
Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors

Abstract
:1. Introduction
2. Results
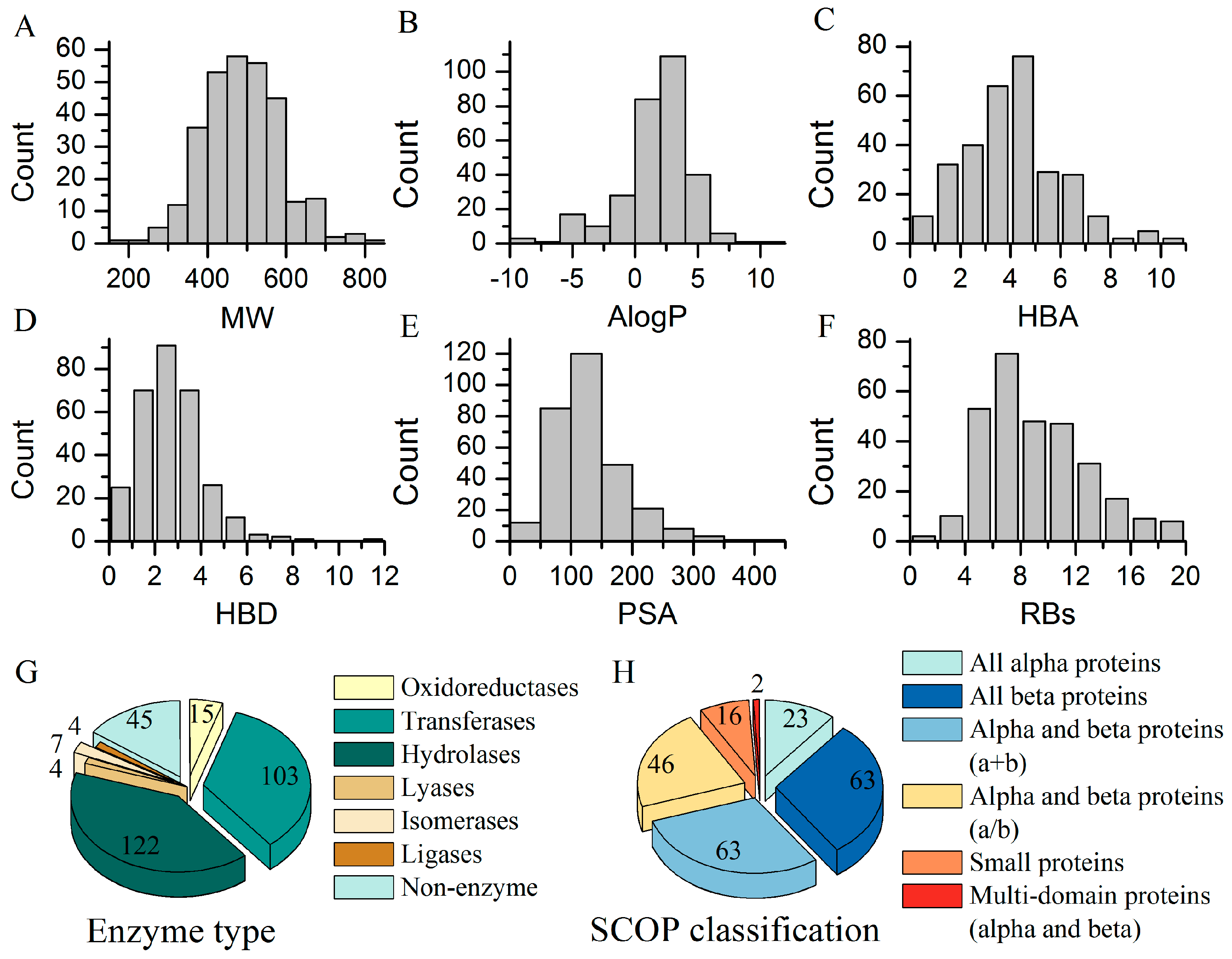
2.1. Summary of BRCD-3D
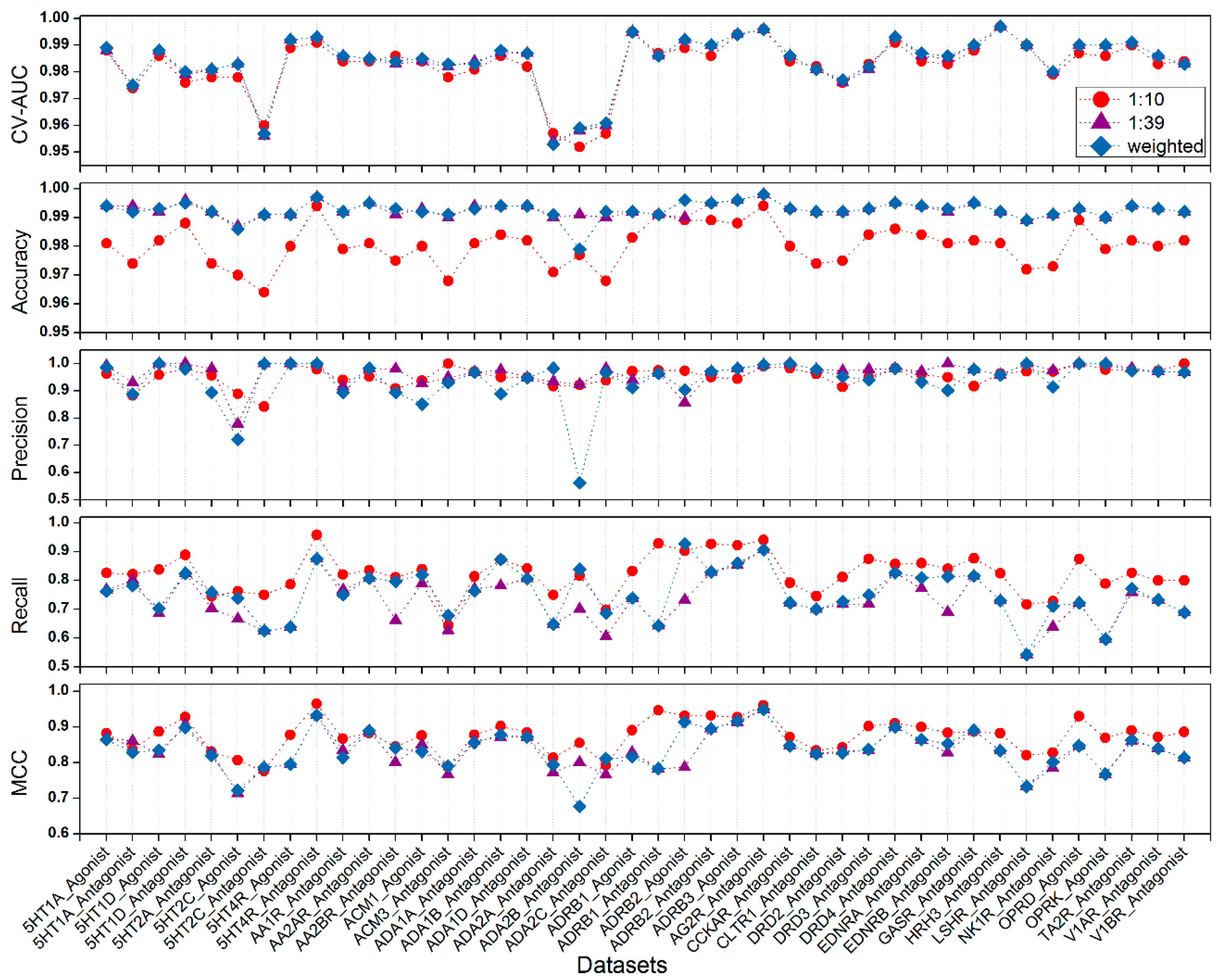
2.2. Evaluation with GLL/GDD (G Protein-Coupled Receptor (GPCR) Ligand Library and the GPCR Decoy Database) Benchmark Data Sets
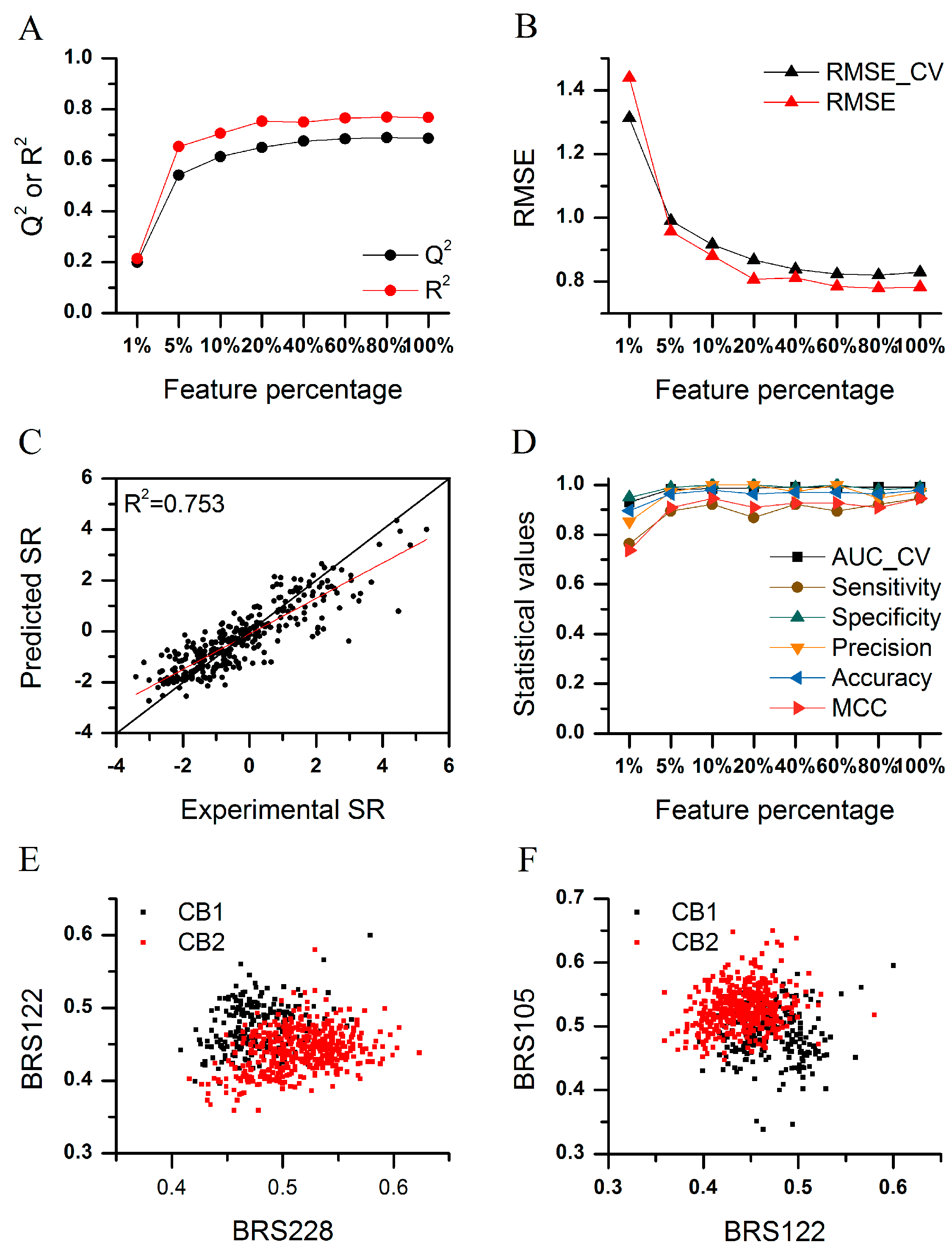
2.3. Feature Selection
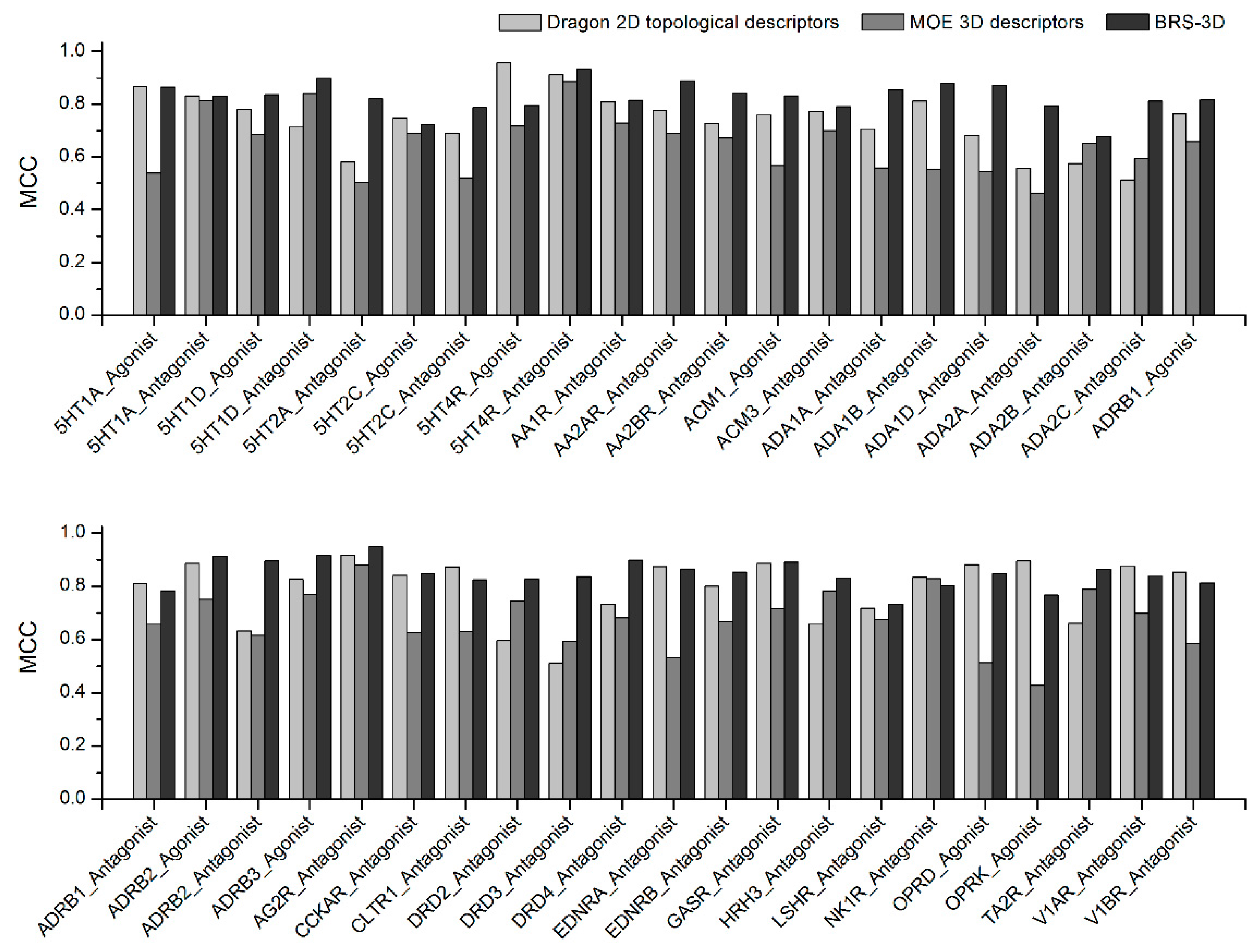
2.4. Comparison with Other Molecular Descriptors
2.5. HDAC1 Inhibitor Screening
2.6. Application of BRS-3D in Subtype Selectivity Predictions
3. Discussion
4. Materials and Methods
4.1. Workflow of BRS-3D-Based Virtual Screening
4.2. Surflex-Sim Superimposition
4.3. Construction of the BRCD-3D
4.4. Calculation of BRS-3D
4.5. The Benchmark Data Sets
4.6. Model Development and Validation
4.7. Feature Selection
4.8. Dragon 2D Descriptors and MOE 3D Descriptors
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Leung, K.H.; Chan, D.S.; Wang, Y.T.; Ma, D.L.; Leung, C.H. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014, 5, e1293. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Sciabola, S.; Carosati, E.; Cucurull-Sanchez, L.; Baroni, M.; Mannhold, R. Novel TOPP descriptors in 3D-QSAR analysis of apoptosis inducing 4-aryl-4h-chromenes: Comparison versus other 2D- and 3D-descriptors. Bioorg. Med. Chem. 2007, 15, 6450–6462. [Google Scholar] [CrossRef] [PubMed]
- Sciabola, S.; Morao, I.; de Groot, M.J. Pharmacophoric fingerprint method (TOPP) for 3D-QSAR modeling: Application to CYP2D6 metabolic stability. J. Chem. Inf. Model. 2007, 47, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Nettles, J.H.; Jenkins, J.L.; Bender, A.; Deng, Z.; Davies, J.W.; Glick, M. Bridging chemical and biological space: “Target fishing” using 2D and 3D molecular descriptors. J. Med. Chem. 2006, 49, 6802–6810. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Perez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.P.; Kuang, G.L.; Xiao, W.; Li, W.H.; Liu, G.X.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [PubMed]
