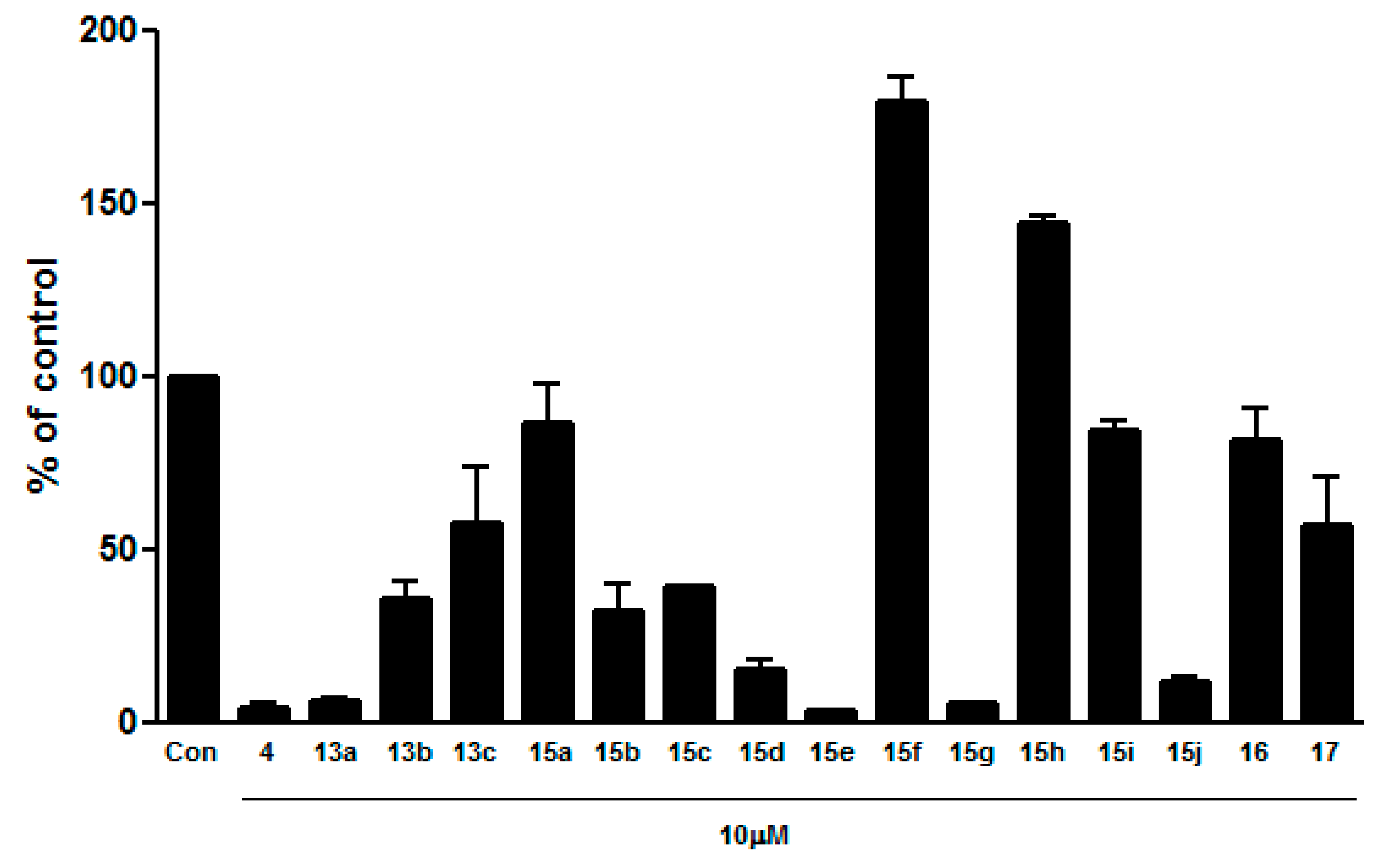
3.2. Chemistry
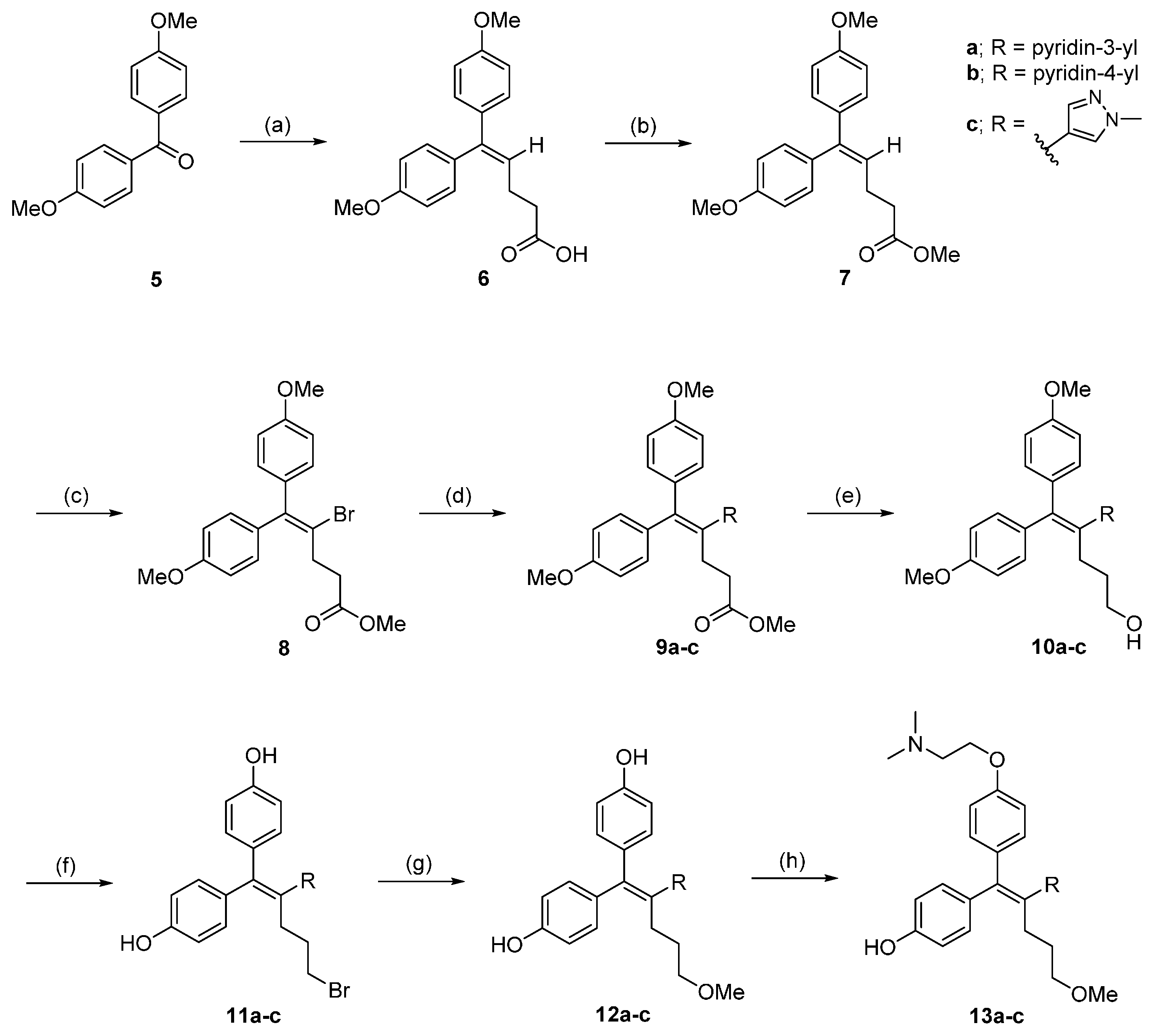
3.2.1. Synthesis of 5,5-bis(4-Methoxyphenyl)pent-4-enoic Acid (6)
In an inert atmosphere, NaH (2.0 g, 49.5 mmol) was added in dry DMSO (50 mL). The mixture was stirred at 70 °C for 1 h. The mixture was cooled to 20 °C and 4-(bromotriphenylphosphoranyl)-butanoic acid (8.5 g, 19.8 mmol) was added in several portions over 5 min. The red solution was stirred at 20 °C for 15 min, then a solution of bis(4-methoxyphenyl)methanone (6.0 g, 24.8 mmol) in dry THF (33 mL) was added at such a rate. The mixture was stirred at rt for 19 h, then it was diluted with ice water and extracted with CH2Cl2. The organic extracts were discarded and the aqueous layer was acidified with 12 N HCl and extracted with CH2Cl2. The organic layer was washed with brine (2 times) and dried over MgSO4. The crude compound was purified by column chromatography to give the desired product (3.1 g, 50% yield).
3.2.2. Synthesis of Methyl 5,5-bis(4-Methoxyphenyl)pent-4-enoate (7)
To a solution of 5,5-bis(4-methoxyphenyl)pent-4-enoic acid (3.1 g, 9.92 mmol) in MeOH (60 mL) stirred at 0 °C, SOCl2 (0.8 mL, 10.7 mmol) was added dropwise. The reaction mixture was heated to reflux for 2 h. The reaction mixture was removed and