1. Introduction
Sophora japonica L. is used in Traditional Chinese Materia Medica (TCMM), and was first officially listed in the Chinese Pharmacopoeia in 1963 [
1,
2,
3,
4,
5,
6,
7,
8,
9]. The dried flower bud of
S. japonica L. is generally called Huaimi (Flos Sophorae Immaturus, FSI) in China. In the south of China more than 13,000 hectares of this species have been artificially planted. Mainly cultivated in Chongqing City, and Quanzhou County, Guilin City, Guangxi Autonomous Region in China, and twice in 2009 and 2015 successively through the examination and approval of varieties by the forestry administration committee Chongqing city, China. The main constituents of
S. japonica L. include rutin, quercetin, genistein, kaempferol, and isorhamnetin, among others [
10,
11,
12,
13,
14,
15]. FSI is used as a hemostatic agent to treat hemorrhoids and hematemesis [
15,
16,
17,
18].
Extraction of herbs for TCMM is a good technique for process engineers in production development and for product quality evaluation in pharmaceutical industry. However, simultaneous extraction of the five aforementioned contents from FSI and Sophora Flower (SF) has not been reported. Various novel techniques have recently been developed to extract one, two, or three constituents from FSI and SF [
8,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61]. These methods include decoction [
49], percolation [
22,
28], reflux [
25,
44,
46,
53], Soxhlet [
27,
44,
60], ultrasonic-assisted extraction [
8,
20,
21,
23,
25,
26,
29,
30,
38,
42,
50,
51,
52,
56,
57,
58,
59,
61], microwave-assisted extraction (MAE) [
24,
25,
32,
33,
34,
35,
36,
45,
47,
55], infrared-assisted extraction [
43], supercritical fluid CO
2 extraction [
41], basic method [
39,
48], and enzymatic method [
37] (
Table 1). However, despite their advantages, these extraction methods require long extraction times [
8,
19,
20,
21,
22,
23,
25,
30,
37,
39,
40,
41,
44,
46,
48,
50,
51,
52,
53,
55,
56,
57,
58], have low efficiency [
1,
19,
20,
22,
25,
26,
28,
39,
41,
44,
45,
47,
50,
60], and/or are expensive [
8,
21,
22,
23,
27,
37,
58]. Thus, developing a reliable, economical, efficient, and ecologically sensitive technique for extracting the constituents of FSI is necessary.
MAE is widely used to extract active constituents from various kinds of plant materials because of its enhanced extraction efficiency compared with other traditional extraction methods [
62,
63,
64,
65,
66]. The MAE system rapidly generates heat and can facilitate the penetration of solvent into raw plant material and intracellular material to improve constituent transfer, reducing extraction time and improving extraction rate [
62,
63]. The efficiency of MAE depends on extraction time, liquid-to-solid ratio, extraction power, and type of extraction solvent [
62,
63,
64,
67]. In this study, we investigated the MAE of major constituents from FSI and SF. Response surface methodology (RSM) was used to improve the extraction yield of constituents by systematically analyzing the effects of extraction parameters on yields. RSM is a collection of statistical, as well as mathematical, techniques and effective for responses that are influenced by various factors and their interactions [
68,
69].
The active compounds of herbs vary according to several factors, including variety, geographic area, nutritional status, harvest time, manufacturing process, and even storage method [
70]. Variations in these factors could result in significant differences in pharmacological activity. Accurate, analytically obtained qualitative and quantitative data are used to evaluate the efficacy and safety of TCMM [
71,
72,
73,
74,
75,
76,
77,
78,
79]. Therefore, developing a reliable and accurate quality control method for TCMM is necessary. Previous, analytical methods developed for determining the quality of a few components in FSI include HPLC-UV [
20,
80,
81,
82], HPLC-DAD [
21,
23,
59], HPLC-DAD-ESI-MS/MS [
83], and capillary electrophoresis [
17,
84,
85]. While these reported methods contribute significantly to the current knowledge of FSI compounds, several drawbacks, such as identification of too few constituents, long analysis times, and high solvent consumption, limit their practical application. Thus, a new analytical method must be developed to qualitatively and quantitatively determine multiple active constituents in FSI.
Ultra-high performance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry (UHPLC-ESI-Q-TOF MS/MS) is a powerful approach that enables simultaneous determination of multiple components [
86,
87,
88,
89,
90,
91,
92]. The chemical structures of FSI constituents could readily be defined by using fragmentation rules, characteristic fragmentation and quasi-molecular ions summarized in the literature, and comparison of retention times and parent and product ions with those of standards.
This study aimed to investigate the significant variables (methanol and ethanol concentrations, particle size, extraction frequency, liquid-to-solid ratio, microwave power, and extraction time) prior to RSM to optimize variables for FSI extraction and developed a simple and accurate UHPLC and LC-ESI-Q-TOF MS/MS method for simultaneously determining five components of FSI.
2. Results and Discussion
2.1 Analysis of Single Factor Test Results
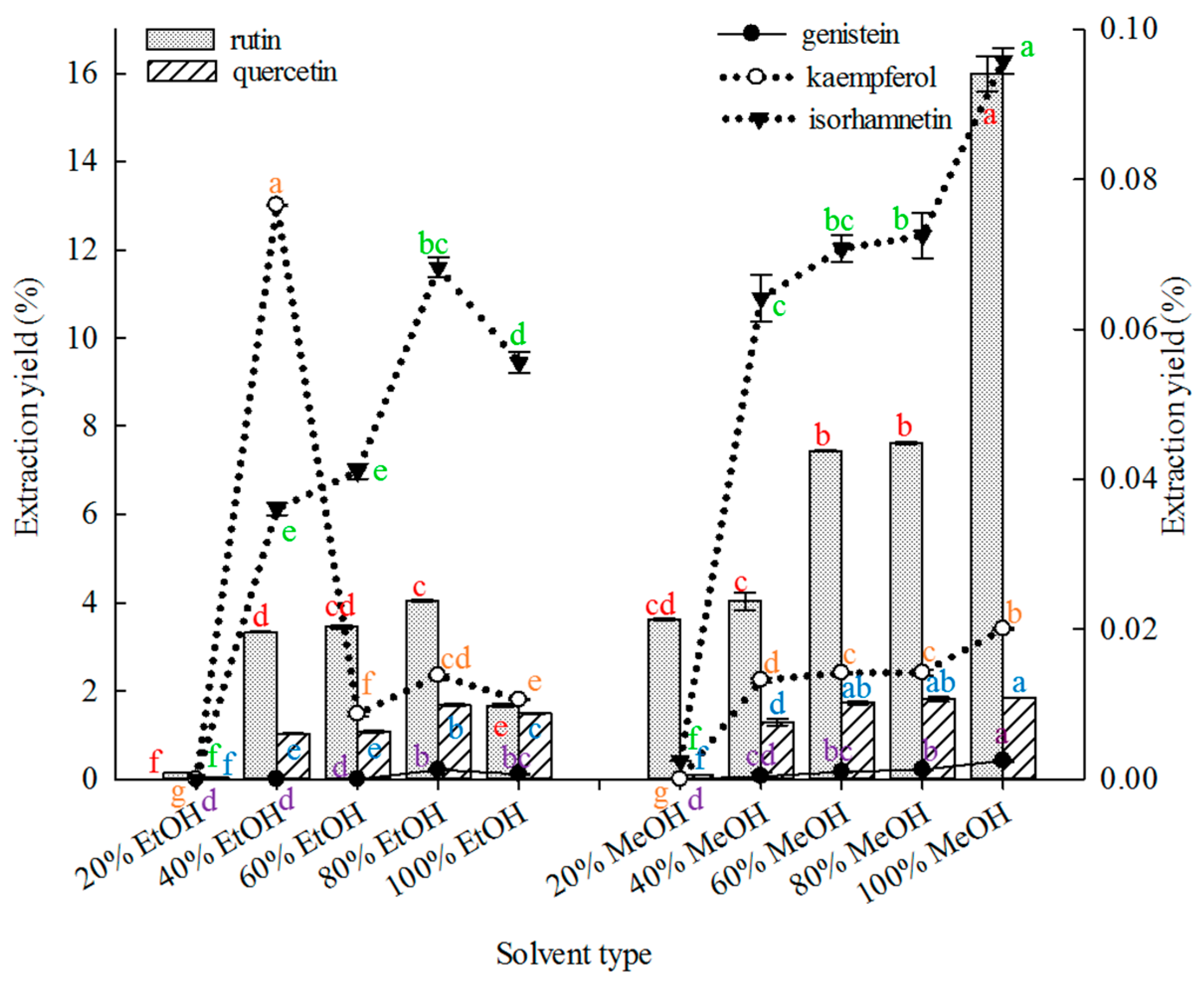
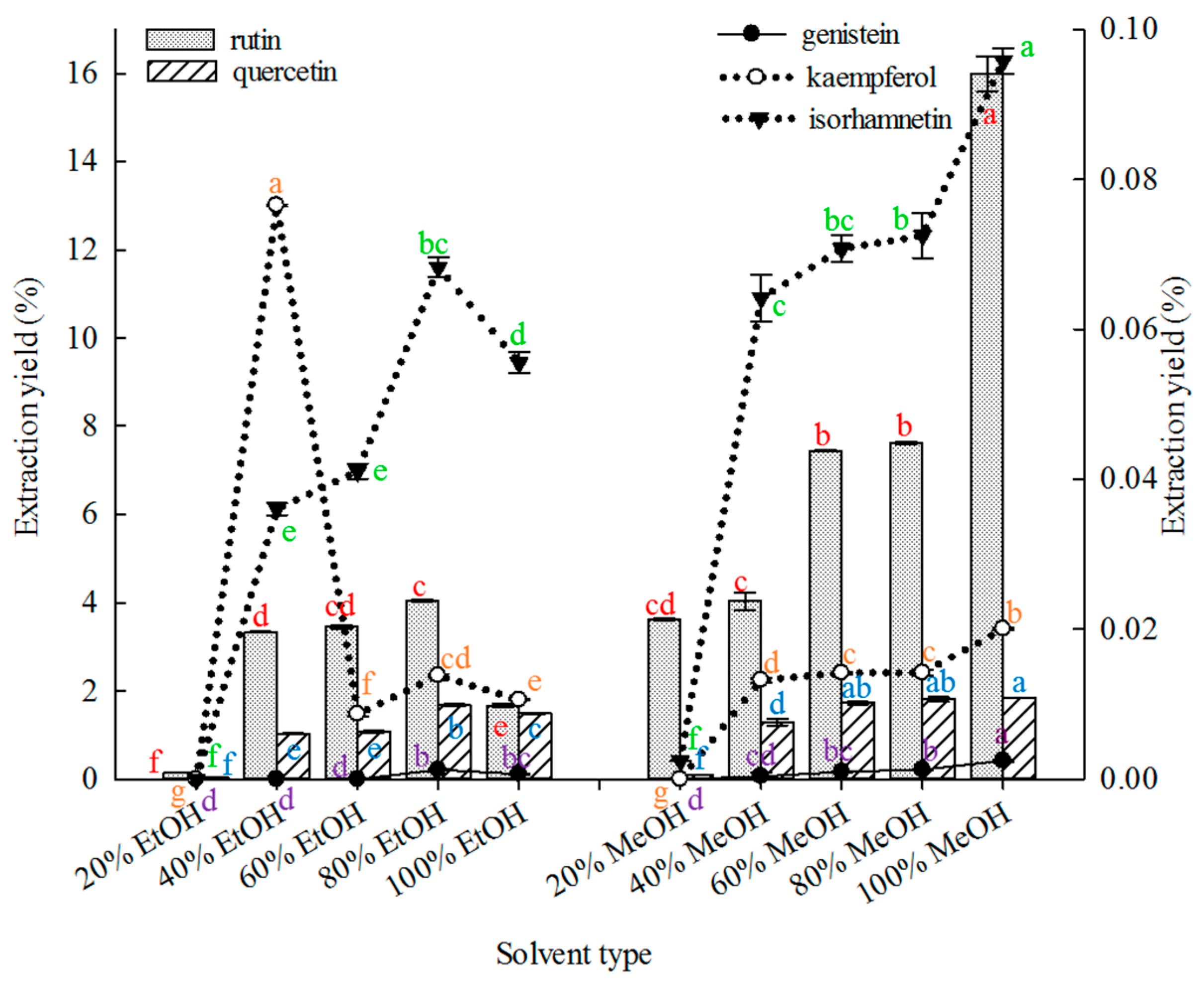
2.1.1. Effect of Solvent Type on Extraction
Solvent selection is important in the extraction of compounds from botanical materials [
69].
Figure 1 shows that under similar extraction conditions. 100% MeOH was superior to other solvents in extracting rutin, genistein and isorhamnetin from FSI. Both 80% MeOH and 100% MeOH were superior to the other solvents in extracting quercetin, but without significant differences in their extraction yields (
p ≤ 0.01). The 40% EtOH solvent was superior to the other solvents in extracting kaempferol.
Thus, 100% methanol was selected as the extraction solvent to extract the five major constituents using MAE in subsequent experiments. Previous studies reported the use of different ethanol and methanol concentrations mainly for extracting rutin or quercetin from SFI [
19,
20,
21,
22,
23,
25,
26,
27,
28,
32,
34,
35,
40,
43,
45,
46,
47,
58,
59,
60]. Water [
31,
33,
36,
42], alkaline solution [
29,
30,
38,
48], cellulase [
37], ether [
41], and sodium hydroxide [
49] have also been reported as extraction solvents for extracting ingredients from SFI. However, ether and alkaline solutions are toxic, water cannot dissolve flavonoid components, and cellulase is expensive. Although the methanol and ethanol concentrations used were somewhat variable, methanol was selected as the best extraction solvent among the extraction solvents listed in
Section 3.2.1.
