
Synthesis and Pharmacochemistry of New Pleiotropic Pyrrolyl Derivatives



Abstract
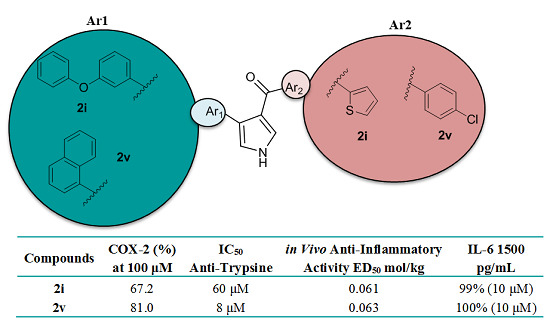
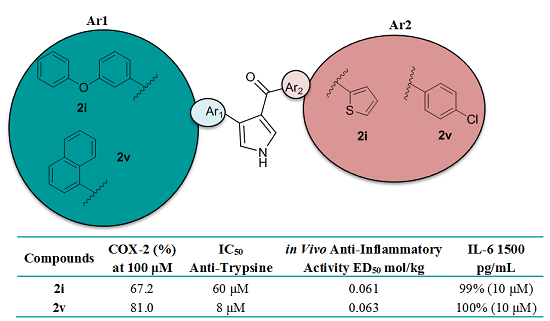
:
1. Introduction
2. Results and Discussion
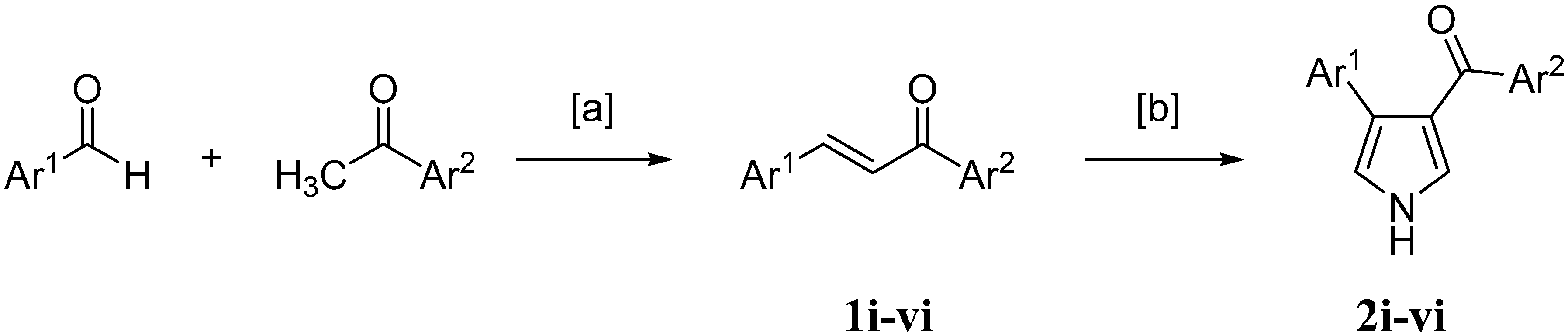
2.1. Chemistry
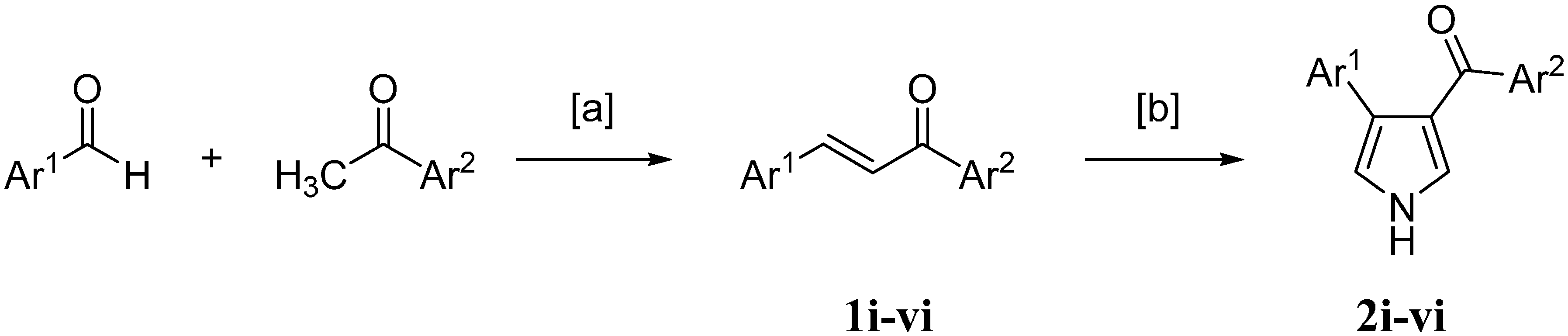

{kind=link}