- Lo Conte, L.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. SCOP database in 2002: Refinements accommodate structural genomics. Nucleic Acids Res. 2002, 30, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004, 32, D226–D229. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chandonia, J.M.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. Data growth and its impact on the SCOP database: New developments. Nucleic Acids Res. 2008, 36, D419–D425. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Hannon, J.; Hoyer, D. Molecular biology of 5-HT receptors. Behav. Brain Res. 2008, 195, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.L.; Du, C.X.; Li, X.; Hu, B.; Kuang, Z.K.; Wang, R.; Feng, S.Y.; Zhang, H.Y.; Kong, D.X. Exploring the biologically relevant chemical space for drug discovery. J. Chem. Inf. Model. 2013, 53, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Available Chemicals Directory (ACD), version 2004.1; MDL Information Systems Inc.: San Leandro, CA, USA, 2004.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- George, S.R.; O’Dowd, B.F.; Lee, S.R. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Lagerstrom, M.C.; Schioth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Heilker, R.; Wolff, M.; Tautermann, C.S.; Bieler, M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov. Today 2009, 14, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kobilka, B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Gatica, E.A.; Cavasotto, C.N. Ligand and decoy sets for docking to G protein-coupled receptors. J. Chem. Inf. Model. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garrido, A.; Helguera, A.M.; Borges, F.; Cordeiro, M.N.D.S.; Rivero, V.; Escudero, A.G. Two new parameters based on distances in a receiver operating characteristic chart for the selection of classification models. J. Chem. Inf. Model. 2011, 51, 2746–2759. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.K.; Feng, S.Y.; Hu, B.; Wang, D.; He, S.B.; Kong, D.X. Predicting subtype selectivity of dopamine receptor ligands with three-dimensional biologically relevant spectrum. Chem. Biol. Drug Des. 2016, 88, 859–872. [Google Scholar] [CrossRef] [PubMed]
- He, S.B.; Ben, H.; Kuang, Z.K.; Wang, D.; Kong, D.X. Predicting subtype selectivity for adenosine receptor ligands with three-dimensional biologically relevant spectrum (BRS-3D). Sci. Rep. 2016, 6, 36595. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.H.; Kruse, C.G. Keynote review: Medicinal chemistry strategies to CB1 cannabinoid receptor antagonists. Drug Discov. Today 2005, 10, 693–702. [Google Scholar] [CrossRef]