added water. The mixture was extracted with EtOAc and washed with aq. K2CO3, brine and dried over MgSO4. The solvent was removed under reduced pressure which was used for the next step without further purification (2.9 g, 91% yield).
3.2.3. Methyl 4-bromo-5,5-bis(4-Methoxyphenyl)pent-4-enoate (8)
To a solution methyl 5,5-bis(4-methoxyphenyl)pent-4-enoate (4.0 g, 12.4 mmol) in CCl4 (45 mL) was added copper(II) bromide (6.91 g, 30.9 mmol) at rt. The reaction mixture was heated to reflux at 83 °C for 12 h, cooled to room temperature and filtered off through a Celite pad. The solvent was removed and reaction mixture was dissolved in EtOAc. The mixture was washed with water, brine and dried over MgSO4. The crude compound was purified by column chromatography to give the brown oil product (4.5 g, 89% yield).
3.2.4. General Synthetic Procedure for 9a–c
To a solution 8 (1 eq) in DMF/H2O (50:1) was added corresponding boronic esters or boronic acid (1.2 eq), PdCl2(dppf)·CH2Cl2 (0.1 eq) and K3PO4 (3 eq) at rt. The reaction mixture was heated to 82 °C for 12 h. The reaction mixture was cooled to rt and was quenched by adding water. The mixture was extracted with EtOAc and washed with brine and dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography to give the desired product.