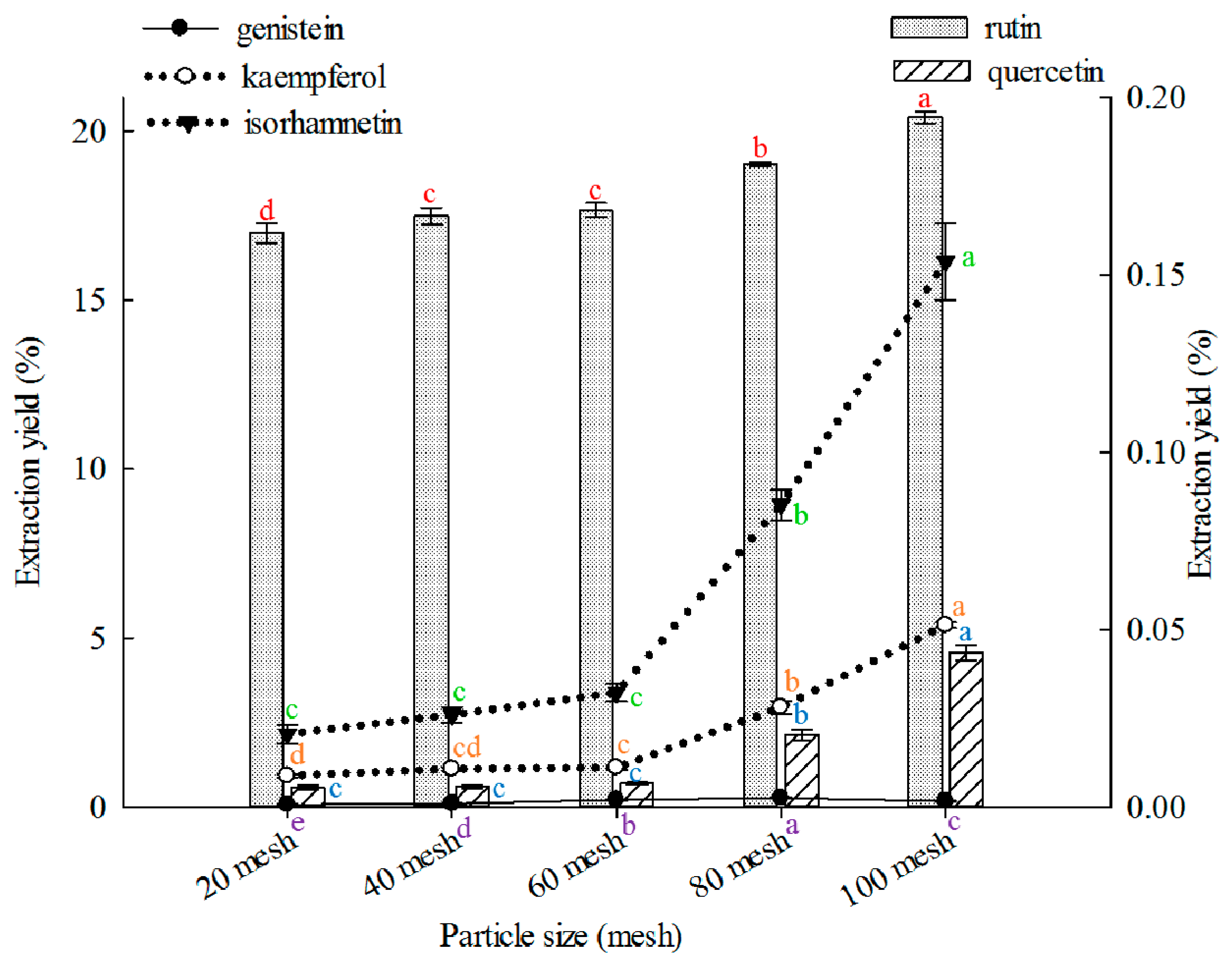
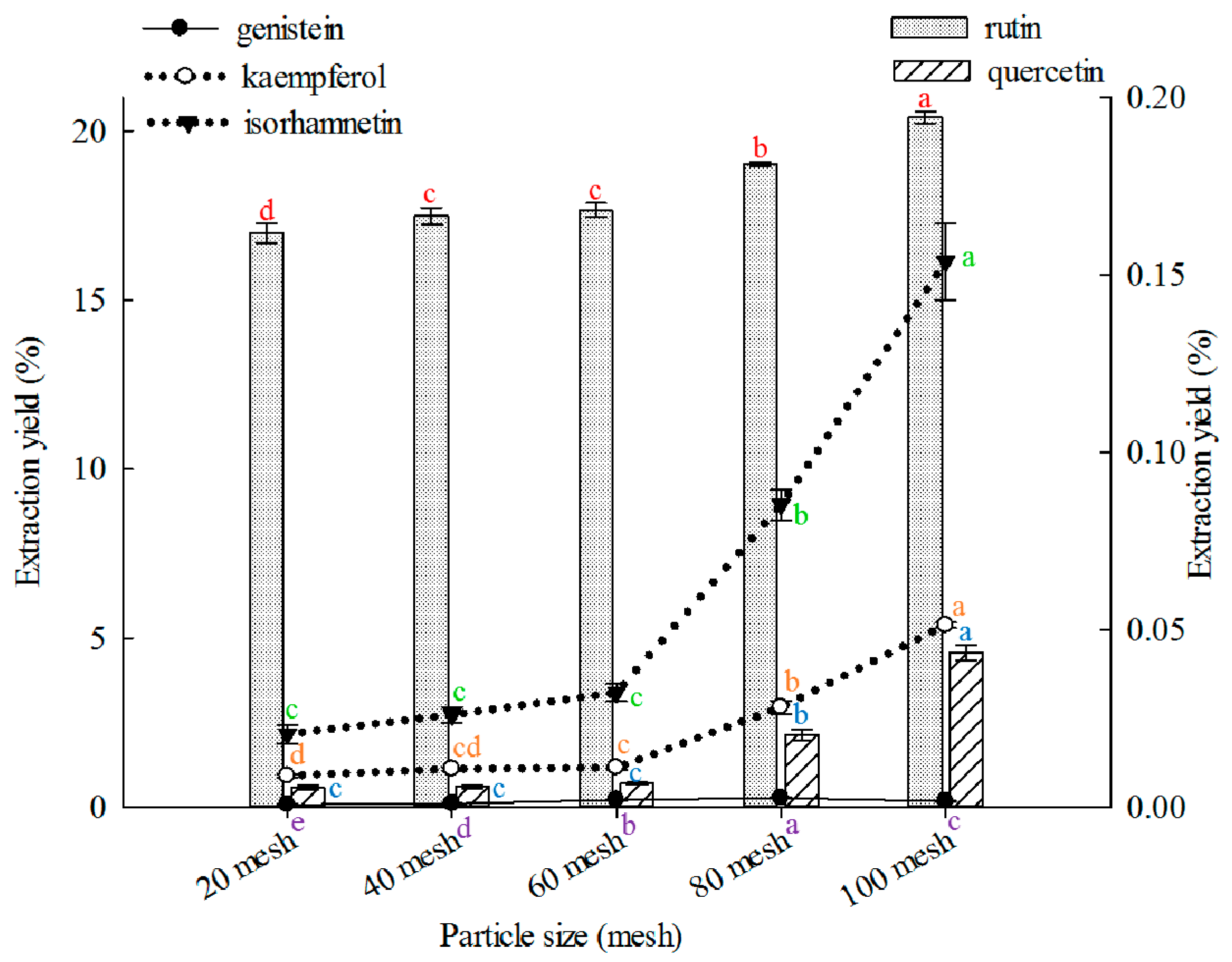
2.1.2. Effect of Particle Size on Extraction
The appropriate particle size is fundamental to obtain optimal extraction, and varied particle sizes can significantly affect extraction yields [
69]. In this study, 100 mesh showed better results than other particle sizes in extracting rutin, quercetin, kaempferol, and isorhamnetin (
Figure 2), and 80 mesh was better than other particle sizes in extracting genistein. Effects of particle size on extraction of rutin, quercetin, genistein, kaempferol, or isorhamnetin from FSI are rarely reported.
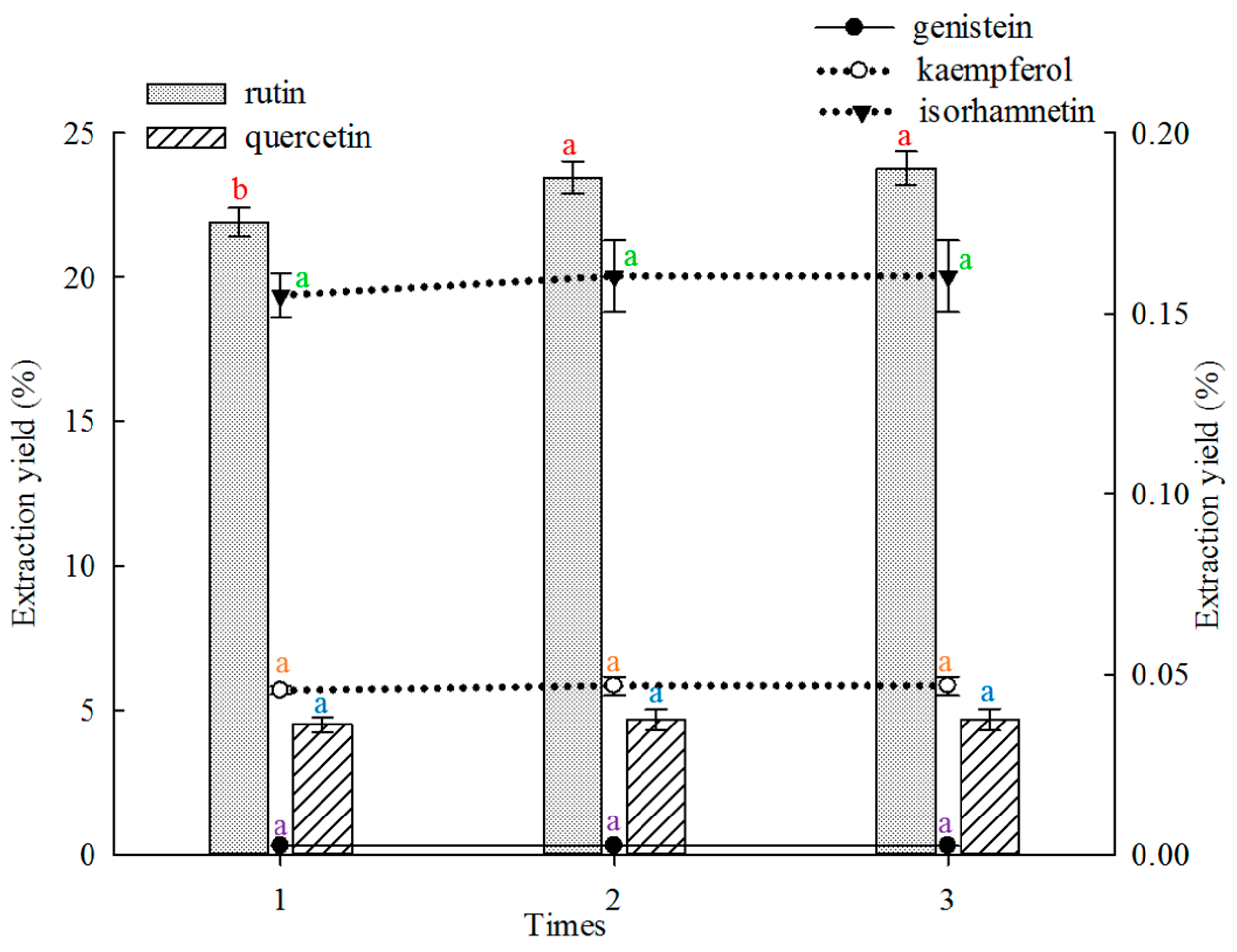
2.1.3. Effect of Frequency on Extraction
The effect of extraction frequency on the extraction yield of the five main constituents from FSI was investigated, and the results are shown in
Figure 3. In this study, twice and thrice extraction showed better results in extracting rutin than single extraction. The twice extraction and thrice extraction did not differ significantly. The extraction yields of quercetin, genistein, kaempferol and isorhamnetin did not differ significantly when the extraction was performed once to thrice.