- Le Foll, B.; Goldberg, S.R. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J. Pharmacol. Exp. Ther. 2005, 312, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, G.T.; Lee, G.P.; Valenzano, K.J. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr. Med. Chem. 2007, 14, 917–936. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Battista, N.; Centonze, D. The endocannabinoid pathway in Huntington’s disease: A comparison with other neurodegenerative diseases. Prog. Neurobiol. 2007, 81, 349–379. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Finazzi-Agro, A.; Bernardi, G.; Maccarrone, M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol. Sci. 2007, 28, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Fliri, A.F.; Loging, W.T.; Thadeio, P.F.; Volkmann, R.A. Analysis of drug-induced effect patterns to link structure and side effects of medicines. Nat. Chem. Biol. 2005, 1, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Fliri, A.F.; Loging, W.T.; Thadeio, P.F.; Volkmann, R.A. Biospectra analysis: Model proteome characterizations for linking molecular structure and biological response. J. Med. Chem. 2005, 48, 6918–6925. [Google Scholar] [CrossRef] [PubMed]
- Fliri, A.F.; Loging, W.T.; Thadeio, P.F.; Volkmann, R.A. Biological spectra analysis: Linking biological activity profiles to molecular structure. Proc. Natl. Acad. Sci. USA 2005, 102, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Petrone, F.M.; Simms, B.; Nigsch, F.; Lounkine, E.; Kutchukian, P.; Cornett, A.; Deng, Z.; Davies, J.W.; Jenkins, J.L.; Glick, M. Rethinking molecular similarity: Comparing compounds on the basis of biological activity. ACS Chem. Biol. 2012, 7, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, A.M.; Kutchukian, P.S.; Lounkine, E.; Luethi, T.; Hamon, J.; Bocker, M.T.; Malik, H.A.; Cowan-Jacob, S.W.; Glick, M. Efficient search of chemical space: Navigating from fragments to structurally diverse chemotypes. J. Med. Chem. 2013, 56, 8879–8891. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, A.M.; Lounkine, E.; Urban, L.; Whitebread, S.; Chen, S.N.; Hughes, K.; Guo, H.Q.; Kutlina, E.; Fekete, A.; Klumpp, M.; et al. A screening pattern recognition method finds new and divergent targets for drugs and natural products. ACS Chem. Biol. 2014, 9, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Helal, K.Y.; Maciejewski, M.; Gregori-Puigjane, E.; Glick, M.; Wassermann, A.M. Public domain HTS fingerprints: Design and evaluation of compound bioactivity profiles from PubChem’s bioassay repository. J. Chem. Inf. Model. 2016, 56, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Foata, N.; Rognan, D. Ranking targets in structure-based virtual screening of three-dimensional protein libraries: Methods and problems. J. Chem. Inf. Model. 2008, 48, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Steindl, T.M.; Schuster, D.; Laggner, C.; Langer, T. Parallel screening: A novel concept in pharmacophore modeling and virtual screening. J. Chem. Inf. Model. 2006, 46, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yuki, H.; Takaya, D.; Sasaki, S.; Tanaka, A.; Honma, T. Application of support vector machine to three-dimensional shape-based virtual screening using comprehensive three-dimensional molecular shape overlay with known inhibitors. J. Chem. Inf. Model. 2012, 52, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Winkler, D.A. Understanding the roles of the “two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chan, D.S.; Leung, C.H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Meslamani, J.; Rognan, D.; Kellenberger, E. sc-PDB: A database for identifying variations and multiplicity of ‘druggable’ binding sites in proteins. Bioinformatics 2011, 27, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Lemmen, C.; Lengauer, T.; Klebe, G. FLEXS: A method for fast flexible ligand superposition. J. Med. Chem. 1998, 41, 4502–4520. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.A.; Gallardo, M.A.; Pickup, B.T. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Morphological similarity: A 3D molecular similarity method correlated with protein-ligand recognition. J. Comput. Aided Mol. Des. 2000, 14, 199–213. [Google Scholar] [CrossRef] [PubMed]
- sc-PDB. An Annotated Database of Druggable Binding Sites from the Protein Data Bank. Available online: http://bioinfo-pharma.u-strasbg.fr/scPDB/ (accessed on 31 August 2013).