Methyl 5,5-bis(4-methoxyphenyl)-4-(pyridin-3-yl)pent-4-enoate (9a). (2 g, 72% yield). 1H-NMR (400 MHz, CDCl3) δ 8.34 (m, 2H), 7.45 (dt, J = 6.3, 1.4 Hz, 1H), 7.12 (m, 3H), 6.89 (d, J = 6.9 Hz, 2H), 6.76 (d, J = 7.0 Hz, 2H), 6.56 (d, J = 7.0 Hz, 2H), 3.83 (s, 3H), 3.68 (s, 3H), 3.56 (s, 3H), 2.83 (m, 2H), 2.32 (m, 2H).
Methyl 5,5-bis(4-methoxyphenyl)-4-(pyridin-4-yl)pent-4-enoate (9b). (0.8 g, 87% yield). 1H-NMR (400 MHz, CDCl3) δ 8.40 (d, J = 6.0 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 6.0 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.76 (d, J = 8.8 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 3.83 (s, 3H), 3.70 (s, 3H), 3.57 (s, 3H), 2.82 (m, 2H), 2.30 (m, 2H).
Methyl 5,5-bis(4-methoxyphenyl)-4-(1-methyl-1H-pyrazol-4-yl)pent-4-enoate (9c). (0.7 g, 72% yield). 1H-NMR (400 MHz, CDCl3) δ 7.07 (d, J = 7.0 Hz, 2H), 7.03 (s, 1H), 6.98 (d, J = 7.0 Hz, 2H), 6.87 (s, 1H), 6.84 (d, J = 7.0 Hz, 2H), 6.73 (d, J = 7.0 Hz, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 3.75 (s, 3H), 3.61 (s, 3H), 2.71 (m, 2H), 2.46 (m, 2H).
3.2.5. General Synthetic Procedure for 10a–c
To a solution of 1 M LiAlH4 solution in THF (1.5 eq) stirred at 0 °C, 9a–c (1 eq) in THF was added dropwise. The solution was stirred at 0 °C for 1 h and was quenched by adding H2O/5 N NaOH/H2O (1:1:3). The reaction mixture was filtered off through celite pad, washed with EtOAc and concentrated in vacuo. The crude compound was purified by column chromatography to give the desired product.
5,5-bis(4-Methoxyphenyl)-4-(pyridin-3-yl)pent-4-en-1-ol (10a). (0.6 g, 85% yield). 1H-NMR (400 MHz, CDCl3) δ 8.35 (d, J = 1.1 Hz, 1H), 8.33 (d, J = 3.8 Hz, 1H), 7.43 (dt, J = 6.3, 1.4 Hz, 1H), 7.16 (d, J = 7.0 Hz, 2H), 7.10 (dd, J = 6.2, 3.9 Hz, 1H), 6.89 (d, J = 7.0 Hz, 2H), 6.77 (d, J = 7.1 Hz, 2H), 6.56 (d, J = 7.0 Hz, 2H), 3.83 (s, 3H), 3.68 (s, 3H), 3.54 (t, J = 5.2 Hz, 2H), 2.56 (m, 2H), 1.60 (m, 2H).
5,5-bis(4-Methoxyphenyl)-4-(pyridin-4-yl)pent-4-en-1-ol (10b). (0.6 g, 74% yield). 1H-NMR (400 MHz, CDCl3) δ 8.37 (d, J = 6.0 Hz, 2H), 7.14 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 6.0 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 3.83 (s, 3H), 3.70 (s, 3H), 3.55 (t, J = 6.8 Hz, 2H), 2.55 (m, 2H), 1.59 (m, 2H).
5,5-bis(4-Methoxyphenyl)-4-(1-methyl-1H-pyrazol-4-yl)pent-4-en-1-ol (10c). (0.9 g, 73% yield). 1H-NMR (400 MHz, CDCl3) δ 7.11 (d, J = 6.9 Hz, 2H), 7.07 (s, 1H), 6.99 (d, J = 7.0 Hz, 2H), 6.85 (d, J = 7.0 Hz, 2H), 6.83 (s, 1H), 6.74 (d, J = 7.0 Hz, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 3.74 (s, 3H), 3.58 (m, 2H), 2.46 (m, 2H), 1.75 (m, 2H).
3.2.6. General Synthetic Procedure for 11a–c
To a solution of 10a–c (1 eq) in CH2Cl2 stirred at 0 °C, 1 M BBr3 solution in CH2Cl2 (10 eq) was added dropwise. The solution was refluxed at 80 °C for 12 h and was quenched by adding H2O at 0 °C. The reaction mixture was diluted with EtOAc and washed with H2O and brine. The organic layer was dried MgSO4 and concentrated in vacuo. The residue was purified by column chromatography to give the desired product.
4,4′-(5-Bromo-2-(pyridin-3-yl)pent-1-ene-1,1-diyl)diphenol (11a). (0.7 g, 86% yield). 1H-NMR (400 MHz, DMSO-d6) δ 9.53 (s, OH), 9.35 (s, OH), 8.59 (d, J = 4.3 Hz, 1H), 8.51 (s, 1H), 8.24 (d, J = 6.5 Hz, 1H), 7.81 (t, J = 6.2 Hz, 1H), 7.02 (d, J = 6.8 Hz, 2H), 6.77 (d, J = 6.8 Hz, 2H), 6.66 (d, J = 5.2 Hz, 2H), 6.49 (d, J = 6.9 Hz, 2H), 3.44 (t, J = 5.2 Hz, 2H), 2.64 (m, 2H), 1.82 (m, 2H).
