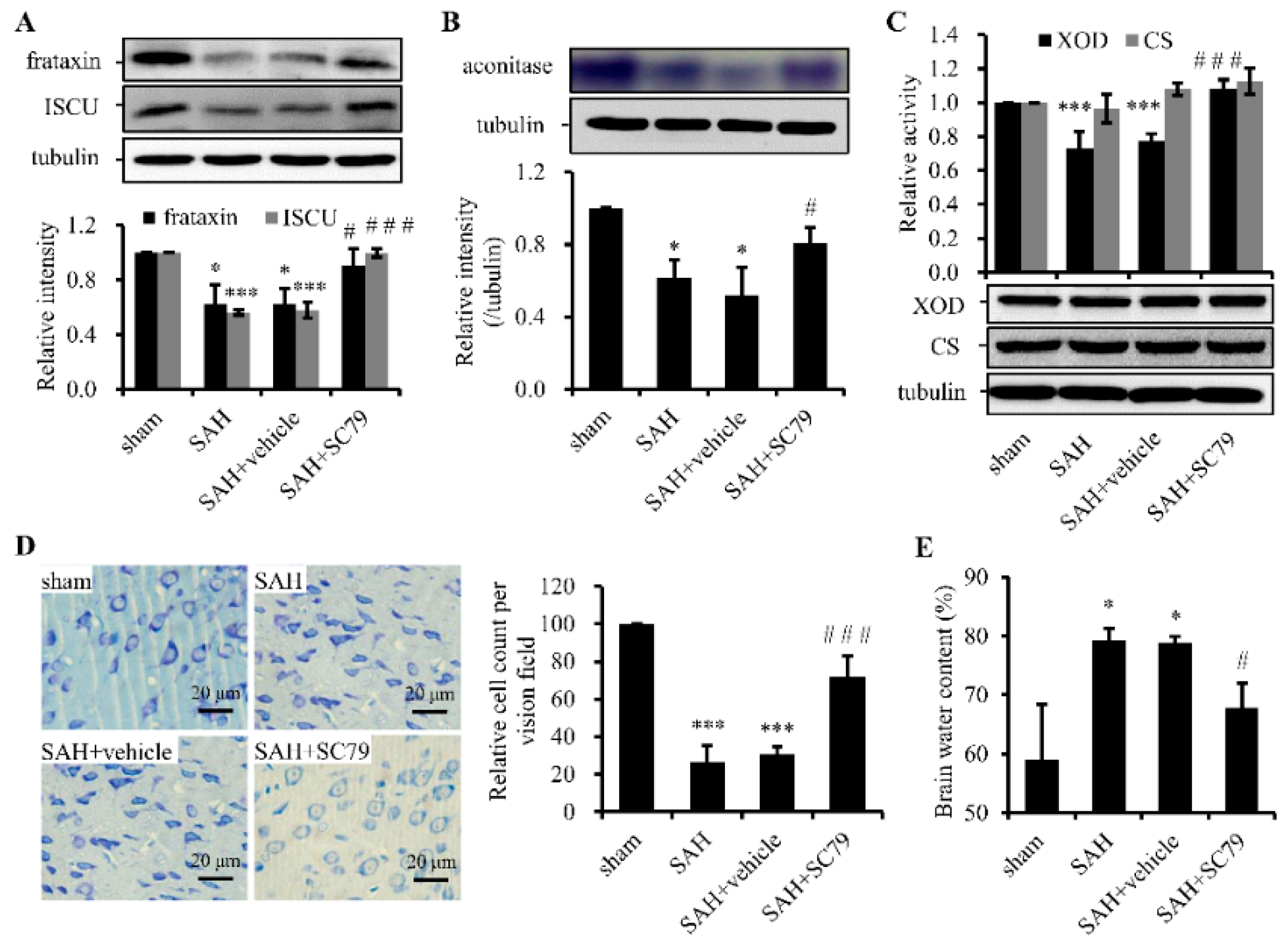
2.3. SC79 Administration Rescues the Fe-S Cluster Biogenesis and Fe-S Cluster-Containing Enzymatic Activities and Alleviates Neuronal Injury after SAH
3. Discussion
4. Experimental Section
4.1. Animal Preparation
4.2. Prechiasmatic Cistern Blood Injection for SAH Model
4.3. Experimental Design and Tissue Preparation
4.4. Iron Perl’s Staining and Ferrozine Iron Assays
4.5. Western Blot Analysis
4.6. Enzymetic Activity Assays
4.7. Measurement of Cellular ROS Production and MDA Levels
4.8. Brain Edema
4.9. Nissl Staining
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Green, D.M.; Burns, J.D.; DeFusco, C.M. ICU management of aneurysmal subarachnoid hemorrhage. J. Intensive Care Med. 2013, 28, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, A.V.; Palade, C.; Voinescu, D.; Nica, D.A. Subarachnoid hemorrhage and cerebral vasospasm—Literature review. J. Med. Life 2013, 6, 120–125. [Google Scholar] [PubMed]
- Yan, H.; Hao, S.; Sun, X.; Zhang, D.; Gao, X.; Yu, Z.; Li, K.; Hang, C.H. Blockage of mitochondrial calcium uniporter prevents iron accumulation in a model of experimental subarachnoid hemorrhage. Biochem. Biophys. Res. Commun. 2015, 456, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Keep, R.F.; He, Y.; Sagher, O.; Hua, Y.; Xi, G. Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Hentze, M.W. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 1995, 14, 2917–2924. [Google Scholar] [PubMed]
- Tampo, Y.; Kotamraju, S.; Chitambar, C.R.; Kalivendi, S.V.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Oxidative stress-induced iron signaling is responsible for peroxide-dependent oxidation of dichlorodihydrofluorescein in endothelial cells: Role of transferrin receptor-dependent iron uptake in apoptosis. Circ. Res. 2003, 92, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Salinas, M.; Martin, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-κB. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chae, S.; Lee, J.H.; Hyun, J.W. The cytoprotective effect of butin against oxidative stress is mediated by the up-regulation of manganese superoxide dismutase expression through a PI3K/Akt/Nrf2-dependent pathway. J. Cell. Biochem. 2012, 113, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Shao, A.; Wang, J.; Chen, S.; Wu, H.; McBride, D.W.; Wu, Q.; Sun, X.; Zhang, J. Neuroprotective effect of hydrogen-rich saline against neurologic damage and apoptosis in early brain injury following subarachnoid hemorrhage: Possible role of the Akt/GSK3β signaling pathway. PLoS ONE 2014, 9, e96212. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Yu, F.S.; Chan, P.H. Akt/GSK3β survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke 2006, 37, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Mondal, S.; Tan, D.W.; Nagata, E.; Takizawa, S.; Sharma, A.K.; Hou, Q.M.; Shanmugasundaram, K.; Prasad, A.; Tung, J.K.; et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 10581–10586. [Google Scholar] [CrossRef] [PubMed]
- Davila, D.; Fernandez, S.; Torres-Aleman, I. Astrocyte resilience to oxidative stress induced by insulin like growth factor I (IGF-I) involves preserved AKT (protein kinase B) activity. J. Biol. Chem. 2015, 291, 2510–2523. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C. Iron promoters of the fenton reaction and lipid-peroxidation can be released from hemoglobin by peroxides. FEBS Lett. 1986, 201, 291–295. [Google Scholar] [CrossRef]