- Pipeline Pilot, version 8.5; Accerlrys Software Inc.: San Diego, CA, USA, 2011.
- Shiraishi, A.; Niijima, S.; Brown, J.B.; Nakatsui, M.; Okuno, Y. Chemical genomics approach for GPCR-ligand interaction prediction and extraction of ligand binding determinants. J. Chem. Inf. Model. 2013, 53, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Computaional Chemistry & Drug Design. Available online: http://cavasotto-lab.net/Databases/GDD/Download/ (accessed on 15 July 2014).
- Hinselmann, G.; Rosenbaum, L.; Jahn, A.; Fechner, N.; Ostermann, C.; Zell, A. Large-scale learning of structure-activity relationships using a linear support vector machine and problem-specific metrics. J. Chem. Inf. Model. 2011, 51, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, R.; Gao, L.; Zhou, D.; Yang, S.; Liu, A.L.; Du, G.H. Predictions of BuChE inhibitors using support vector machine and naive Bayesian classification techniques in drug discovery. J. Chem. Inf. Model. 2013, 53, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, K.; Bajorath, J. Comparison of confirmed inactive and randomly selected compounds as negative training examples in support vector machine-based virtual screening. J. Chem. Inf. Model. 2013, 53, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, K.; Bajorath, J. Prediction of compounds with closely related activity profiles using weighted support vector machine linear combinations. J. Chem. Inf. Model. 2013, 53, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Khanna, M.; Jo, I.; Wang, F.; Ashpole, N.M.; Hudmon, A.; Meroueh, S.O. Target-specific support vector machine scoring in structure-based virtual screening: Computational validation, in vitro testing in kinases, and effects on lung cancer cell proliferation. J. Chem. Inf. Model. 2011, 51, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Alexander, D.L.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Boulesteix, A.L.; Zeileis, A.; Hothorn, T. Bias in random forest variable importance measures: Illustrations, sources and a solution. BMC Bioinform. 2007, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragon (for Windows), version 5.4; Talete srl: Milano, Italy, 2006.
- Molecular Operating Environment (MOE), version 2009.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2009.
- Sample Availability: Not available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Data Sets | CV AUC | Accuray | Precision | Recall | MCC |
|---|---|---|---|---|---|---|
| 1 | 5HT1A_Agonist | 0.989 | 0.994 | 0.986 | 0.763 | 0.865 |
| 2 | 5HT1A_Antagonist | 0.975 | 0.992 | 0.888 | 0.782 | 0.829 |
| 3 | 5HT1D_Agonist | 0.988 | 0.993 | 1.000 | 0.703 | 0.835 |
| 4 | 5HT1D_Antagonist | 0.980 | 0.995 | 0.981 | 0.825 | 0.898 |
| 5 | 5HT2A_Antagonist | 0.981 | 0.992 | 0.894 | 0.759 | 0.820 |
| 6 | 5HT2C_Agonist | 0.983 | 0.986 | 0.721 | 0.738 | 0.722 |
| 7 | 5HT2C_Antagonist | 0.957 | 0.991 | 1.000 | 0.625 | 0.787 |
| 8 | 5HT4R_Agonist | 0.992 | 0.991 | 1.000 | 0.638 | 0.795 |
| 9 | 5HT4R_Antagonist | 0.993 | 0.997 | 1.000 | 0.875 | 0.933 |
| 10 | AA1R_Antagonist | 0.986 | 0.992 | 0.894 | 0.750 | 0.814 |
| 11 | AA2AR_Antagonist | 0.985 | 0.995 | 0.983 | 0.808 | 0.889 |
| 12 | AA2BR_Antagonist | 0.984 | 0.993 | 0.894 | 0.797 | 0.841 |
| 13 | ACM1_Agonist | 0.985 | 0.992 | 0.851 | 0.820 | 0.831 |
| 14 | ACM3_Antagonist | 0.983 | 0.991 | 0.930 | 0.678 | 0.790 |