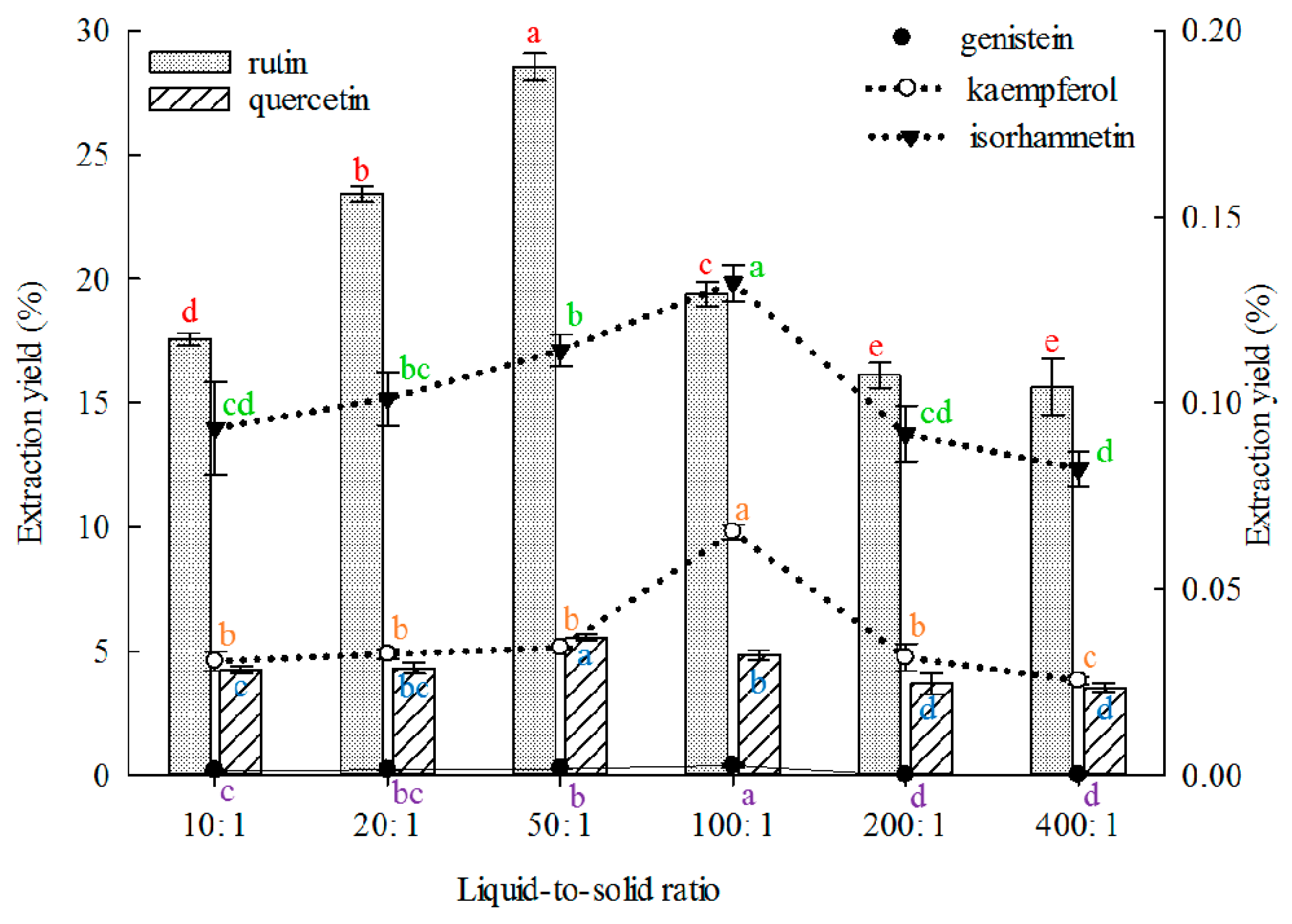
2.1.4. Effect of Liquid-to-Solid Ratio on Extraction
The solvent volume must be sufficient to ensure complete immersion of materials for efficient extraction. Extraction solvent deficiency can lead to incomplete extraction of ingredients, but redundant solvent may also lead to lower extraction yields and solvent waste [
69]. Therefore, the liquid-to-solid ratio must be appropriate. The effect of liquid-to-solid ratio on the extraction yield of the five constituents was investigated, and the results are shown in
Figure 4. The 50:1 (
v/
m) proportion showed better results than other liquid-to-solid ratios in extracting rutin and quercetin. The 100:1 proportions were better than other liquid-to-solid ratios in extracting genistein, kaempferol and isorhamnetin. The maximum and minimum liquid-to-solid ratios of 500:1 and 4:1, respectively, have been reported for the extraction of rutin or quercetin from FSI, but with low extraction yields [
21,
47].
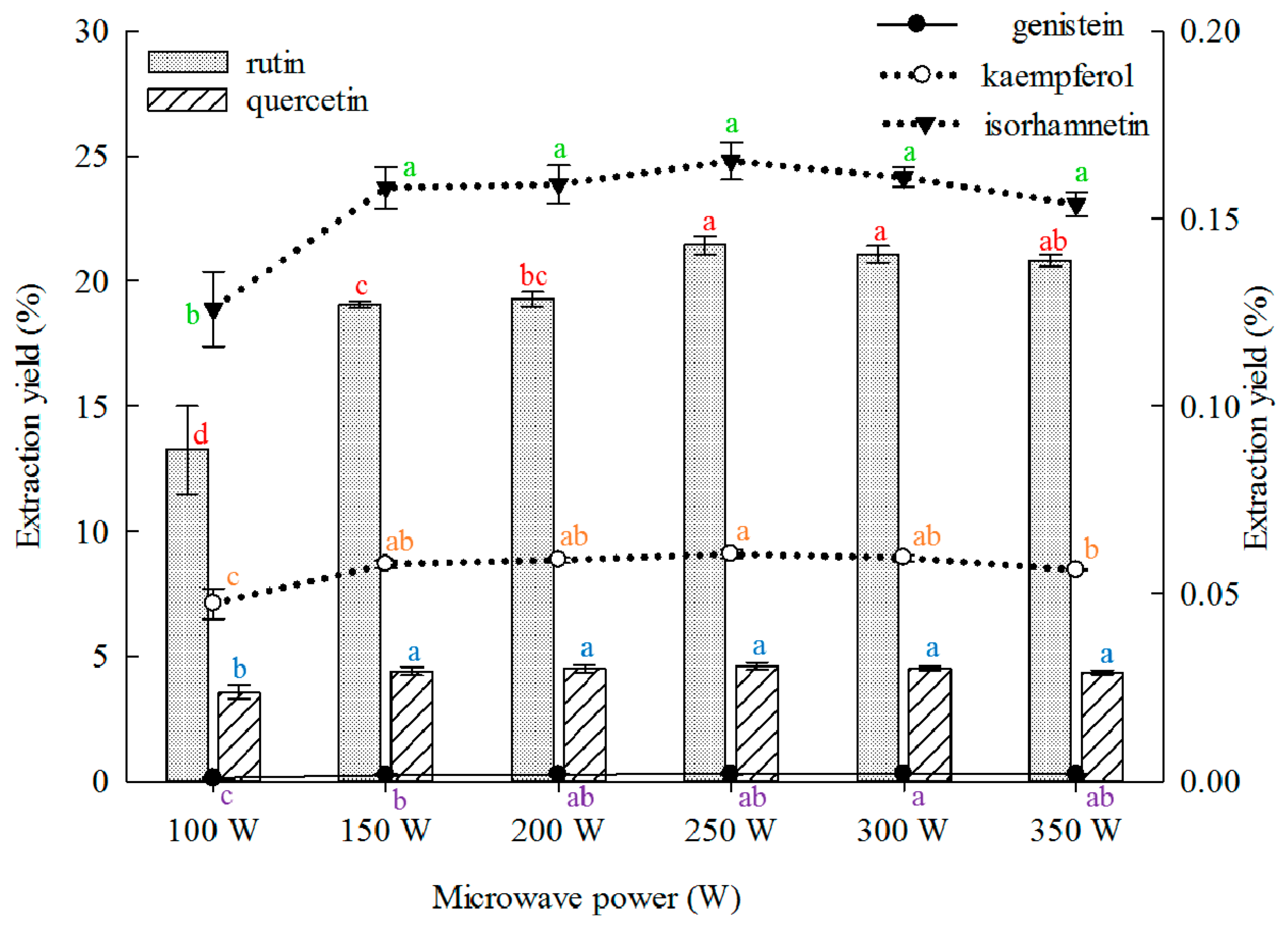
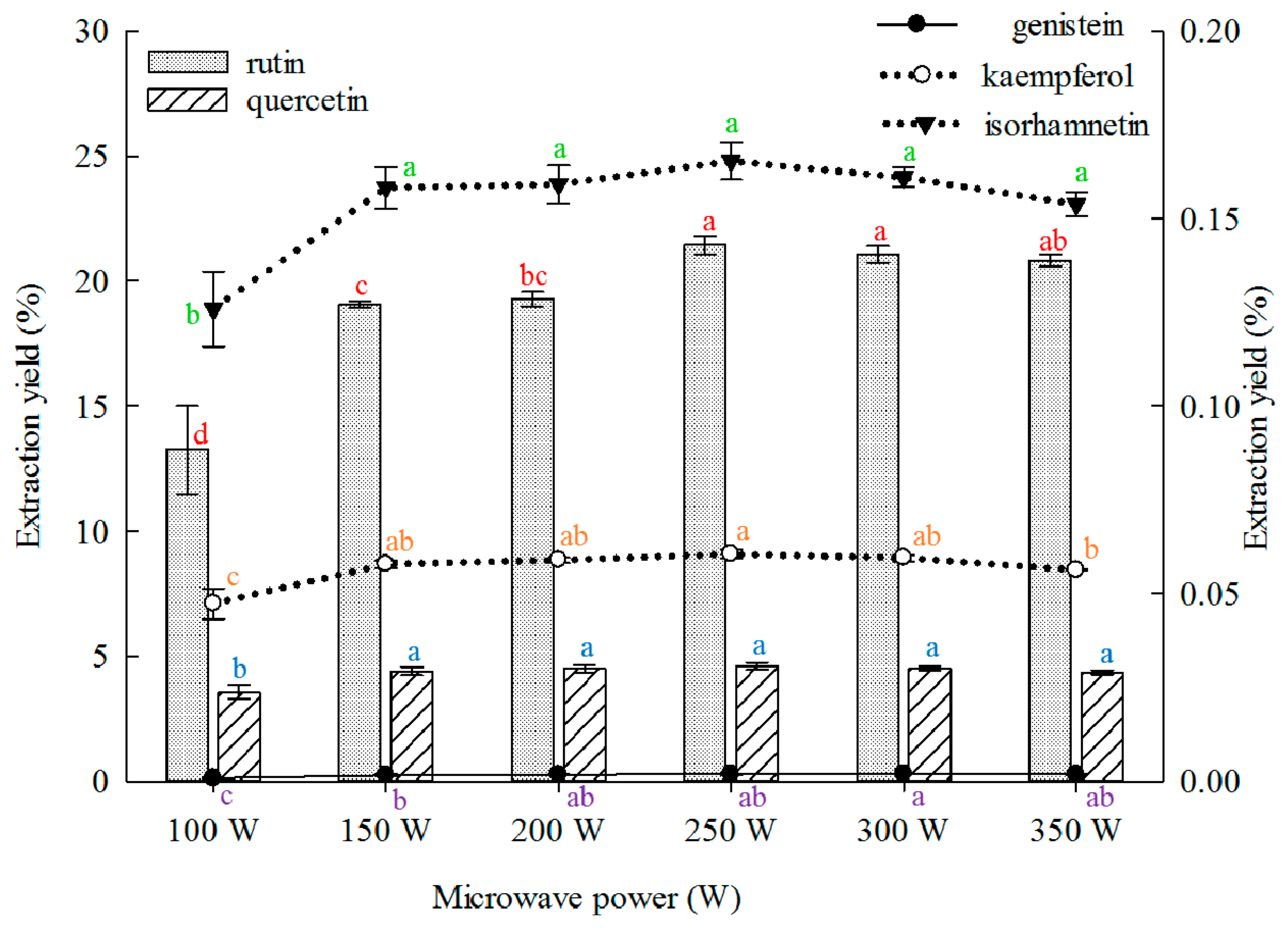
2.1.5. Effect of Microwave Power on Extraction
Low microwave power reduces extraction yield. However, excessively high power results in energy wastage. Therefore, the optimal microwave power should be determined. The effect of microwave power on the extraction yield of the five main constituents from the FSI was investigated, and the results are shown in
Figure 5. The extraction yields of rutin, quercetin, kaempferol, and isorhamnetin from the FSI evidently increased with increasing microwave power, but no increase was observed above 250 W. The extraction yield of genistein continued to increase with the microwave power and peaked when the power was 300 W. Subsequently, a reduction in yield was observed. Previous study on the effects of microwave power for rutin and quercetin extraction from FSI are available, but microwave power was high (350–480 W) and the extraction yield was low (rutin, 14.66%–21.97%; quercetin, 0.46%–0.61%) [
31,
36,
45,
47].
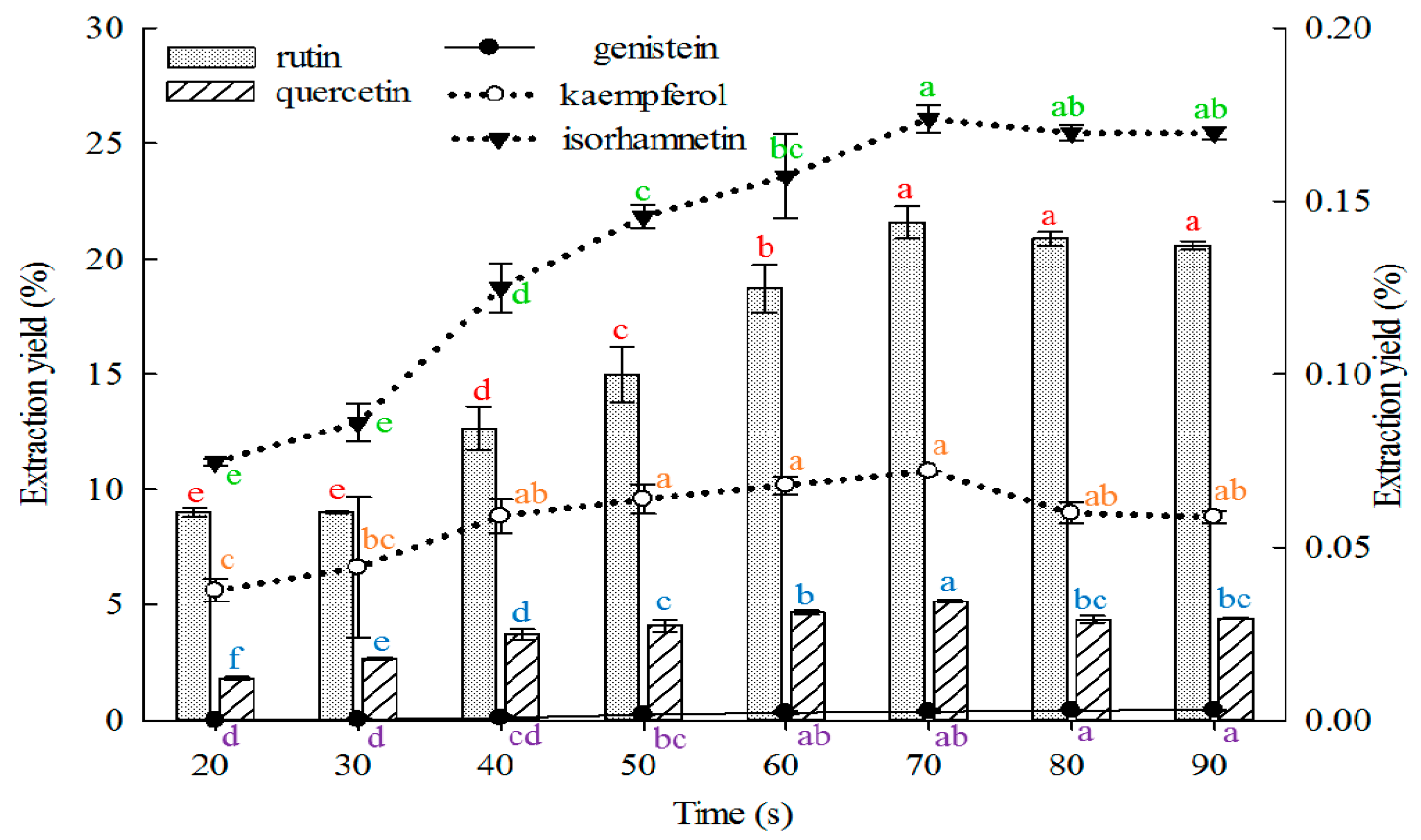
2.1.6. Effect of Time on Extraction
Longer time indicates greater contact between the solvent and sample contact. This phenomenon may accelerate the absorption of solvent, soften the plant tissues, and weaken the cell wall integrity. Moreover, ingredient solubility may be enhanced, thus larger amounts of substances are distributed to the solvent. However, excessive extraction time may lead to lower process efficiency and wasted time. Therefore, the extraction time must be appropriate. The effects of extraction time on the extraction yields of the five constituents from FSI are shown in
Figure 6.