4,4′-(5-Bromo-2-(pyridin-4-yl)pent-1-ene-1,1-diyl)diphenol (11b). (0.7 g, 99% yield). 1H-NMR (400 MHz, DMSO-d6) δ 9.59 (s, OH), 9.47 (s, OH), 8.61 (d, J = 4.6 Hz, 2H), 7.60 (d, J = 3.5 Hz, 2H), 7.02 (d, J = 7.7 Hz, 2H), 6.78 (d, J = 6.8 Hz, 2H), 6.66 (d, J = 6.9 Hz, 2H), 6.50 (d, J = 6.8 Hz, 2H), 3.43 (t, J = 5.2 Hz, 2H), 2.66 (m, 2H), 1.79 (m, 2H).
4,4′-(5-Bromo-2-(1-methyl-1H-pyrazol-4-yl)pent-1-ene-1,1-diyl)diphenol (11c). (0.2 g, 39% yield). 1H-NMR (400 MHz, DMSO-d6) δ 7.27 (s, 1H), 6.92 (d, J = 6.7 Hz, 2H), 6.81 (d, J = 6.7 Hz, 2H), 6.71 (s, 1H), 6.69 (d, J = 7.0 Hz, 2H), 6.62 (d, J = 6.7 Hz, 2H), 3.68 (s, 3H), 3.44 (t, J = 5.3 Hz, 2H), 2.41 (m, 2H), 1.91 (m, 2H).
3.2.7. General Synthetic Procedure for 12a–c
To a solution of 11a–c (1 eq) in MeOH stirred at 0 °C, 5 M NaOCH3 solution in MeOH (10 eq) was added dropwise. The solution was stirred at rt for 12 h and quenched by adding 6 N HCl at 0 °C. The reaction mixture was diluted with MeOH and filtered. The residue was purified by column chromatography to give the desired product.
4,4′-(5-Methoxy-2-(pyridin-3-yl)pent-1-ene-1,1-diyl)diphenol (12a). (0.4 g, 90% yield). 1H-NMR (400 MHz, DMSO-d6) δ 9.53 (s, OH), 9.43 (s, OH), 8.60 (d, J = 5.6 Hz, 1H), 8.51 (s, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.81 (dd, J = 8.0, 5.6 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.49 (d, J = 11.4 Hz, 2H), 3.21 (t, J = 6.0 Hz, 2H), 3.17 (s, 3H), 2.55 (m, 2H), 1.50 (m, 2H).
4,4′-(5-Methoxy-2-(pyridin-4-yl)pent-1-ene-1,1-diyl)diphenol (12b). (0.4 g, 82% yield). 1H-NMR (400 MHz, DMSO-d6) δ 9.60 (s, OH), 9.48 (s, OH), 8.62 (d, J = 5.0 Hz, 2H), 7.63 (d, J = 4.7 Hz, 2H), 7.01 (d, J = 6.8 Hz, 2H), 6.78 (d, J = 6.9 Hz, 2H), 6.65 (d, J = 6.9 Hz, 2H), 6.51 (d, J = 6.9 Hz, 2H), 3.21 (t, J = 5.0 Hz, 2H), 3.13 (s, 3H), 2.58 (m, 2H), 1.47 (m, 2H).
4,4′-(5-Methoxy-2-(1-methyl-1H-pyrazol-4-yl)pent-1-ene-1,1-diyl)diphenol (12c). (0.2 g, 91% yield). 1H-NMR (400 MHz, DMSO-d6) δ 7.24 (s, 1H), 6.92 (d, J = 6.6 Hz, 2H), 6.79 (d, J = 6.6 Hz, 2H), 6.69 (d, J = 6.6 Hz, 2H), 6.67 (s, 1H), 6.61 (d, J = 6.6 Hz, 2H), 3.68 (s, 3H), 3.21 (t, J = 5.1 Hz, 2H), 3.14 (s, 1H), 2.30 (m, 2H), 1.59 (m, 2H).
3.2.8. General Synthetic Procedure for 13a–c
To a solution of 12a–c (1 eq) in acetone/H2O (10:1) was added 2-chloro-N,N-dimethylethylamine hydrochloride (2 eq) and K2CO3 (2 eq). The mixture was refluxed at 63 °C for 4 h and concentrated under reduced pressure. The crude E/Z mixture (1:1) was purified by preparative HPLC using eluent A (0.1% TFA H2O) and eluent B (0.1% TFA ACN) (A/B = 70/30) to give the desired Z isomer product.