| 15 | ADA1A_Antagonist | 0.983 | 0.993 | 0.968 | 0.763 | 0.856 |
| 16 | ADA1B_Antagonist | 0.988 | 0.994 | 0.889 | 0.873 | 0.878 |
| 17 | ADA1D_Antagonist | 0.987 | 0.994 | 0.948 | 0.807 | 0.872 |
| 18 | ADA2A_Antagonist | 0.953 | 0.991 | 0.983 | 0.648 | 0.794 |
| 19 | ADA2B_Antagonist | 0.959 | 0.979 | 0.562 | 0.839 | 0.677 |
| 20 | ADA2C_Antagonist | 0.961 | 0.992 | 0.967 | 0.686 | 0.811 |
| 21 | ADRB1_Agonist | 0.995 | 0.992 | 0.912 | 0.738 | 0.816 |
| 22 | ADRB1_Antagonist | 0.986 | 0.991 | 0.964 | 0.643 | 0.783 |
| 23 | ADRB2_Agonist | 0.992 | 0.996 | 0.904 | 0.927 | 0.914 |
| 24 | ADRB2_Antagonist | 0.990 | 0.995 | 0.971 | 0.829 | 0.895 |
| 25 | ADRB3_Agonist | 0.994 | 0.996 | 0.982 | 0.860 | 0.917 |
| 26 | AG2R_Antagonist | 0.996 | 0.998 | 0.996 | 0.907 | 0.949 |
| 27 | CCKAR_Antagonist | 0.986 | 0.993 | 1.000 | 0.722 | 0.847 |
| 28 | CLTR1_Antagonist | 0.981 | 0.992 | 0.979 | 0.701 | 0.825 |
| 29 | DRD2_Antagonist | 0.977 | 0.992 | 0.951 | 0.726 | 0.827 |
| 30 | DRD3_Antagonist | 0.982 | 0.993 | 0.941 | 0.750 | 0.837 |
| 31 | DRD4_Antagonist | 0.993 | 0.995 | 0.982 | 0.827 | 0.899 |
| 32 | EDNRA_Antagonist | 0.987 | 0.994 | 0.932 | 0.809 | 0.865 |
| 33 | EDNRB_Antagonist | 0.986 | 0.993 | 0.902 | 0.814 | 0.853 |
| 34 | GASR_Antagonist | 0.990 | 0.995 | 0.979 | 0.816 | 0.891 |
| 35 | HRH3_Antagonist | 0.997 | 0.992 | 0.958 | 0.730 | 0.833 |
| 36 | LSHR_Antagonist | 0.990 | 0.989 | 1.000 | 0.543 | 0.733 |
| 37 | NK1R_Antagonist | 0.980 | 0.991 | 0.914 | 0.711 | 0.802 |
| 38 | OPRD_Agonist | 0.990 | 0.993 | 1.000 | 0.722 | 0.847 |
| 39 | OPRK_Agonist | 0.990 | 0.990 | 1.000 | 0.596 | 0.768 |
| 40 | TA2R_Antagonist | 0.991 | 0.994 | 0.974 | 0.772 | 0.864 |
| 41 | V1AR_Antagonist | 0.986 | 0.993 | 0.971 | 0.733 | 0.840 |
| 42 | V1BR_Antagonist | 0.983 | 0.992 | 0.969 | 0.689 | 0.813 |
| Data Sets | Accuracy | Precision | Recall | MCC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dragon 2D | MOE 3D | BRS-3D | Dragon 2D | MOE 3D | BRS-3D | Dragon 2D | MOE 3D | BRS-3D | Dragon 2D | MOE 3D | BRS-3D | |
| 1 | 0.993 | 0.951 | 0.994 | 0.849 | 0.330 | 0.986 | 0.889 | 0.932 | 0.763 | 0.866 | 0.539 | 0.865 |
| 2 | 0.992 | 0.991 | 0.992 | 0.819 | 0.814 | 0.888 | 0.851 | 0.822 | 0.782 | 0.831 | 0.813 | 0.829 |
| 3 | 0.987 | 0.977 | 0.993 | 0.671 | 0.526 | 1.000 | 0.919 | 0.919 | 0.703 | 0.779 | 0.686 | 0.835 |
| 4 | 0.981 | 0.992 | 0.995 | 0.568 | 0.831 | 0.981 | 0.921 | 0.857 | 0.825 | 0.715 | 0.840 | 0.898 |
| 5 | 0.964 | 0.945 | 0.992 | 0.401 | 0.300 | 0.894 | 0.883 | 0.903 | 0.759 | 0.581 | 0.502 | 0.820 |
| 6 | 0.987 | 0.980 | 0.986 | 0.744 | 0.571 | 0.721 | 0.762 | 0.857 | 0.738 | 0.747 | 0.691 | 0.722 |
| 7 | 0.983 | 0.949 | 0.991 | 0.636 | 0.317 | 1.000 | 0.766 | 0.906 | 0.625 | 0.690 | 0.519 | 0.787 |
| 8 | 0.998 | 0.979 | 0.991 | 0.939 | 0.541 | 1.000 | 0.979 | 0.979 | 0.638 | 0.957 | 0.719 | 0.795 |
| 9 | 0.995 | 0.994 | 0.997 | 0.855 | 0.863 | 1.000 | 0.979 | 0.917 | 0.875 | 0.912 | 0.886 | 0.933 |
| 10 | 0.990 | 0.986 | 0.992 | 0.774 | 0.705 | 0.894 | 0.857 | 0.769 | 0.750 | 0.810 | 0.729 | 0.814 |
| 11 | 0.989 | 0.982 | 0.995 | 0.756 | 0.602 | 0.983 | 0.808 | 0.808 | 0.808 | 0.776 | 0.689 | 0.889 |
| 12 | 0.987 | 0.978 | 0.993 | 0.794 | 0.539 | 0.894 | 0.676 | 0.865 | 0.797 | 0.726 | 0.672 | 0.841 |
| 13 | 0.986 | 0.962 | 0.992 | 0.662 | 0.383 | 0.851 | 0.888 | 0.882 | 0.820 | 0.760 | 0.567 | 0.831 |