The extraction yields of rutin, quercetin, kaempferol, and isorhamnetin increased as the extraction time was prolonged from 20 s to 70 s and peaked at 70 s. However, extraction yields of genistein increased as the extraction time was increased from 20 s to 80 s and peaked at 80 and 90 s, but without a statistically significant difference. Thus, extraction time is shorter than those reported in previous reports [
8,
19,
20,
21,
22,
23,
25,
30,
37,
39,
40,
41,
44,
46,
48,
50,
51,
52,
53,
55,
56,
57,
58].
2.2. Model Fitting of Parameters Based on the Extraction Yields of the Five Constituents
The responses of the five extracts in each run are presented in
Table 2. The regression coefficients and results from ANOVA of the second-order polynomial models (Y = A
0 + A
1X
1 + A
2X
2 + A
3X
3 + A
11X
12 + A
22X
22 + A
33X
32 + A
12X
1X
2 + A
13X
1X
3 + A
23X
2X
3) for five extracts are summarized in
Table 3. Regression parameters of the surface response analysis of the linear and quadratic models, and their corresponding interaction terms showed significant differences (
p ≤ 0.0001,
p ≤ 0.01 or
p ≤ 0.05). The fitness of the model was evaluated through the lack of fit test (
p < 0.05), which indicates the adequacy of the model to predict the variation accurately [
93]. The models were used to construct three-dimensional response surface plots to predict the relationship between independent and dependent variables.
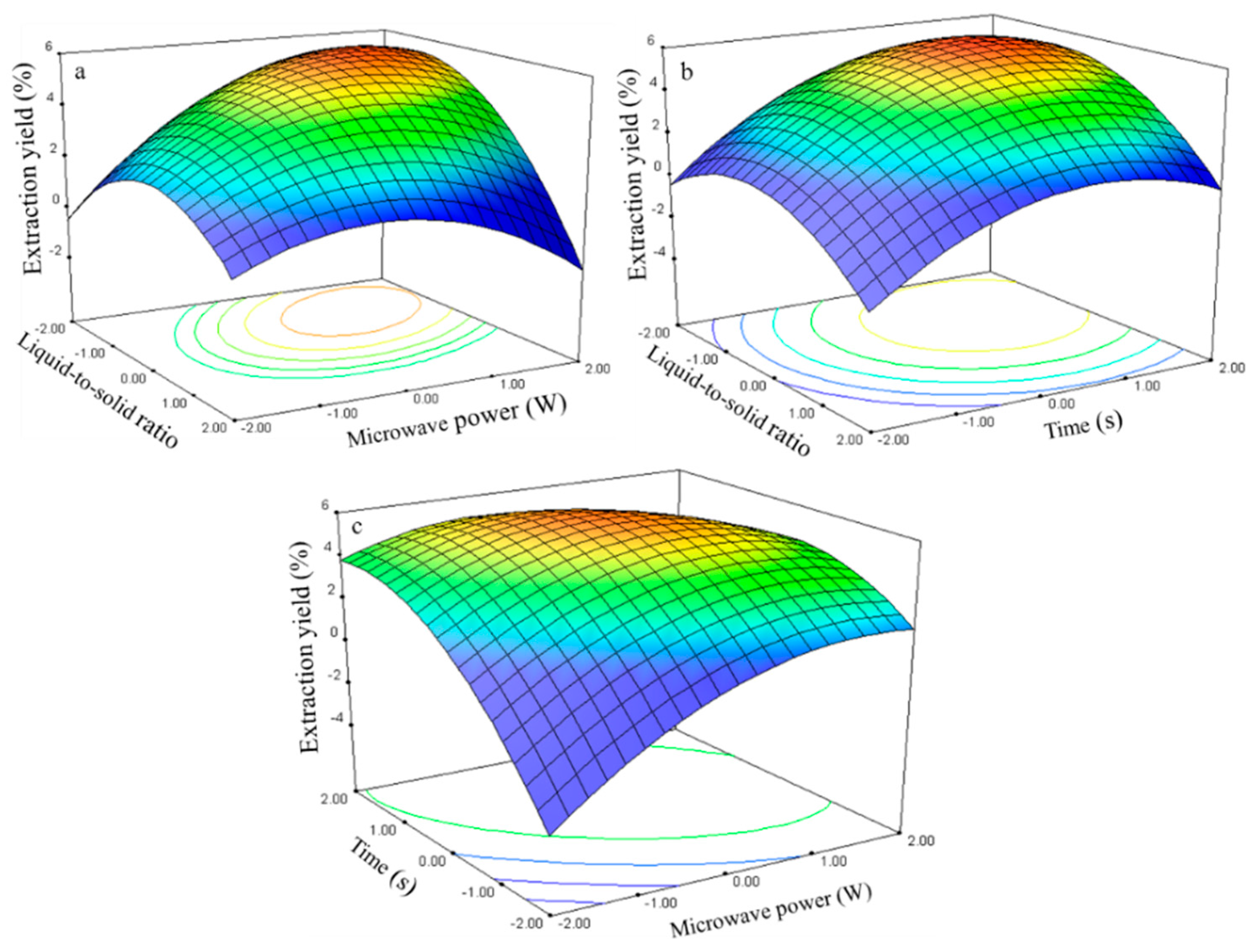
2.2.1. Effect of Process Variables on the Extraction Yield of Rutin
The experimental data were examined through regression analysis, and the coefficients of the model were evaluated for significance. Liquid-to-solid ratio (
X1), microwave power (
X2), and time (
X3) significantly affected the extraction yield of rutin (
Y1,
Table 3), with corresponding contribution rates of 2.88, 2.90, and 2.92. These results indicate that microwave power and extraction time exhibited the greatest effect on the extraction yield of rutin.
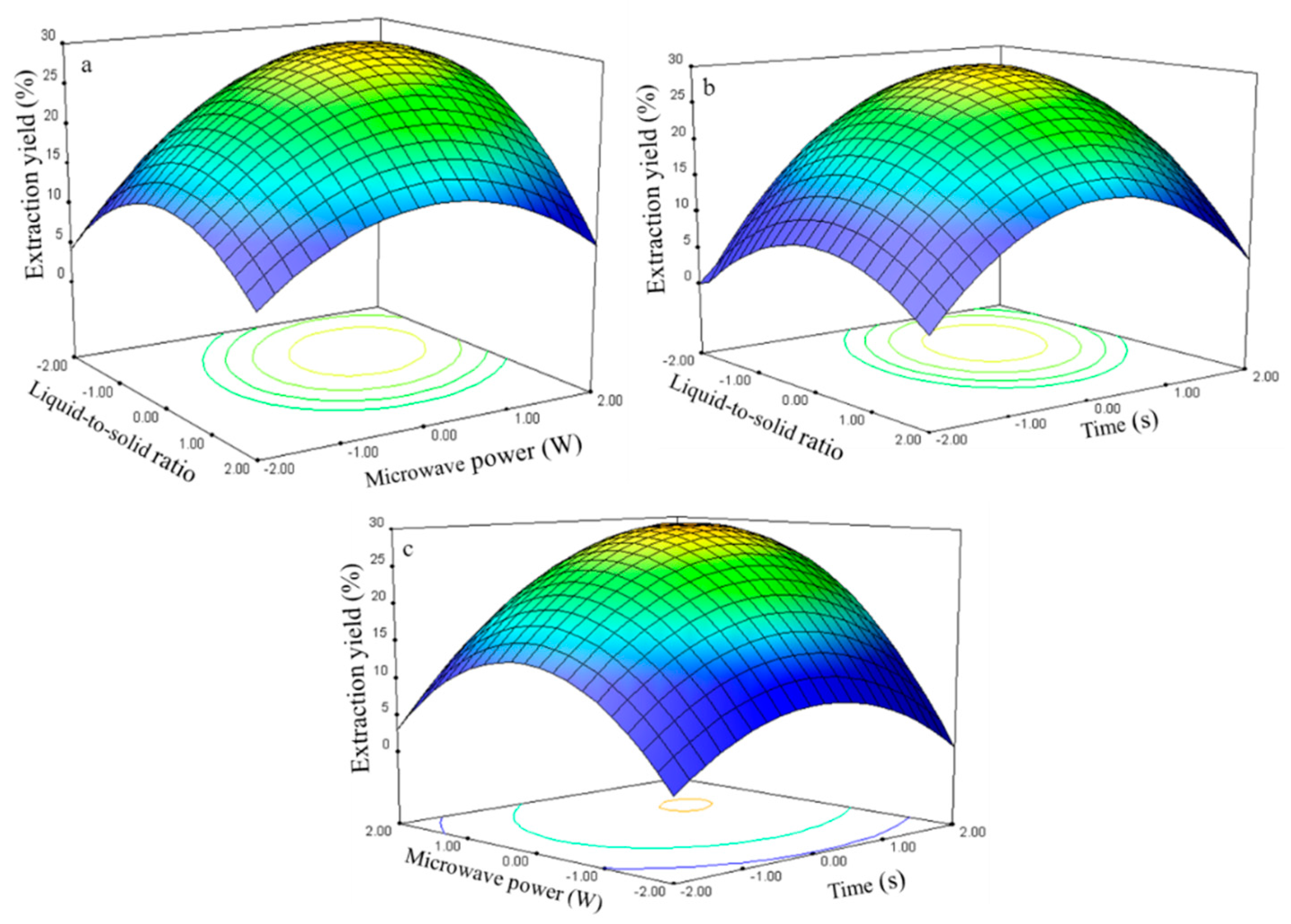
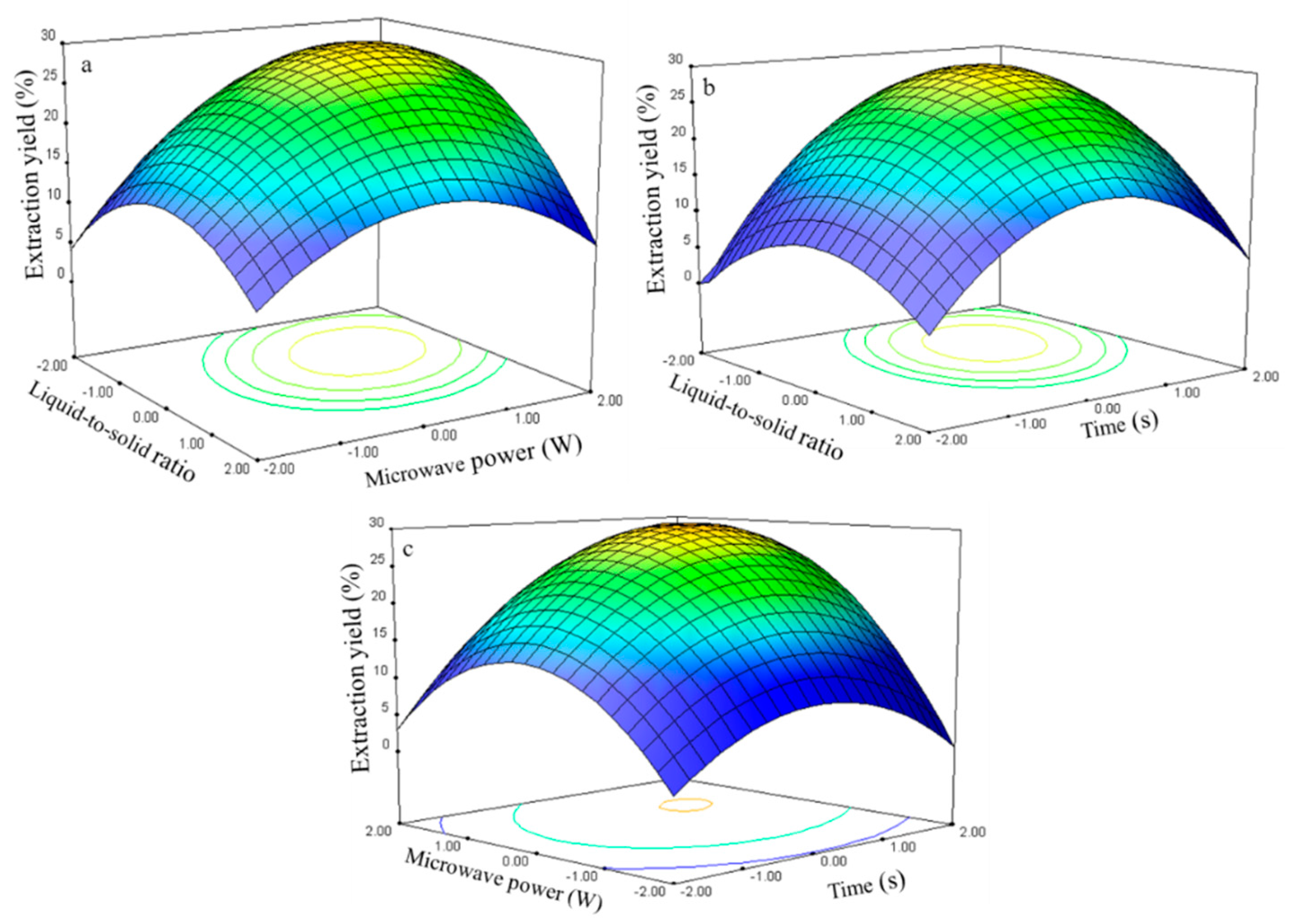
The three-dimensional response surface plots in
Figure 7 illustrate the relationship between the extraction yield of rutin and experimental variables.