- Ayer, R.E.; Zhang, J.H. Oxidative stress in subarachnoid haemorrhage: Significance in acute brain injury and vasospasm. Acta Neurochir. Suppl. 2008, 104, 33–41. [Google Scholar] [PubMed]
- Jonker, F.A.M.; van Hensbroek, M.B. Anaemia, iron deficiency and susceptibility to infections. J. Infect. 2014, 69, S23–S27. [Google Scholar] [CrossRef] [PubMed]
- Szalay, F. Hemochromatosis: One form of iron-overload diseases. Orv. Hetil. 2013, 154, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Echigo, R.; Shimohata, N.; Karatsu, K.; Yano, F.; Kayasuga-Kariya, Y.; Fujisawa, A.; Ohto, T.; Kita, Y.; Nakamura, M.; Suzuki, S.; et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J. Transl. Med. 2012, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Dancis, A. Life without Fe-S clusters. Mol. Microbiol. 2015, 99, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Muhlenhoff, U.; Richhardt, N.; Ristow, M.; Kispal, G.; Lill, R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 2002, 11, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Ai, J.; Sabri, M.; Tariq, A.; Macdonald, R.L. Learning deficits after experimental subarachnoid hemorrhage in rats. Neuroscience 2010, 169, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yan, H.; Li, H.; Hao, S.; Zhuang, Z.; Liu, M.; Sun, Q.; Yang, Y.; Zhou, M.; Li, K.; et al. TGFB-activated kinase 1 (Tak1) inhibition by 5Z-7-oxozeaenol attenuates early brain injury after experimental subarachnoid hemorrhage. J. Biol. Chem. 2015, 290, 19900–19909. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.S.; Kang, B.H.; Krzeminski, J.; Amin, S.; Aliaga, C.; Zhu, J.J.; McDevitt, E.I.; Kocher, S.; Richie, J.P.; Isom, H.C. 3,5,5-Trimethyl-hexanoyl-ferrocene diet protects mice from moderate transient acetaminophen-induced hepatotoxicity. Toxicol. Sci. 2011, 124, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Riemer, J.; Hoepken, H.H.; Czerwinska, H.; Robinson, S.R.; Dringen, R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004, 331, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Xu, F.; Li, K. Production and application of polyclonal antibody against mouse frataxin. Chin. J. Biotechnol. 2013, 29, 1313–1322. [Google Scholar]
- Sample Availability: Samples of the compound SC79 are not available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, S.; Song, C.; Shang, L.; Yu, J.; Qiao, T.; Li, K. Phosphorylation of Akt by SC79 Prevents Iron Accumulation and Ameliorates Early Brain Injury in a Model of Experimental Subarachnoid Hemorrhage. Molecules 2016, 21, 325. https://doi.org/10.3390/molecules21030325
Hao S, Song C, Shang L, Yu J, Qiao T, Li K. Phosphorylation of Akt by SC79 Prevents Iron Accumulation and Ameliorates Early Brain Injury in a Model of Experimental Subarachnoid Hemorrhage. Molecules. 2016; 21(3):325. https://doi.org/10.3390/molecules21030325
Chicago/Turabian StyleHao, Shuangying, Chuanhui Song, Longcheng Shang, Jiang Yu, Tong Qiao, and Kuanyu Li. 2016. "Phosphorylation of Akt by SC79 Prevents Iron Accumulation and Ameliorates Early Brain Injury in a Model of Experimental Subarachnoid Hemorrhage" Molecules 21, no. 3: 325. https://doi.org/10.3390/molecules21030325