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-methoxy-2-(pyridin-3-yl)pent-1-en-1-yl)phenol (13a). (5 mg, 5% yield). 1H-NMR (400 MHz, CD3OD) δ 8.23 (d, J = 3.1 Hz, 1H), 8.18 (s, 1H), 7.68 (d, J = 6.4 Hz, 1H), 7.28 (dd, J = 6.2, 4.0 Hz, 1H), 7.16 (d, J = 6.9 Hz, 2H), 6.96 (d, J = 6.9 Hz, 2H), 6.68 (d, J = 6.8 Hz, 2H), 6.46 (d, J = 6.8 Hz, 2H), 4.14 (t, J = 4.3 Hz, 2H), 3.28 (t, J = 5.0 Hz, 2H), 3.21 (s, 3H), 2.81 (t, J = 4.3 Hz, 2H), 2.57 (m, 2H), 2.39 (s, 6H), 1.58 (m, 2H). MS m/z (ESI) 433.2 (M + 1)+ (calcd for C27H32N2O3H+ 432.56).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-methoxy-2-(pyridin-4-yl)pent-1-en-1-yl)phenol (13b). (12 mg, 5% yield). 1H-NMR (400 MHz, CD3OD) δ 8.32 (d, J = 4.9 Hz, 2H), 7.21 (d, J = 4.9 Hz, 2H), 7.17 (d, J = 6.9 Hz, 2H), 6.99 (d, J = 7.0 Hz, 2H), 6.72 (d, J = 6.9 Hz, 2H), 6.50 (d, J = 6.9 Hz, 2H), 4.18 (t, J = 4.3 Hz, 2H), 3.30 (t, J = 5.3 Hz, 2H), 3.25 (s, 3H), 2.87 (t, J = 4.3 Hz, 2H), 2.60 (m, 2H), 2.43 (s, 6H), 1.60 (m, 2H). MS m/z (ESI) 433.2 (M + 1)+ (calcd for C27H32N2O3H+ 433.56).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-methoxy-2-(1-methyl-1H-pyrazol-4-yl)pent-1-en-1-yl)-phenol (13c). (3 mg, 2% yield). 1H-NMR (400 MHz, CD3OD) δ 7.16 (s, 1H), 7.09 (d, J = 6.7 Hz, 2H), 6.91 (s, 1H), 6.90 (d, J = 6.7 Hz, 2H), 6.85 (d, J = 6.6 Hz, 2H), 6.63 (d, J = 6.6 Hz, 2H), 4.11 (d, J = 4.3 Hz, 2H), 3.73 (s, 3H), 3.35 (m, 2H), 3.25 (s, 3H), 2.80 (t, J = 4.2 Hz, 2H), 2.42 (m, 2H), 2.37 (s, 6H), 1.70 (m, 2H). MS m/z (ESI) 436.3 (M + 1)+ (calcd for C26H33N3O3H+ 436.57).
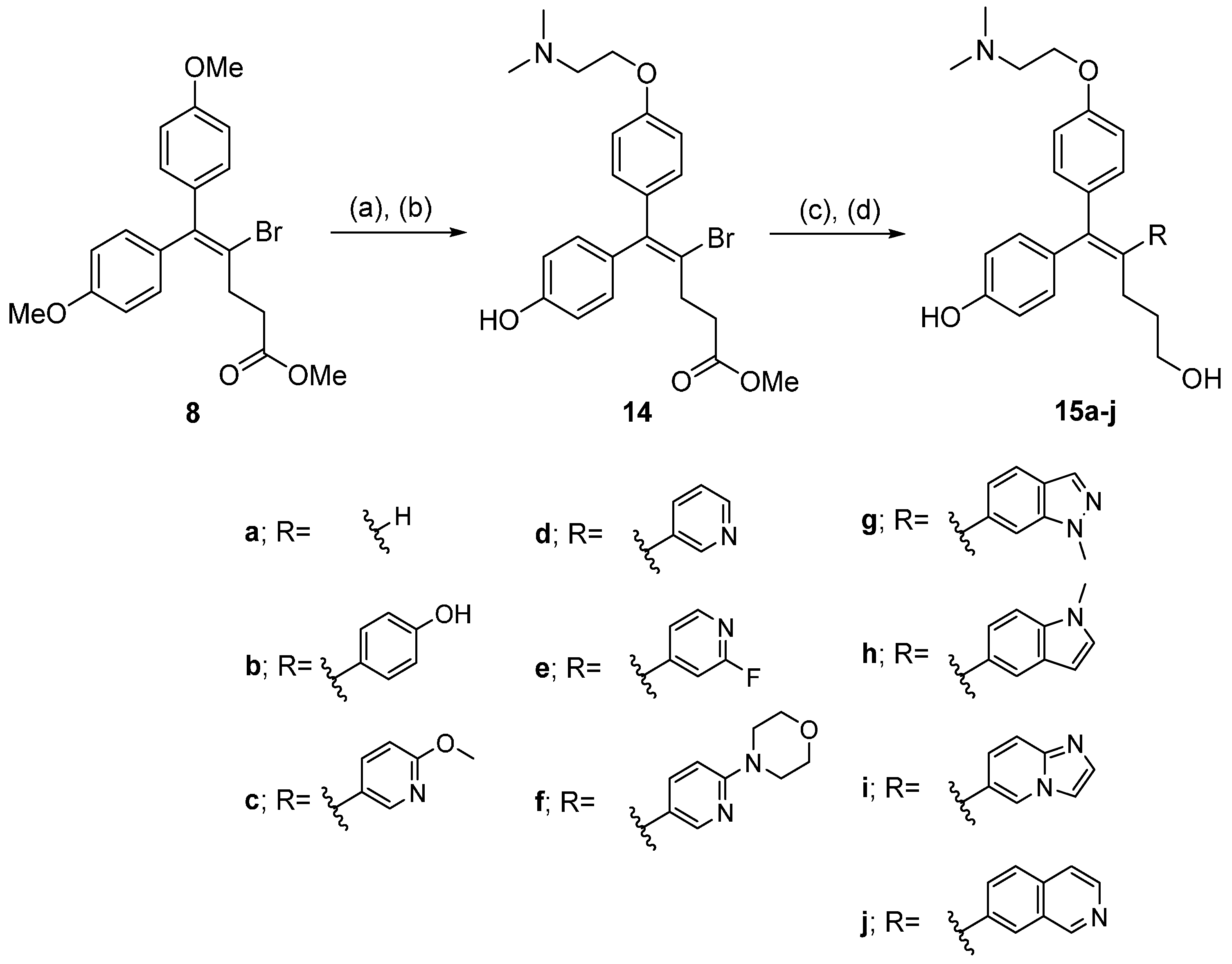
(Z)-Methyl 4-bromo-5-(4-(2-(dimethylamino)ethoxy)phenyl)-5-(4-hydroxyphenyl)pent-4-enoate (14). To a solution of 8 (4.1 g, 10.1 mmol) in CH2Cl2 (80 mL) was added 1M BBr3 in CH2Cl2 (30.3 mL, 30.3 mmol) was added dropwise at 0 °C. The mixture was stirred at rt for 3 h and quenched by adding sat. NaHCO3 solution at 0 °C. The mixture was extracted with CH2Cl2 and extracted with CH2Cl2. The combined organic layer was concentrated under reduced pressure and crude residue was purified by column chromatography. (2.4 g, 63% yield). To a solution of 2-(dimethylamino)ethanol (0.64 mL, 6.42 mmol) and triphenylphosphine (1.7 g, 6.42 mmol) in CH2Cl2 (50 mL) stirred at 0 °C. DIAD (1.3 mL, 6.42 mmol) was added dropwise. After 10 min, a solution of methyl 4-bromo-5,5-bis(4-hydroxyphenyl)pent-4-enoate (2.4 g, 6.42 mmol) in CH2Cl2 (50 mL) was added slowly to the reaction mixture at 0 °C. The reaction mixture was stirred for 12 h at rt and was removed under reduced pressure. The crude E/Z mixture (1:1) was purified by column chromatography. (0.99 g, 34% yield). 1H-NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.0 Hz, 2H), 6.98 (m, 4H), 6.72 (d, J = 8.0 Hz, 2H), 4.34 (t, J = 4.0 Hz, 2H), 3.63 (s, 3H), 3.58 (t, J = 4.0 Hz, 2H), 2.98 (s, 6H), 2.87 (m, 2H), 2.62 (m, 2H).
(E)-Methyl 4-bromo-5-(4-(2-(dimethylamino)ethoxy)phenyl)-5-(4-hydroxyphenyl)pent-4-enoate. 1H-NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.0 Hz, 2H), 7.01 (m, 4H), 6.70 (d, J = 8.0 Hz, 2H), 4.36 (t, J = 4.0 Hz, 2H), 3.63 (s, 3H), 3.59 (t, J = 4.0 Hz, 2H), 2.98 (s, 6H), 2.82 (m, 2H), 2.62 (m, 2H).
3.2.9. General Synthetic Procedure for 15a–j
To a solution of 14 (1 eq) in DMF was added corresponding boronic esters or boronic acid (1.5 eq), Pd(dppf)Cl2·CH2Cl2 (0.1 eq) and 2 M Na2CO3 (3 eq). The mixture was refluxed at 85 °C for 4 h. The reaction mixture was cooled to rt and was quenched by adding water. The mixture was extracted with EtOAc and dried over MgSO4. The crude compound was purified by column chromatography. The obtained product was dissolved in THF and added 1 M LiAlH4 in THF (1.5 eq) dropwise at 0 °C. The solution was stirred at 0 °C for 1 h and was quenched by adding H2O/5 N NaOH/H2O (1:1:3). The reaction mixture was filtered off through a Celite pad, washed with EtOAc and concentrated in vacuo. The crude compound was purified by preparative HPLC using eluent A (0.1% TFA H2O) and eluent B (0.1% TFA ACN) (A/B = 70/30) to give the desired product.
(E)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxypent-1-en-1-yl)phenol (15a). (7 mg, 22% yield). 1H-NMR (400 MHz, CD3OD) δ 7.14 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.4 Hz, 2H), 5.94 (t, J = 7.2 Hz, 1H), 4.20 (t, J = 5.6 Hz, 2H), 3.53 (t, J = 6.4 Hz, 2H), 3.16 (t, J = 5.2 Hz, 2H), 2.65 (s, 6H), 2.16 (m, 2H), 1.64 (m, 2H). MS m/z (ESI) 342.2 (M + 1)+ (calcd for C21H27NO3H+ 342.45).