Figure 7a illustrates the interaction effect of liquid-to-solid ratio and power on the extraction yield of rutin when the time was set to its 0 level (70 s). Rutin yields gradually increased with liquid-to-solid ratio and microwave power and peaked at approximately 50:1 to 75:1 and 250 W to 300 W. However, extraction yield of rutin began to decrease after further increase in these parameters.
The interaction effects between the liquid-to-solid ratio and the time on the rutin extraction yield when the power was set to its 0 level (250 W) are presented in
Figure 7b. The rutin yield increased and peaked at 50:1 to 100:1 from 75 s to 80 s.
The interaction effects of power and time at 75:1 (0 level) on the rutin extraction yield are presented in
Figure 7c. Strong interaction was observed when the power was set from 275 W to 300 W and the time ranged from 75 s to 80 s, which contributed to the increased extraction yield.
The rutin regression model for the statistical frequency method of analysis with 95% confidence interval was obtained (X1: –1.47 to –0.58, X2: 0.72 to 1.26, and X3: 0.64 to 1.18) when the extraction yield was >26.80% (n = 21). Therefore, the optimal conditions were 46.33–60.60, 286.05–312.85W, and 76.35–81.83 s.
2.2.2. Effect of Process Variables on the Extraction Yield of Quercetin
The quercetin extraction yield results are presented in
Table 2. The regression analysis results indicate that the main extraction parameters of quercetin are the liquid-to-solid ratio (
X1), microwave power (
X2), and time (
X3). The relationships between the quercetin extraction yield (
Y2,
Table 3) and the variables are shown in
Figure 8. The contributions of the liquid-to-solid ratio, microwave power, and extraction time were 2.79, 2.95, and 2.80, respectively. Microwave power had the largest impact on the quercetin extraction yield.
The effect of the liquid-to-solid ratio and microwave power on the quercetin extraction yield at a constant time (0 level) is shown in
Figure 8a. The quercetin extraction yield gradually increased with the ratio and power and peaked at approximately 45–55 and 260–300 W, respectively. The quercetin extraction yield began to decrease beyond 55 and 300 W. The appropriate extraction time (75–80 s) had positive effects on the extraction yield, as shown in the response surface plots for the time effect on the extraction yield (
Figure 8b,c) at constant microwave power and liquid-to-solid ratio.
The quercetin regression model for the statistical frequency method of analysis with 95% confidence interval was obtained (X1: −1.11 to −0.53, X2: 0.23 to 0.95 and X3: 0.51 to 1.10) when the extraction yield was >5% (n = 20). Thus, the optimal conditions were 47.28–61.70, 261.35–297.30 W, and 75.10–80.63 s.
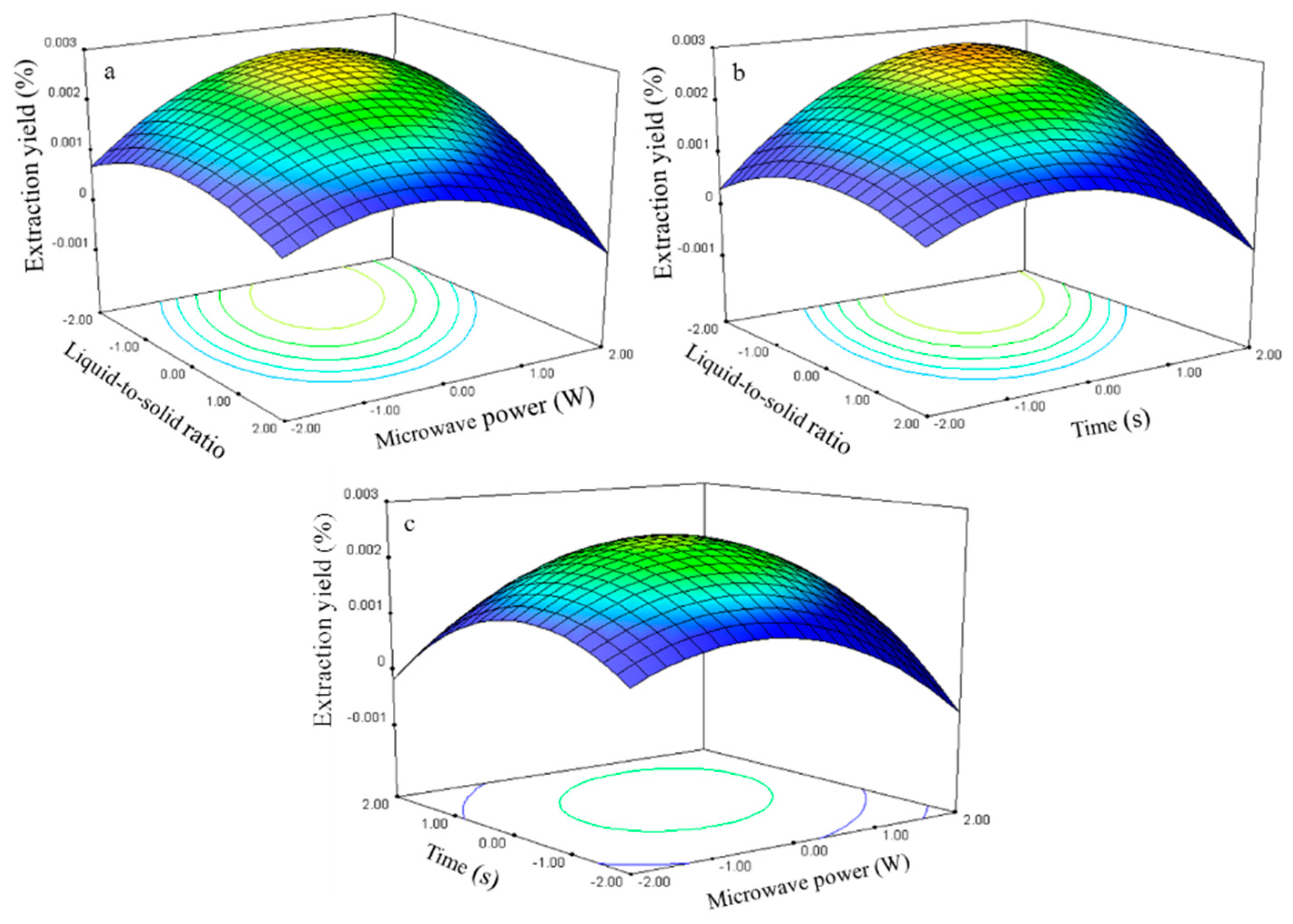
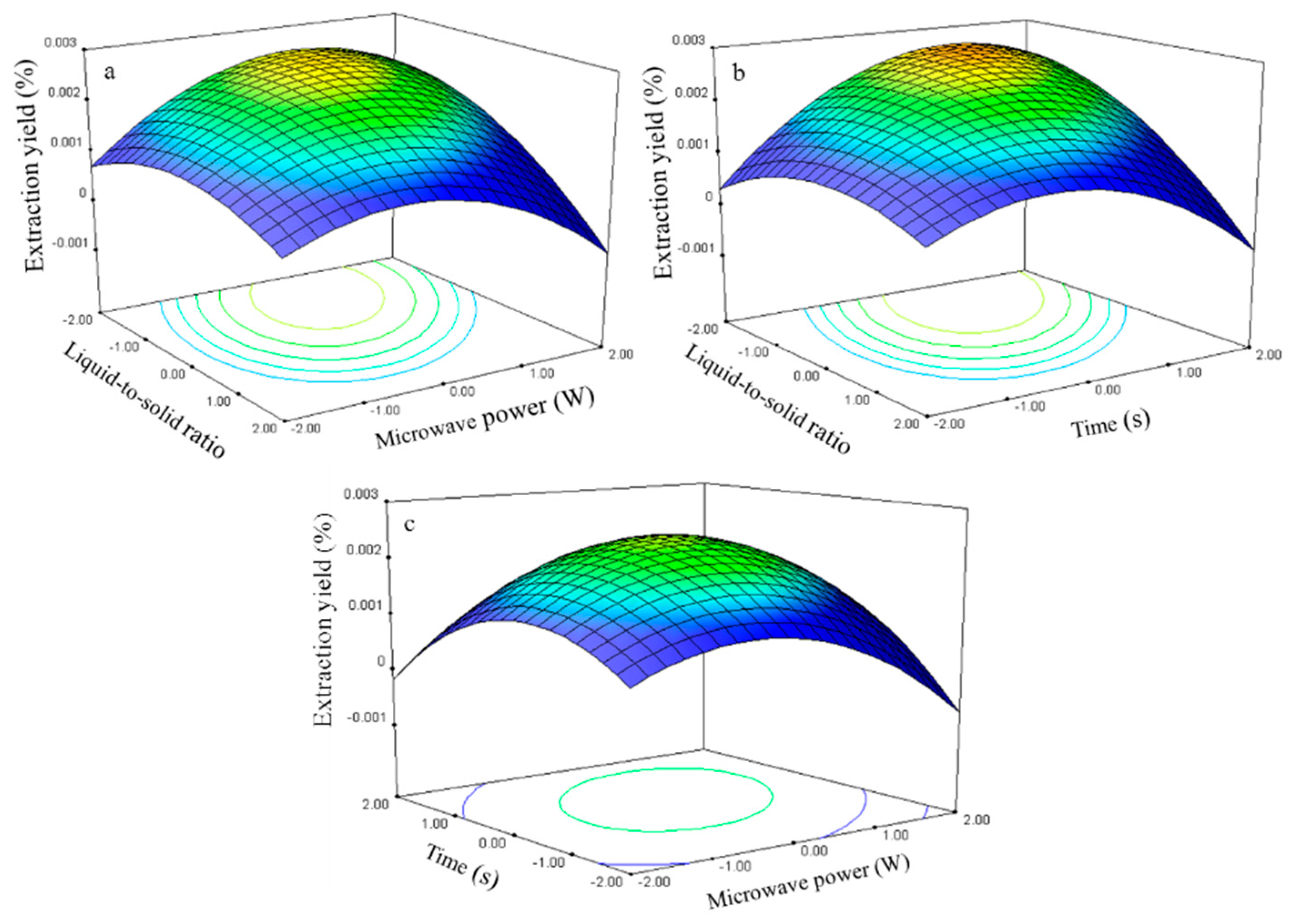
2.2.3. Effect of Process Variables on the Extraction Yield of Genistein
The genistein extraction yield is presented in
Table 2. The regression analysis shows that the extraction yield (
Y3,
Table 3) was significantly affected by the liquid-to-solid ratio (
X1), microwave power (
X2), and time (
X3), with corresponding contribution rates of 2.88, 1.91, and 2.86, respectively. The liquid-to-solid ratio and extraction time exhibited the largest impact on the genistein extraction yield.
The relationship between the genistein extraction yield and the process variables is depicted in
Figure 9. The liquid-to-solid ratio and microwave power effects on the extraction yield at 0 level fixed time are shown in
Figure 9a. The genistein extraction yield gradually increased with the ratio and power and peaked at approximately 50–60 and 270–300 W. Further increases in these parameters resulted in decreased genistein extraction yield.
The response surface plots for the time effect on the extraction yield (
Figure 9b,c) at constant microwave power and liquid-to-solid ratio show that an appropriate extraction time (75–80 s) had positive effects on the extraction yield.
The genistein regression model for the statistical frequency method of analysis with 95% confidence interval was obtained (X1: −1.31 to −0.79, X2: 0.30 to 0.96, and X3: 0.51 to 1.10) when the extraction yield >0.002% (n = 23). Thus, the optimal conditions were 42.33–55.20, 264.75–297.90 W, and 75.07–80.66 s.