| 14 | 0.988 | 0.982 | 0.991 | 0.714 | 0.605 | 0.930 | 0.847 | 0.831 | 0.678 | 0.772 | 0.700 | 0.790 |
| 15 | 0.980 | 0.960 | 0.993 | 0.557 | 0.373 | 0.968 | 0.915 | 0.881 | 0.763 | 0.705 | 0.558 | 0.856 |
| 16 | 0.990 | 0.955 | 0.994 | 0.758 | 0.349 | 0.889 | 0.882 | 0.927 | 0.873 | 0.812 | 0.554 | 0.878 |
| 17 | 0.977 | 0.955 | 0.994 | 0.528 | 0.347 | 0.948 | 0.904 | 0.904 | 0.807 | 0.681 | 0.544 | 0.872 |
| 18 | 0.957 | 0.938 | 0.991 | 0.359 | 0.270 | 0.983 | 0.909 | 0.864 | 0.648 | 0.556 | 0.462 | 0.794 |
| 19 | 0.973 | 0.982 | 0.979 | 0.471 | 0.632 | 0.562 | 0.736 | 0.690 | 0.839 | 0.576 | 0.651 | 0.677 |
| 20 | 0.955 | 0.978 | 0.992 | 0.335 | 0.542 | 0.967 | 0.837 | 0.674 | 0.686 | 0.513 | 0.593 | 0.811 |
| 21 | 0.986 | 0.974 | 0.992 | 0.655 | 0.494 | 0.912 | 0.905 | 0.905 | 0.738 | 0.763 | 0.658 | 0.816 |
| 22 | 0.989 | 0.977 | 0.991 | 0.702 | 0.522 | 0.964 | 0.952 | 0.857 | 0.643 | 0.812 | 0.659 | 0.783 |
| 23 | 0.995 | 0.987 | 0.996 | 0.900 | 0.694 | 0.904 | 0.878 | 0.829 | 0.927 | 0.886 | 0.752 | 0.914 |
| 24 | 0.974 | 0.971 | 0.995 | 0.452 | 0.429 | 0.971 | 0.917 | 0.917 | 0.829 | 0.633 | 0.616 | 0.895 |
| 25 | 0.990 | 0.986 | 0.996 | 0.738 | 0.665 | 0.982 | 0.938 | 0.907 | 0.860 | 0.827 | 0.770 | 0.917 |
| 26 | 0.996 | 0.994 | 0.998 | 0.877 | 0.843 | 0.996 | 0.967 | 0.927 | 0.907 | 0.918 | 0.881 | 0.949 |
| 27 | 0.992 | 0.972 | 0.993 | 0.857 | 0.467 | 1.000 | 0.833 | 0.875 | 0.722 | 0.841 | 0.627 | 0.847 |
| 28 | 0.994 | 0.968 | 0.992 | 0.857 | 0.441 | 0.979 | 0.896 | 0.940 | 0.701 | 0.873 | 0.632 | 0.825 |
| 29 | 0.964 | 0.987 | 0.992 | 0.403 | 0.699 | 0.951 | 0.925 | 0.811 | 0.726 | 0.597 | 0.746 | 0.827 |
| 30 | 0.948 | 0.975 | 0.993 | 0.313 | 0.500 | 0.941 | 0.891 | 0.734 | 0.750 | 0.510 | 0.594 | 0.837 |
| 31 | 0.981 | 0.975 | 0.995 | 0.575 | 0.502 | 0.982 | 0.955 | 0.955 | 0.827 | 0.733 | 0.683 | 0.899 |
| 32 | 0.994 | 0.951 | 0.994 | 0.864 | 0.329 | 0.932 | 0.890 | 0.912 | 0.809 | 0.874 | 0.531 | 0.865 |
| 33 | 0.990 | 0.975 | 0.993 | 0.758 | 0.505 | 0.902 | 0.858 | 0.912 | 0.814 | 0.801 | 0.668 | 0.853 |
| 34 | 0.994 | 0.981 | 0.995 | 0.861 | 0.582 | 0.979 | 0.921 | 0.904 | 0.816 | 0.887 | 0.717 | 0.891 |
| 35 | 0.977 | 0.988 | 0.992 | 0.524 | 0.736 | 0.958 | 0.857 | 0.841 | 0.730 | 0.660 | 0.781 | 0.833 |
| 36 | 0.986 | 0.985 | 0.989 | 0.733 | 0.744 | 1.000 | 0.717 | 0.630 | 0.543 | 0.718 | 0.677 | 0.733 |
| 37 | 0.992 | 0.992 | 0.991 | 0.805 | 0.830 | 0.914 | 0.872 | 0.839 | 0.711 | 0.834 | 0.830 | 0.802 |
| 38 | 0.994 | 0.946 | 0.993 | 0.867 | 0.307 | 1.000 | 0.903 | 0.917 | 0.722 | 0.882 | 0.513 | 0.847 |
| 39 | 0.995 | 0.935 | 0.990 | 0.869 | 0.251 | 1.000 | 0.930 | 0.807 | 0.596 | 0.896 | 0.428 | 0.768 |
| 40 | 0.973 | 0.988 | 0.994 | 0.479 | 0.721 | 0.974 | 0.945 | 0.876 | 0.772 | 0.662 | 0.789 | 0.864 |
| 41 | 0.994 | 0.987 | 0.993 | 0.870 | 0.800 | 0.971 | 0.889 | 0.622 | 0.733 | 0.876 | 0.699 | 0.840 |
| 42 | 0.992 | 0.969 | 0.992 | 0.768 | 0.435 | 0.969 | 0.956 | 0.822 | 0.689 | 0.853 | 0.585 | 0.813 |
| No. | Target | Target Name | Ligand Type | Ligand Count | Decoy Count |
|---|---|---|---|---|---|
| 1 | 5HT1A | 5-hydroxytryptamine receptor 1A | Agonist | 952 | 37,128 |
| 2 | 5HT1A | 5-hydroxytryptamine receptor 1A | Antagonist | 506 | 19,734 |
| 3 | 5HT1D | 5-hydroxytryptamine receptor 1D | Agonist | 558 | 21,762 |
| 4 | 5HT1D | 5-hydroxytryptamine receptor 1D | Antagonist | 315 | 12,285 |