(Z)-4,4′-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxypent-1-ene-1,2-diyl)diphenol (15b). (12 mg, 31% yield). 1H-NMR (400 MHz, CD3OD) δ 7.15 (d, J = 8.4 Hz, 2H), 6.99 (m, 4H), 6.92 (m, 4H), 6.82 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 6.66 (m, 4H), 6.58 (m, 4H), 6.43 (d, J = 8.8 Hz, 2H), 4.32 (t, J = 4.8 Hz, 2H), 4.18 (t, J = 5.2 Hz, 2H), 3.47 (m, 2H), 3.40 (m, 6H), 2.89 (s, 6H), 2.84 (s, 6H), 2.46 (m, 4H), 1.55 (m, 4H). MS m/z (ESI) 434.2 (M + 1)+ (calcd for C27H31NO4H+ 434.55).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(6-methoxypyridin-3-yl)pent-1-en-1-yl)phenol (15c). (21 mg, 74% yield). 1H-NMR (400 MHz, CD3OD) δ 7.77 (dd, J = 8.9, 1.7 Hz, 2H), 7.51 (dd, J = 6.9, 1.9 Hz, 2H), 7.17 (d, J = 6.9 Hz, 2H), 7.01 (m, 4H), 6.84 (d, J = 7.0 Hz, 2H), 6.77 (d, J = 6.8 Hz, 2H), 6.69 (m, 4H), 6.48 (d, J = 6.9 Hz, 2H), 4.30 (t, J = 4.0 Hz, 2H), 4.17 (t, J = 4.0 Hz, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.44 (m, 4H), 3.39 (m, 2H), 3.32 (m, 2H), 2.83 (s, 6H), 2.78 (s, 6H), 2.51 (m, 4H), 1.56 (m, 4H). MS m/z (ESI) 449.2 (M + 1)+ (calcd for C27H32N2O4H+ 449.56).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(pyridin-3-yl)pent-1-en-1-yl)phenol (15d). (2 mg, 19% yield). 1H-NMR (400 MHz, CD3OD) δ 8.21 (d, J = 3.2 Hz, 1H), 8.16 (s, 1H), 7.68 (d, J = 5.9 Hz, 1H), 7.28 (d, J = 4.2 Hz, 1H), 7.04 (d, J = 6.4 Hz, 2H), 6.78 (t, J = 7.0 Hz, 4H), 6.64 (d, J = 6.6 Hz, 2H), 4.00 (t, J = 4.2 Hz, 2H), 3.43 (t, J = 5.2 Hz, 2H), 2.81 (t, J = 4.0 Hz, 2H), 2.58 (m, 2H), 2.38 (s, 6H), 1.55 (m, 2H). MS m/z (ESI) 419.2 (M + 1)+ (calcd for C26H30N2O3H+ 419.54).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-2-(2-fluoropyridin-4-yl)-5-hydroxypent-1-en-1-yl)phenol (15e). (12 mg, 40% yield). 1H-NMR (400 MHz, CD3OD) δ 7.93 (m, 2H), 7.15 (d, J = 8.7 Hz, 2H), 7.03 (m, 4H), 6.96 (d, J = 8.7 Hz, 2H), 6.81 (m, 6H), 6.70 (m, 4H), 6.50 (d, J = 8.6 Hz, 2H), 4.16 (t, J = 5.3 Hz, 2H), 4.04 (t, J = 5.3 Hz, 2H), 3.44 (m, 4H), 2.92 (t, J = 5.3 Hz, 2H), 2.86 (t, J = 5.3 Hz, 2H), 2.58 (m, 4H), 2.46 (s, 6H), 2.42 (s, 6H), 1.54 (m, 4H). MS m/z (ESI) 437.2 (M + 1)+ (calcd for C26H29FN2O3H+ 437.53).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(6-morpholinopyridin-3-yl)pent-1-en-1-yl)phenol (15f). (7 mg, 57% yield). 1H-NMR (400 MHz, CD3OD) δ 8.54 (s, OH), 7.76 (d, J = 1.7 Hz, 1H), 7.42 (dd, J = 7.0, 1.8 Hz, 1H), 7.00 (d, J = 6.8 Hz, 2H), 6.84 (d, J = 7.0 Hz, 2H), 6.75 (d, J = 6.8 Hz, 2H), 6.67 (m, 3H), 4.09 (t, J = 4.2 Hz, 2H), 3.75 (t, J = 3.7 Hz, 4H), 3.42 (t, J = 5.3 Hz, 2H), 3.37 (t, J = 3.9 Hz, 4H), 3.07 (t, J = 3.7 Hz, 2H), 2.58 (s, 6H), 2.49 (m, 2H), 1.56 (m, 2H). MS m/z (ESI) 504.3 (M + 1)+ (calcd for C30H37N3O4H+ 504.64).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(1-methyl-1H-indazol-6-yl)pent-1-en-1-yl)phenol (
15g). (6 mg, 99% yield).
1H-NMR (400 MHz, CD
3OD) δ 7.87 (s, 1H), 7.50 (d,
J = 7.6 Hz, 1H), 7.30 (s, 1H), 7.06 (d,
J = 8.4 Hz, 2H), 6.94 (d,
J = 8.3 Hz, 1H), 6.83 (d,
J = 8.7 Hz, 2H), 6.78 (d,
J = 8.4 Hz, 2H), 6.60 (d,
J = 8.7 Hz, 2H), 4.06 (t,
J = 4.6 Hz, 2H), 3.93 (s, 3H), 3.43 (t,
J = 6.6 Hz, 2H), 3.11 (m, 2H), 2.64–2.61 (m, 7H), 1.57 (m, 2H).