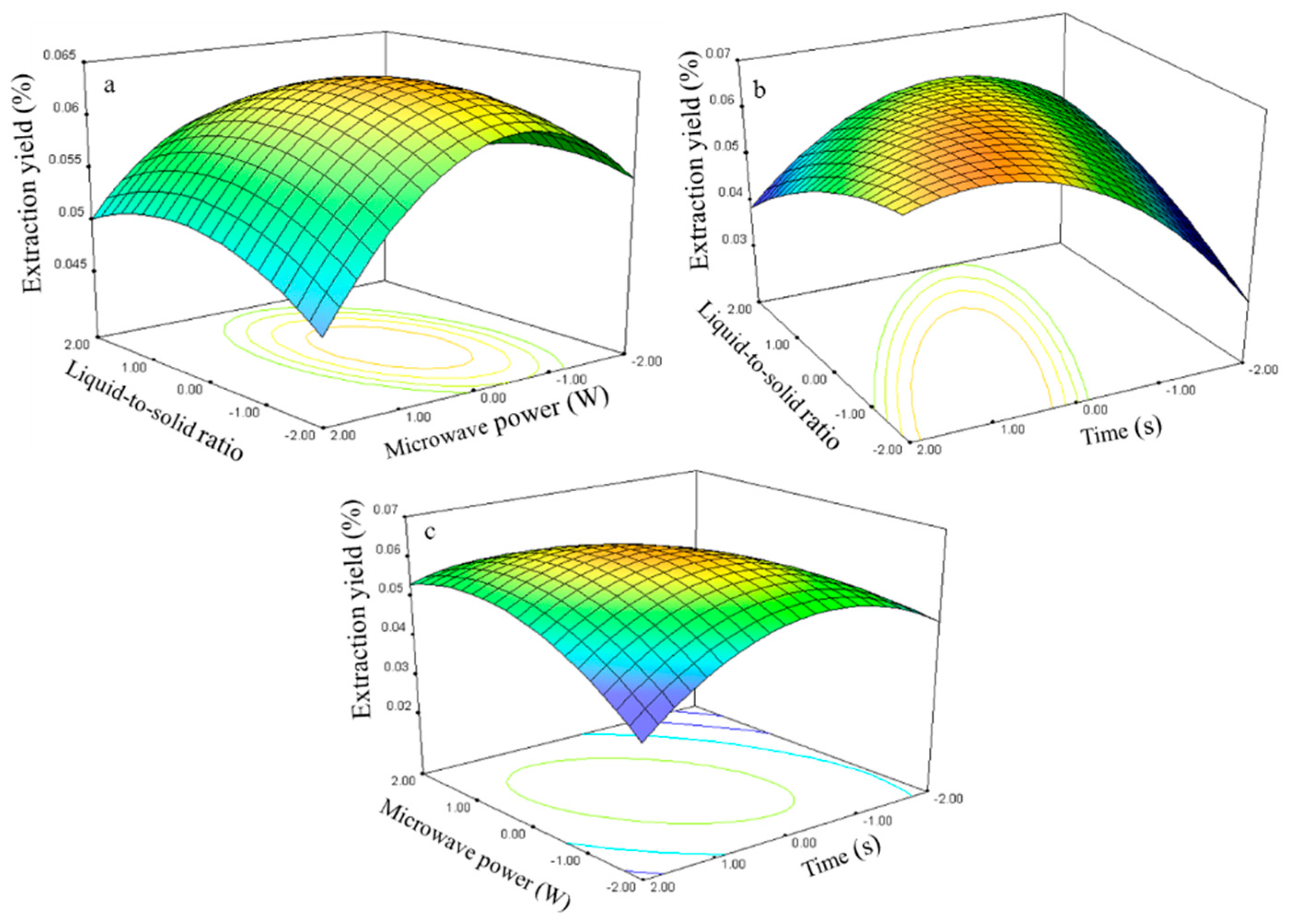
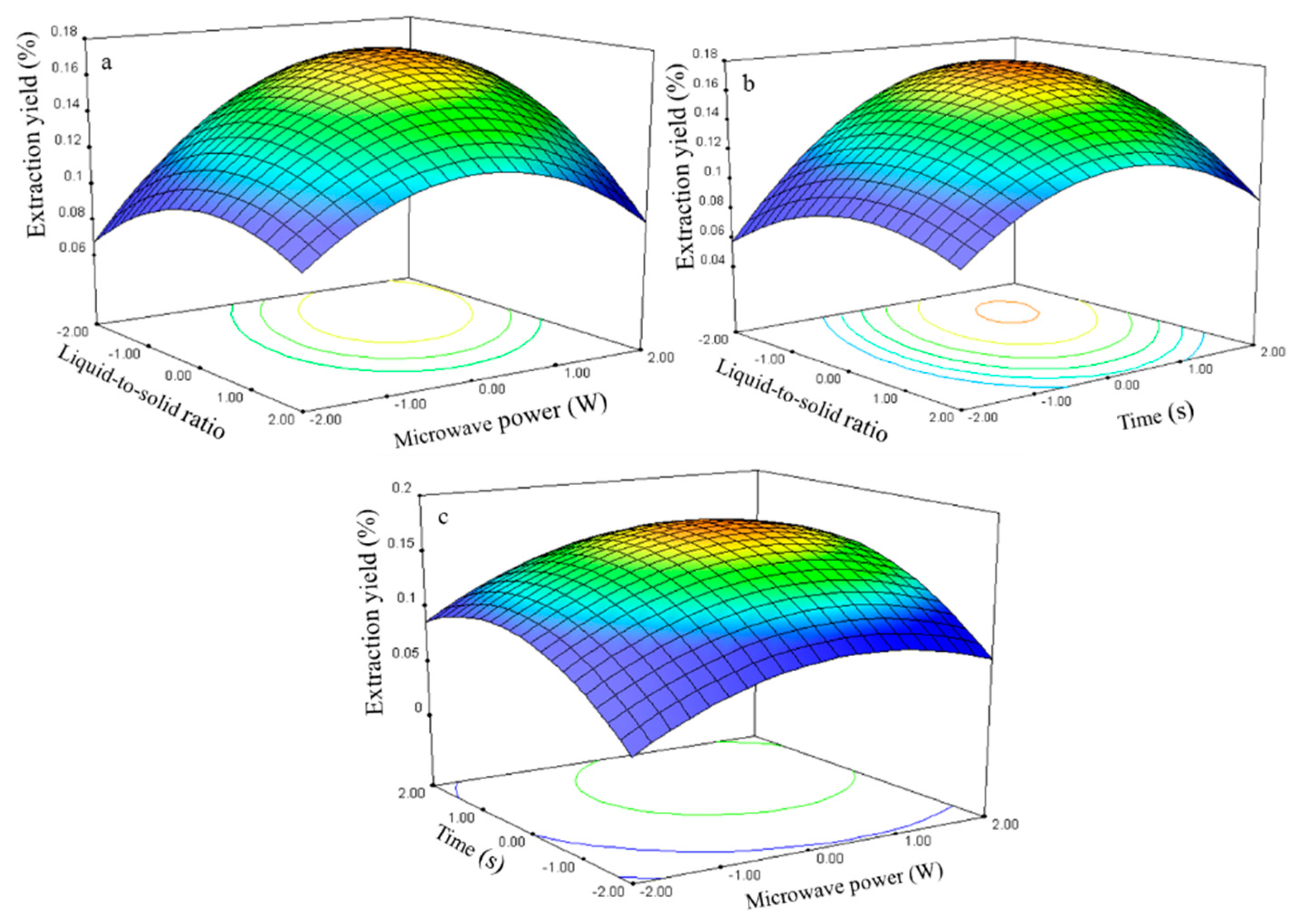
2.2.4. Effect of Process Variables on the Extraction Yield of Kaempferol
The kaempferol extraction yield is presented in
Table 2. The regression analysis shows that the extraction yield (
Y4,
Table 3) was significantly affected by the liquid-to-solid ratio (
X1), microwave power (
X2), and time (
X3), with corresponding contribution rates of 1.06, 1.93, and 2.81. The extraction time had the largest impact on the kaempferol extraction yield.
The relationship between the kaempferol extraction yield and the process variables is depicted in
Figure 10. The ratio and power effects on the extraction yield at 0 level fixed time are shown in
Figure 10a.
The extraction yield gradually increased with the ratio and power and peaked at approximately 50–75 and 250–300 W. Further increases in these parameters resulted in decreased kaempferol extraction yields. The response surface plots for the time effect on the extraction yield (
Figure 10b,c) at constant microwave power and constant liquid-to-solid ratio show that an appropriate extraction time (75–80 s) had positive effects on the extraction yield.
The kaempferol regression model for the statistical frequency method of analysis with 95% confidence interval was obtained (X1: −1.14 to −0.17, X2: 0.11 to 0.78, and X3: 0.44 to 1.12) when the extraction yield was >0.06% (n = 22). Thus, the optimal conditions were 46.5–70.75, 255.45–288.30 W, and 74.35–81.18 s.
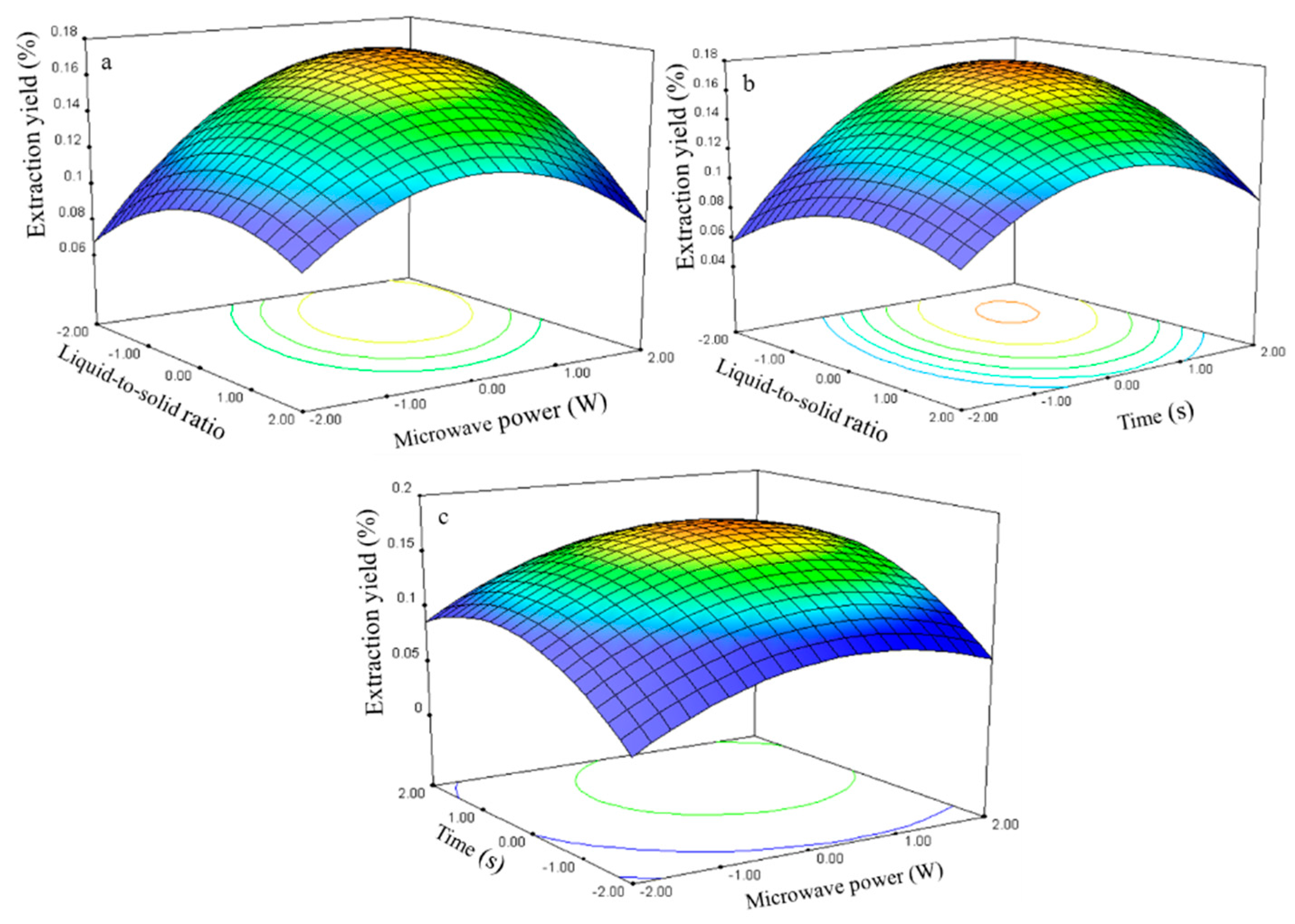
2.2.5. Effect of Process Variables on the Extraction Yield of Isorhamnetin
A regression analysis was performed using the experimental data, and the model coefficients were evaluated for significance. The liquid-to-solid ratio (
X1), microwave power (
X2), and time (
X3) significantly affected the isorhamnetin extraction yield (
Y5,
Table 3), with corresponding contribution rates of 2.85, 2.44, and 2.45. The liquid-to-solid ratio had the largest impact on the isorhamnetin extraction yield.
The three-dimensional response surface plots (
Figure 11) illustrate the relationship between the isorhamnetin extraction yield and experimental variables. These plots present the response as a function of two factors with another variable constant at its 0 level.
Figure 11a shows the interaction effect between the ratio and power when the time was set at its 0 level (70 s) in the isorhamnetin extraction. The isorhamnetin extraction yield gradually increased with the ratio and power and peaked at approximately 40–55 and 275–300 W.
The interaction effect between liquid-to-solid ratio and time at 0 level power (250 W) on isorhamnetin extraction is presented in
Figure 11b. The response surface plot shows that the extraction yield of isorhamnetin increased and reached the maximum level at 50–60 for the time interval of 75–80 s.
The interaction effect between power and time at the 0 level liquid-to-solid ratio (75) on isorhamnetin extraction yield is presented in
Figure 11c. Strong interaction was observed when the power was set from 250 W to 300 W and time ranged from 70 s to 80 s, which contributed to the increase in extraction yield.
The isorhamnetin regression model for the statistical frequency method of analysis with 95% confidence interval was obtained (X1: –1.33 to –0.81, X2: 0.58 to 1.15, and X3: 0.58 to 1.15) when the extraction yield was >0.16% (n = 21). Thus, the optimal conditions were 41.83–54.70, 278.80–307.35 W, and 75.76–81.47 s.
2.3. Optimization of the Extraction Process
Table 4 indicates the optimum microwave-assisted conditions for the extraction of the five major constituents from FSI using RSM. Thirty accurately weighed samples (
i.e., 0.5 g samples filtered through a 100 mesh sieve) were added to 25 mL 100% MeOH and divided into six groups. A sample set was extracted under the optimum single ingredient conditions (obtained using statistical software), and the predicted results fitted well with the experimental results (
Table 4).