| 5 | 5HT2A | 5-hydroxytryptamine receptor 2A | Antagonist | 725 | 28,275 |
| 6 | 5HT2C | 5-hydroxytryptamine receptor 2C | Agonist | 209 | 8151 |
| 7 | 5HT2C | 5-hydroxytryptamine receptor 2C | Antagonist | 318 | 12,402 |
| 8 | 5HT4R | 5-hydroxytryptamine receptor 4 | Agonist | 235 | 9165 |
| 9 | 5HT4R | 5-hydroxytryptamine receptor 4 | Antagonist | 241 | 9399 |
| 10 | AA1R | Adenosine receptor A1 | Antagonist | 280 | 10,920 |
| 11 | AA2AR | Adenosine receptor A2a | Antagonist | 361 | 14,079 |
| 12 | AA2BR | Adenosine receptor A2b | Antagonist | 370 | 14,430 |
| 13 | ACM1 | Muscarinic acetylcholine receptor M1 | Agonist | 806 | 31,434 |
| 14 | ACM3 | Muscarinic acetylcholine receptor M3 | Antagonist | 295 | 11,505 |
| 15 | ADA1A | Alpha-1A adrenergic receptor | Antagonist | 588 | 22,932 |
| 16 | ADA1B | Alpha-1B adrenergic receptor | Antagonist | 550 | 21,450 |
| 17 | ADA1D | Alpha-1D adrenergic receptor | Antagonist | 568 | 22,152 |
| 18 | ADA2A | Alpha-2A adrenergic receptor | Antagonist | 440 | 17,160 |
| 19 | ADA2B | Alpha-2B adrenergic receptor | Antagonist | 437 | 17,043 |
| 20 | ADA2C | Alpha-2C adrenergic receptor | Antagonist | 433 | 16,887 |
| 21 | ADRB1 | Beta-1 adrenergic receptor | Agonist | 209 | 8151 |
| 22 | ADRB1 | Beta-1 adrenergic receptor | Antagonist | 211 | 8229 |
| 23 | ADRB2 | Beta-2 adrenergic receptor | Agonist | 206 | 8034 |
| 24 | ADRB2 | Beta-2 adrenergic receptor | Antagonist | 204 | 7956 |
| 25 | ADRB3 | Beta-3 adrenergic receptor | Agonist | 643 | 25,077 |
| 26 | AG2R | Type-1 angiotensin II receptor | Antagonist | 1502 | 58,578 |
| 27 | CCKAR | Cholecystokinin receptor type A | Antagonist | 360 | 14,040 |
| 28 | CLTR1 | Cysteinyl leukotriene receptor 1 | Antagonist | 333 | 12,987 |
| 29 | DRD2 | D2 dopamine receptor | Antagonist | 529 | 20,631 |
| 30 | DRD3 | D3 dopamine receptor | Antagonist | 317 | 12,363 |
| 31 | DRD4 | D4 dopamine receptor | Antagonist | 665 | 25,935 |
| 32 | EDNRA | Endothelin-1 receptor | Antagonist | 676 | 26,364 |
| 33 | EDNRB | Endothelin B receptor | Antagonist | 561 | 21,879 |
| 34 | GASR | Gastrin/cholecystokinin type B receptor | Antagonist | 567 | 22,113 |
| 35 | HRH3 | Histamine H3 receptor | Antagonist | 313 | 12,207 |
| 36 | LSHR | Lutropin-choriogonadotropic hormone receptor | Antagonist | 230 | 8970 |
| 37 | NK1R | Substance-P receptor | Antagonist | 900 | 35,100 |
| 38 | OPRD | Delta-type opioid receptor | Agonist | 361 | 14,079 |
| 39 | OPRK | Kappa-type opioid receptor | Agonist | 284 | 11,076 |
| 40 | TA2R | Thromboxane A2 receptor | Antagonist | 725 | 28,275 |
| 41 | V1AR | Vasopressin V1a receptor | Antagonist | 225 | 8775 |
| 42 | V1BR | Vasopressin V1b receptor | Antagonist | 225 | 8775 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, B.; Kuang, Z.-K.; Feng, S.-Y.; Wang, D.; He, S.-B.; Kong, D.-X. Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors. Molecules 2016, 21, 1554. https://doi.org/10.3390/molecules21111554
Hu B, Kuang Z-K, Feng S-Y, Wang D, He S-B, Kong D-X. Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors. Molecules. 2016; 21(11):1554. https://doi.org/10.3390/molecules21111554
Chicago/Turabian StyleHu, Ben, Zheng-Kun Kuang, Shi-Yu Feng, Dong Wang, Song-Bing He, and De-Xin Kong. 2016. "Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors" Molecules 21, no. 11: 1554. https://doi.org/10.3390/molecules21111554