13C-NMR (100 MHz, CD
3OD) δ 157.59, 156.08, 142.01, 140.09, 139.71, 139.01, 136.32, 134.57, 132.03, 131.62, 130.25, 123.53, 122.14, 119.89, 114.54, 113.02, 109.45, 64.11, 61.59, 57.32, 43.90, 33.92, 32.23, 31.75. MS
m/
z (ESI) 472.3 (M + 1)
+ (calcd for C
29H
33N
3O
3H
+ 472.60), (
Supplementary Materials; Figures S9–S12).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(1-methyl-1H-indol-6-yl)pent-1-en-1-yl)phenol (15h). (3 mg, 6% yield). 1H-NMR (400 MHz, CD3OD) δ 7.33 (d, J = 1.0 Hz, 1H), 7.19 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 1H), 7.05 (d, J = 3.1 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 6.91 (dd, J = 8.5, 1.5 Hz, 1H), 6.65 (d, J = 8.7 Hz, 2H), 6.34 (d, J = 8.7 Hz, 2H), 6.28 (d, J = 3.0 Hz, 1H), 4.36 (t, J = 5.0 Hz, 2H), 3.7 (s, 3H), 3.58 (t, J = 5.0 Hz, 2H), 3.38 (t, J = 6.8 Hz, 2H), 2.97 (s, 6H), 2.51 (m, 2H), 1.55 (m, 2H). MS m/z (ESI) 471.3 (M + 1)+ (calcd for C30H34N2O3H+ 471.61).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(imidazo[1,2-a]pyridin-6-yl)pent-1-en-1-yl)phenol (15i). (6 mg, 44% yield). 1H-NMR (400 MHz, CD3OD) δ 8.55 (s, OH), 8.22 (s, 1H), 7.69 (s, 1H), 7.50 (s, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.12 (d, J = 8.9 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.3 Hz, 2H), 4.17 (m, 2H), 3.45 (m, 4H), 2.84 (s, 6H), 2.57 (m, 2H), 1.61 (m, 2H). MS m/z (ESI) 458.2 (M + 1)+ (calcd for C28H31N3O3H+ 458.57).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-5-hydroxy-2-(isoquinolin-7-yl)pent-1-en-1-yl)phenol (15j). (3 mg, 15% yield). 1H-NMR (400 MHz, CD3OD) δ 9.03 (s, 1H), 8.53 (s, OH), 8.32 (d, J = 5.8 Hz, 1H), 7.87 (s, 1H), 7.69 (m, 2H), 7.53 (dd, J = 8.5, 1.5 Hz, 1H), 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 8.6 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 3.99 (t, J = 5.2 Hz, 2H), 3.43 (t, J = 6.6 Hz, 2H), 2.90 (t, J = 5.2 Hz, 2H), 2.68 (m, 2H), 2.45 (s, 6H), 1.57 (m, 2H). MS m/z (ESI) 469.2 (M + 1)+ (calcd for C30H32N2O3H+ 469.60).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)-3-(trifluoromethyl)phenyl)-5-hydroxy-2-(pyridin-3-yl)pent-1-en-1-yl)phenol (16). To a solution of 13a (66.5 mg, 0.15 mmol) and zinc trifluoromethanesulfinate (127 mg, 0.38 mmol) in CH2Cl2 (2.5 mL)/H2O (1.0 mL) was added TFA (12 μL, 0.15 mmol) and tert-butyl hydroperoxide (70% in H2O) (64 μL, 0.46 mmol) at 0 °C. The reaction mixture was stirred at rt for 12 h. Upon consumption of the starting material, the reaction was partitioned between CH2Cl2 (2 mL) and sat. NaHCO3 solution (2 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 and dried over MgSO4. The crude product was purified by preparative HPLC using eluent A (0.1% TFA H2O) and eluent B (0.1% TFA ACN) (A/B = 70/30) to give the desired product. (5 mg, 7% yield). 1H-NMR (400 MHz, CD3OD) δ 8.24 (s, 1H), 8.18 (s, 1H), 7.70 (d, J = 6.2 Hz, 1H), 7.28 (m, 3H), 6.94 (d, J = 6.7 Hz, 1H), 6.83 (d, J = 6.8 Hz, 2H), 6.71 (d, J = 6.8 Hz, 2H), 4.11 (t, J = 4.0 Hz, 2H), 3.28 (m, 2H), 3.21 (s, 3H), 3.16 (m, 2H), 2.65 (s, 6H), 2.57 (m, 2H), 1.58 (m, 2H). MS m/z (ESI) 487.2 (M + 1)+ (calcd for C27H29F3N2O3H+ 487.54).
(Z)-4-(1-(4-(2-(Dimethylamino)ethoxy)phenyl)-2-(6-isopropylpyridin-3-yl)-5-methoxypent-1-en-1-yl)phenol (17). To a solution of 13a (55.9 mg, 0.13 mmol) and zinc isopropylsulfinate (90 mg, 0.32 mmol) in DMSO (1.5 mL) was added TFA (10 μL, 0.13 mmol) and tert-butyl hydroperoxide (70% in H2O) (54 μL, 0.39 mmol) at 0 °C. The reaction mixture was stirred at rt for 12 h. Upon consumption of the starting material, the reaction was partitioned between EtOAc (5 mL) and sat. NaHCO3 solution (5 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc and dried over MgSO4. The crude product was purified by preparative HPLC using eluent A (0.1% TFA H2O) and eluent B (0.1% TFA ACN) (A/B = 70/30) to give the desired product. (3 mg, 4% yield). 1H-NMR (400 MHz, CD3OD) δ 8.04 (s, OH), 7.60 (d, J = 6.1 Hz, 1H), 7.17 (d, J = 6.1 Hz, 2H), 7.03 (d, J = 6.1 Hz, 2H), 6.79 (m, 3H), 6.64 (d, J = 6.7 Hz, 2H), 6.45 (d, J = 6.4 Hz, 1H), 3.99 (t, J = 4.1 Hz, 2H), 3.27 (m, 2H), 3.21 (s, 3H), 2.94 (m, 1H), 2.77 (m, 2H), 2.55 (m, 2H), 2.36 (s, 6H), 1.57 (m, 2H), 1.22 (t, J = 5.3 Hz, 6H). MS m/z (ESI) 475.3 (M + 1)+ (calcd for C30H38N2O3H+ 475.65).


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}