The following optimum extraction conditions were obtained on the basis of the statistics frequency method: liquid-to-solid ratio, 50; microwave power, 287 W; and extraction time, 80 s. Six accurately weighed samples (
i.e., 0.5 g samples filtered through a 100 mesh sieve) were added to 25 mL of extracted 100% MeOH. The optimum extraction conditions were obtained through the statistics frequency method (
Table 4). The calculated extraction yields of the statistical frequency condition were compared with the actual extraction yields under optimum conditions.
The results show that the significant results obtained in the genistein extraction yields are insignificant unlike the other four constituents. Therefore, the optimum extraction conditions for the simultaneous extraction of the five ingredients can be obtained through the statistical frequency method. The optimum extraction condition for one ingredient can be obtained through the single-component method.
With the development of the “green chemistry” and “green extraction” concept during the past years, reduce energy consumption, safe, robust, and environmentally friendly extraction techniques are becoming more toward and popular [
94,
95]. Green extraction is based on the discovery and design of extraction processes, which will reduce energy consumption, allows use of alternative solvents and renewable natural products, and ensure a safe and high quality of extract/product [
94,
95]. The principles of green extraction are renewable plant resource instead of non-renewable resources, green and alternative solvents, reduce energy consumption and heat production, safe and robust to extraction processes [
94]. In the present study, six factors, namely extraction solvent type, sample particle size, frequency, liquid-to-solid ratio, microwave power, and extraction time, were investigated first to optimize the extraction solvent type, sample particle size, and frequency. Additionally, the levels of the response surface experimental design factors (liquid-to-solid ratio, microwave power, and time) were determined according to the liquid-to-solid ratio, microwave power, and extraction time optimized in the single factor tests. Higher yields of the constituents were green extracted from FSI using a renewable raw material, green extraction solvent, less solvent, lower extraction power, reduce energy consumption, more economical, and simultaneously and considerably decreasing the extraction time. TCMM and natural product green extraction, according to the principle of green extraction, is a new concept to meet the future trends [
94,
95].
2.4. Optimization of UHPLC-ESI-Q-TOF MS/MS Conditions
Isocratic and gradient LC were tested to optimize the separation conditions of all constituents. Various UHPLC conditions, such as mobile phase, gradient program, column, column temperature, and flow rate were optimized systematically in a preliminary test to improve the separation efficiency of analytes over short analysis times. Two analytical columns, namely, an Acquity UPLC® BEH C18 column (100 mm × 2.1 mm, 1.7 μm, Waters, Wexford, Ireland) and a Kinetex C18 column (100 mm × 2.1 mm, 2.6 μm, Phenomenex, Torrance, CA, USA), were compared. Results showed that although all of the compounds could be separated satisfactorily, chromatograms with better peak shapes and shorter analysis times were obtained with the Acquity UPLC® BEH C18 column (100 mm × 2.1 mm, 1.7 μm). Thus, this column was selected as the analytical column in subsequent experiments.
Different mobile phases (water/acetonitrile, water/methanol, 0.1% formic acid/acetonitrile, 0.1% formic acid/methanol, 0.2% phosphate/acetonitrile, 0.2% phosphate/methanol, 0.5% acetic acid/acetonitrile, 0.5% acetic acid/methanol), flow rates (0.10, 0.12, 0.15, 0.20, 0.25, 0.30, 0.35, 0.40, and 0.45 mL/min), and column temperatures (30, 35, 40, 45, 50, 55, and 60 °C) were also examined and compared. Considering the results obtained, we selected the isocratic mobile phase consisting of 0.1% aqueous formic acid and acetonitrile (71:29,
v/
v), column temperature of 40 °C, and flow rate of 0.35 mL/min (
Figure 12) for subsequent investigations.
2.5. Qualitative Analysis
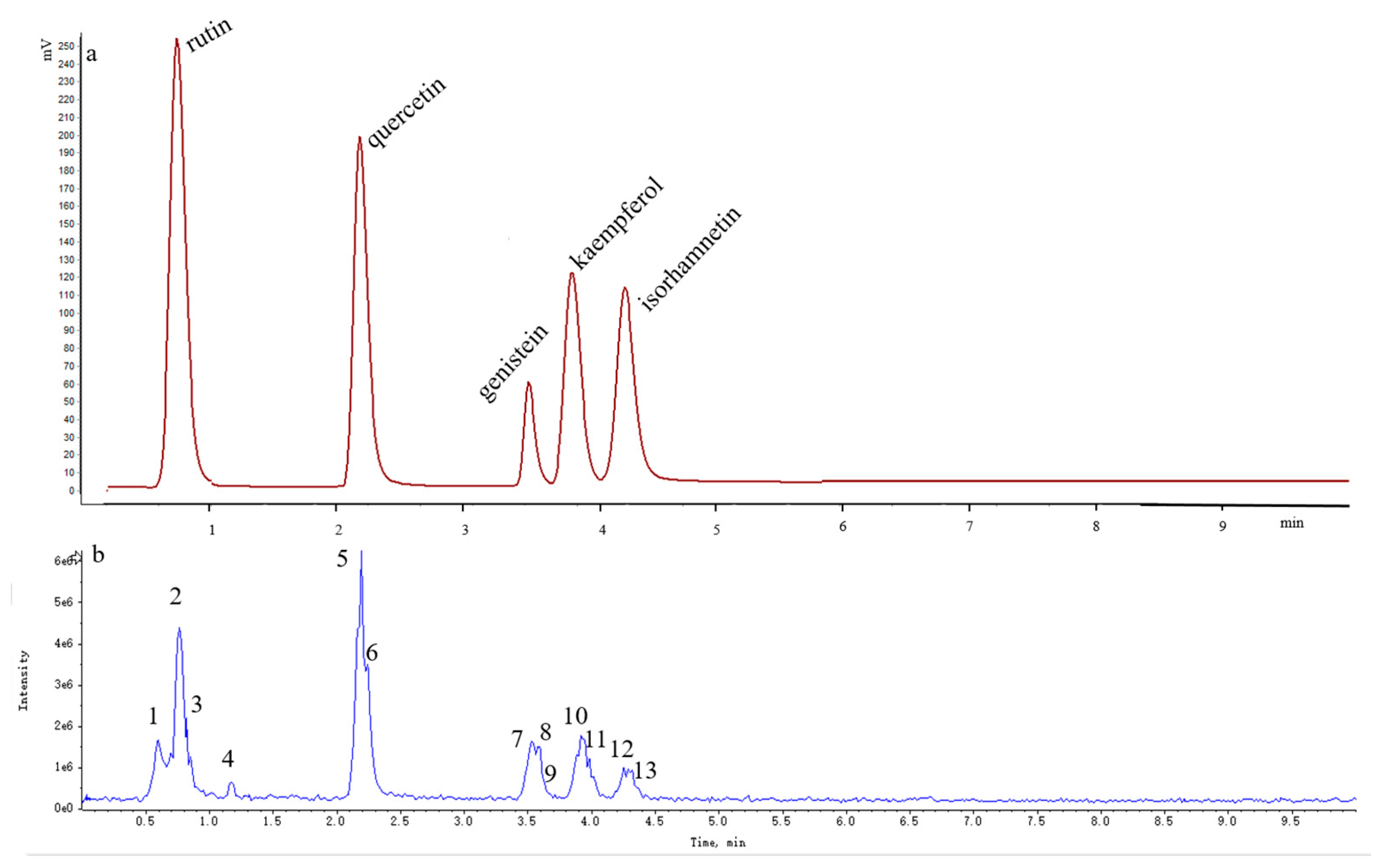
Over 13 peaks were detected within 10 min in the mass spectrometry total ion current (TIC) chromatograms obtained in positive and negative modes. The molecular weights of these 13 compounds were determined on the basis of their positive and negative ion mass spectra.
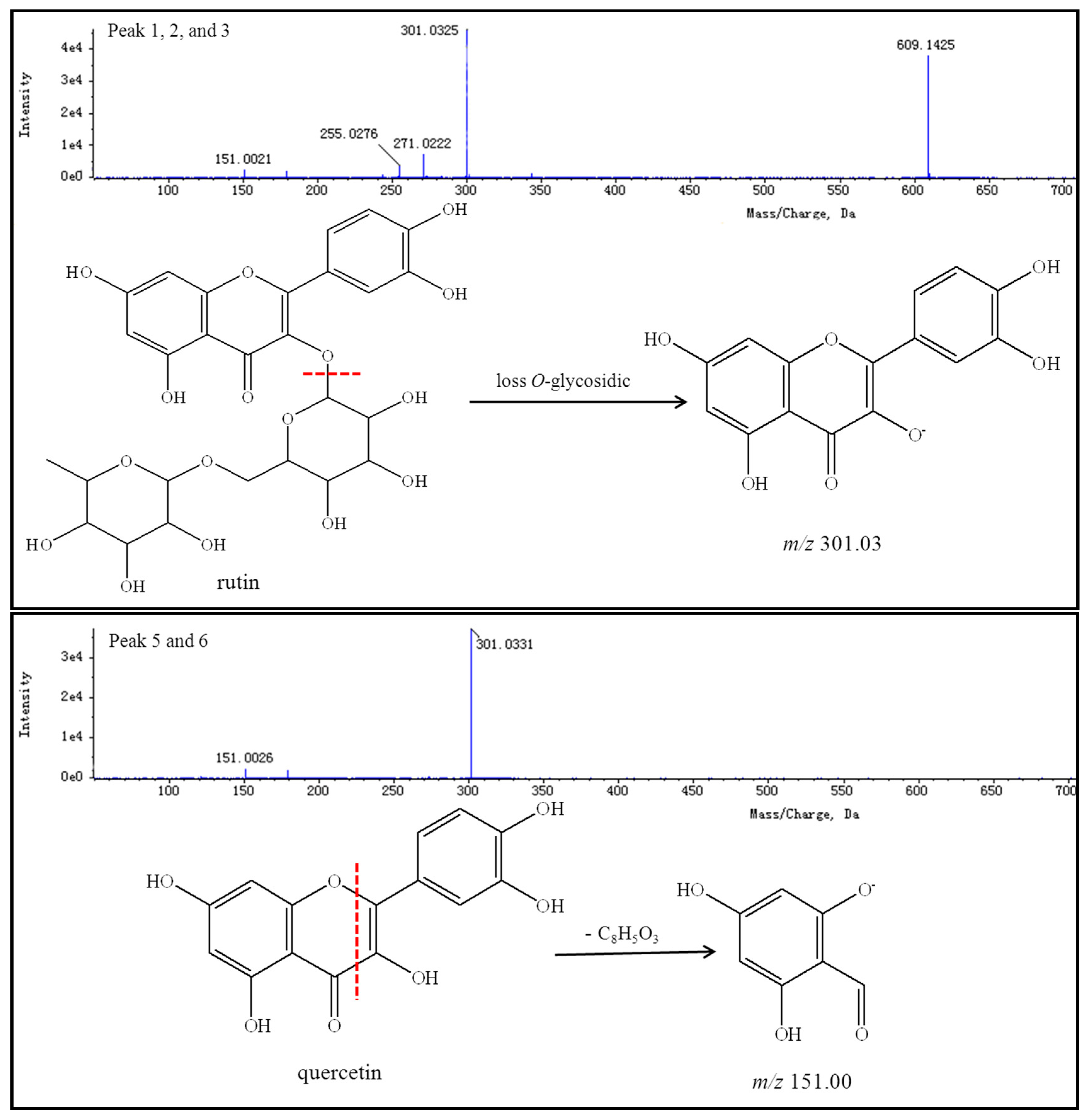
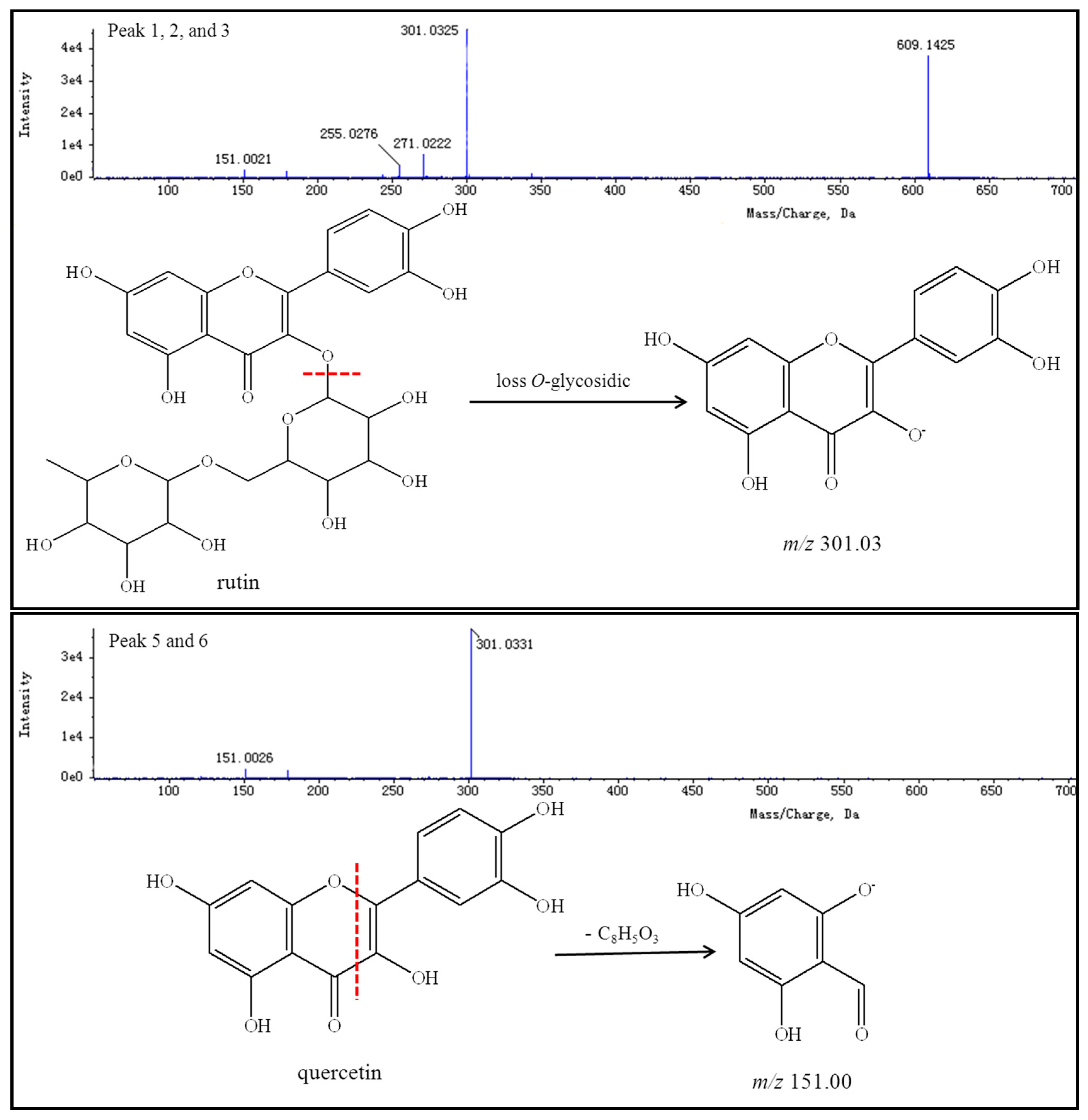
Thirteen peaks, including those of rutin, quercetin, genistein, kaempferol, isorhamnetin, and their isomers, were identified using authentic standards and simultaneously quantified in the FSI extracts. The retention times, formulas, and MS/MS fragmentation data of the 13 compounds are summarized in
Table 5. Possible structures and fragmentation schemes are shown in
Figure 13; this information was deduced by carefully studying the MS and MS/MS spectral data of each compound and comparing findings with literature values. Peaks 1–3 reveal a group of isomers with a molecular weight of 610 and chemical formula of C
27H
30O
16. The MS
2 spectra of compounds
1–
3 showed that the most abundant fragment peak at
m/
z 301.03 may be attributed to elimination of
O-glycosidic groups from the precursor ion at
m/
z 609.14. Peaks 1–3 were unambiguously identified as rutin (including its isomers) by comparison with literature values [
92,
96].
Peaks 5 and 6 indicate a group of isomers with a molecular weight of 302 and chemical formula of C
15H
10O
7. The MS
2 spectra of compounds
5 and
6 showed the most abundant fragment peaks at
m/
z 151.00 (negative ion mode) and
m/
z 153.02 (positive ion mode) were appeared. Peaks 5 and 6 were identified as quercetin (including its isomers) by comparison with literature values [
83,
92,
96].
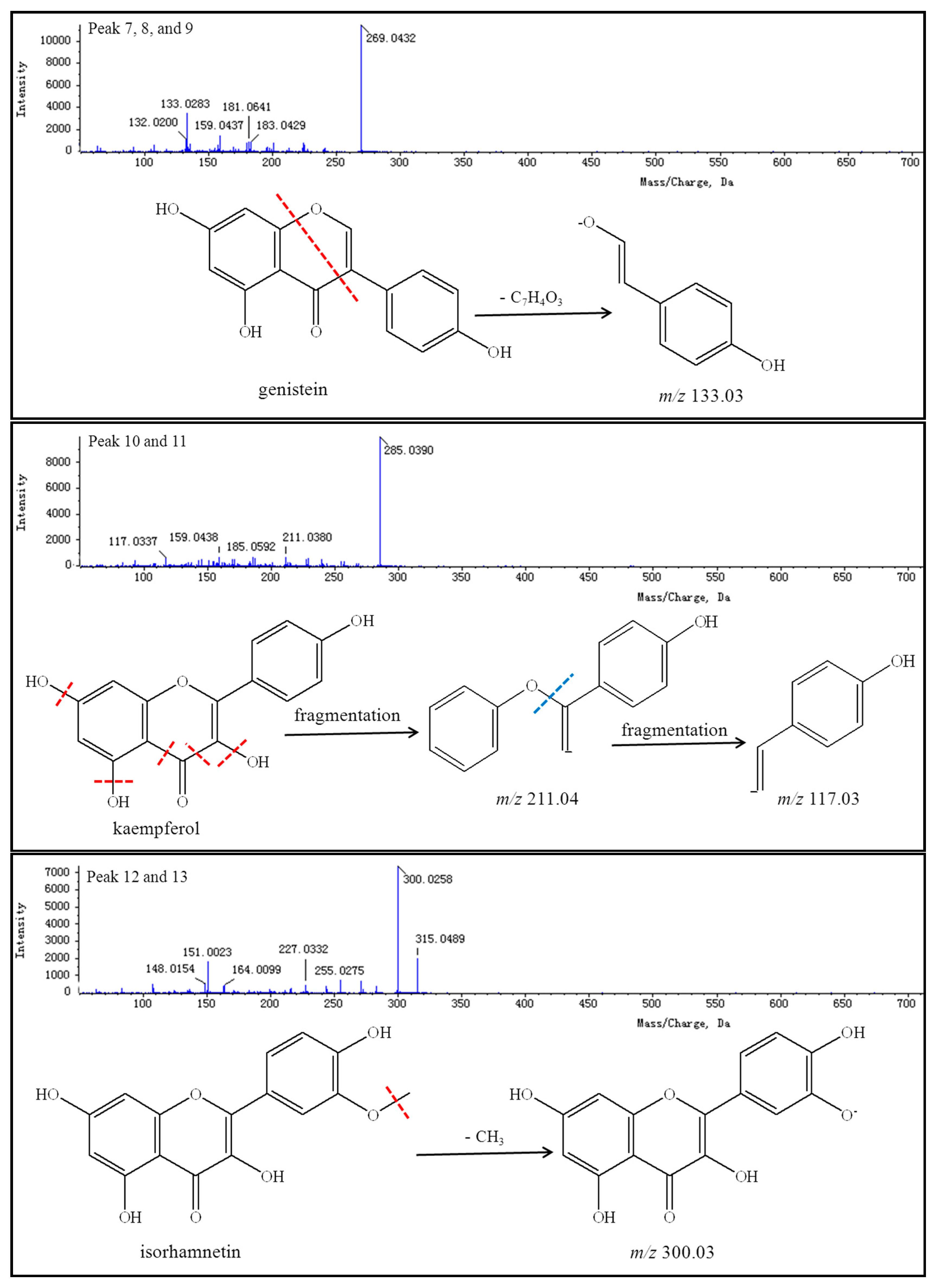
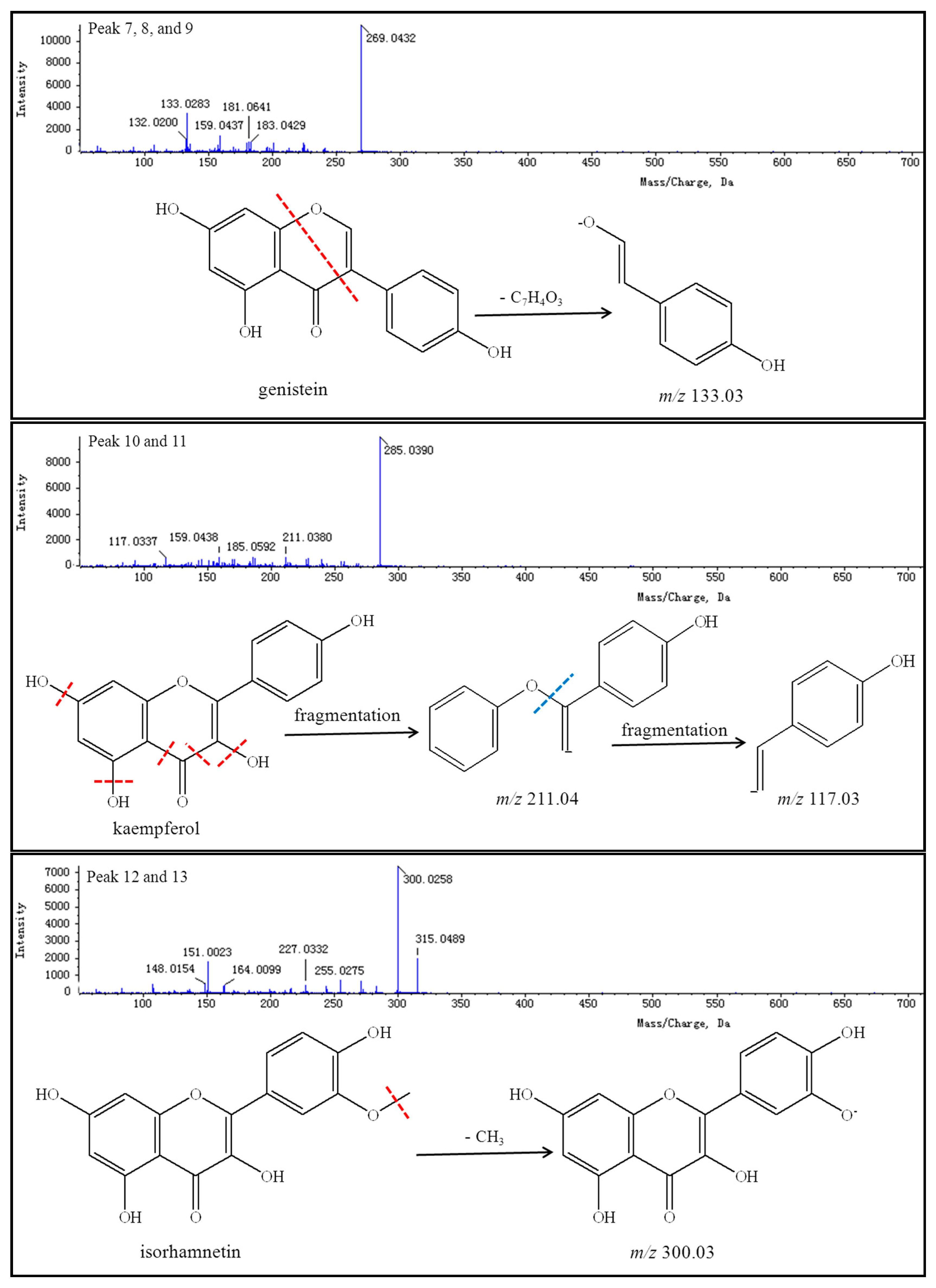
Peaks 7–9 reveal a group of isomers with a molecular weight of 270 and chemical formula of C
15H
10O
5. The MS
2 spectra of compounds
7–
9 showed that most the abundant fragment peak at
m/
z 133.03 may be attributed to cleavage of C-O and C-C bonds from the precursor ion at
m/
z 269.04. Peaks 7–9 were finally identified as genistein (including its isomers) by comparison with literature values [
96].
Peaks 10 and 11 reflect a group of isomers with a molecular weight of 286 and chemical formula of C
15H
10O
6. The MS
2 spectra of compounds
10 and
11 showed the most abundant fragment peaks at
m/
z 211.04 and
m/
z 117.03 (negative ion mode). Peaks 10 and 11 were subsequently identified as kaempferol (including its isomers) by comparison with literature values [
96].
Peaks 12 and 13 demonstrate a group of isomers with a molecular weight of 316 and chemical formula of C
16H
12O
7. The MS
2 spectra of compounds
12 and
13 showed that the most abundant fragment peak at
m/
z 300.03 (negative ion mode) may be attributed to the loss of a methyl radical from the precursor ion at
m/
z 315.05. Peaks 12 and 13 were thus identified as isorhamnetin (including its isomers) by comparison with literature values [
83,
92].
Considering that rutin, quercetin, genistein, kaempferol, and isorhamnetin present extensive biological activities, developing a quality control method based on the content of these compounds is necessary.
2.6. Quantitative Analysis
The UHPLC method was fully validated for quantitative determination. Linear regression equations, correlation coefficients, and ranges of calibration curves for the listed compounds are shown in
Table 6. Correlation coefficients
R2 > 0.9990 indicated excellent correlations between the concentrations of the investigated compounds and their
PAs within the tested ranges. Constituent contents were calculated using the relevant calibration curves. The LODs and LOQs of the five compounds were in the range of 13.7661–2684.4763 ng/mL and 24.2013–8948.2543 ng/mL, respectively. Analytical variations in intra- and inter-day (retention time and area) RSD% were less than 2.8330% for all five constituents, as shown in
Table 7. The
PA RSD% of all five constituents after 0, 1, 2, 4, 8, 12, 24, 48, 72, and 96 h was less than 0.4841%; these results imply that the extract solutions are very stable. The RSD% (retention time and area) of six samples from the same batch (repeatability) of material was less than 0.3748% for all five compounds (
Table 7). The recoveries of the five compounds exceeded 95.1217%, and RSD% less than 1.8041% (
Table 8). These results indicate that the proposed UHPLC method is sensitive, repeatable, and accurate for the quantitative analysis of active compounds in FSI.
4. Conclusions
RSM was successfully applied in this study to optimize the microwave-assisted extraction of the five major constituents from FSI. The extraction solvent type, sample particle size, times, liquid-to-solid ratio, microwave power, and extraction time played significant roles in the constituent extraction. Higher yields of the constituents were extracted from FSI using a green extraction solvent, less solvent, and lower extraction power, and simultaneously and considerably decreasing the extraction time. The statistical frequency method indicates that the optimum extraction conditions for the simultaneous extraction of the five components were 100% MeOH, 100 mesh, 50:1, 287 W, and 80 s.
The present study described the development and evaluation of a relatively simple UHPLC and LC-ESI-Q-TOF MS/MS method for the simultaneous analysis of rutin, quercetin, genistein, kaempferol, and isorhamnetin with high sensitivity. The proposed method is simple and rapid, provides high precision, sensitivity, accuracy, and reliability, and is appropriate for detection work. Separation of five compounds of interest was completed within 5 min, and well-resolved peaks were obtained. Run times were significantly shortened. The method developed in this project will be very useful for future studies.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}