{kind=link}
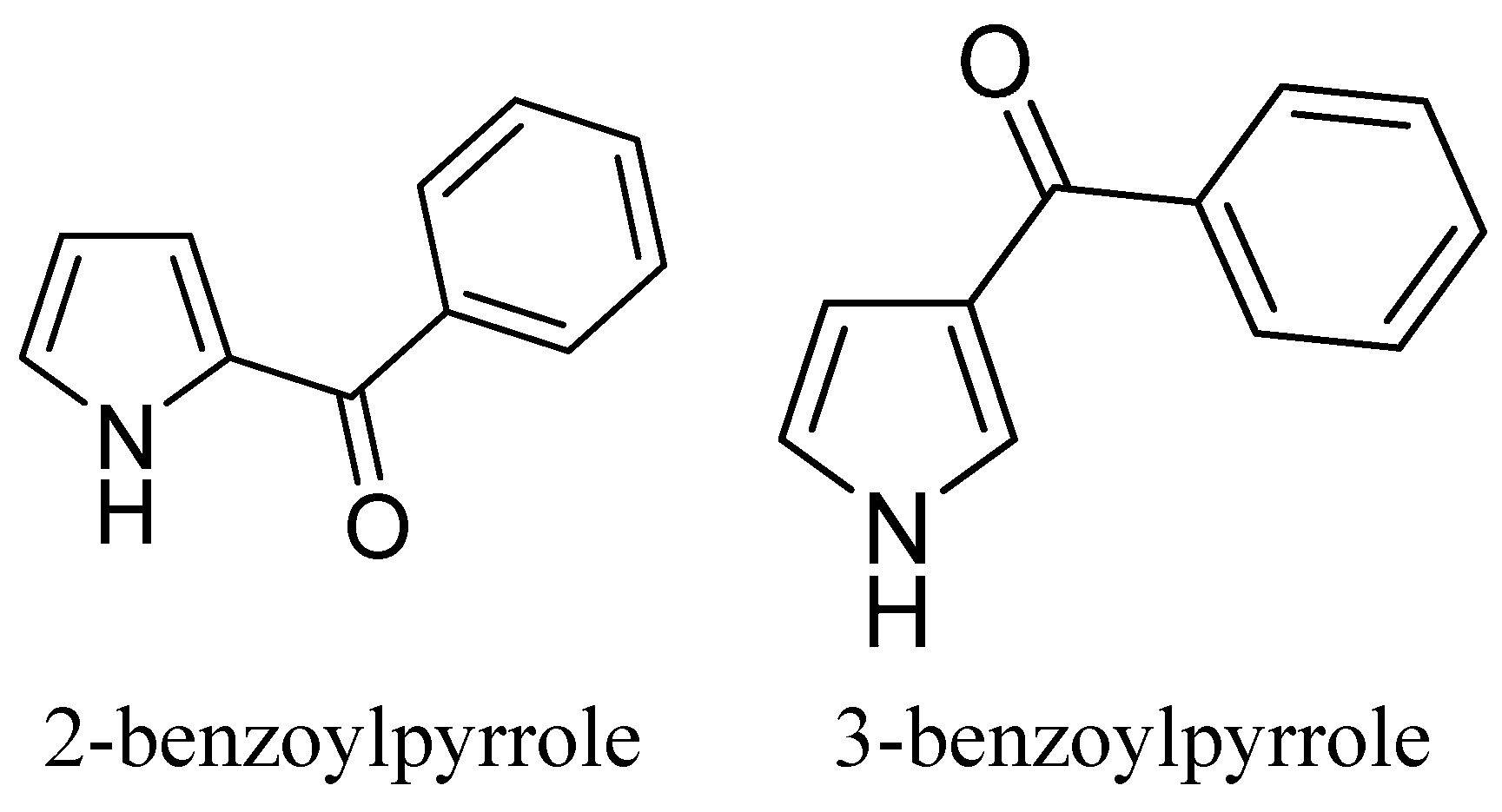{kind=link}
| No. | Ar1 | Ar2 | Rf # | m.p. (°C) | Yield (%) |
|---|---|---|---|---|---|
| 1i |  |  | 0.7 | 48–50 | 57 |
| 1ii [39] |  |  | 0.6 | 113–115 | 75 |
| 1iii |  |  | 0.8 | 141–143 | 88 |
| 1iv [40] |  |  | 0.8 | 48–50 | 78 |
| 1v [41] |  |  | 0.7 | 48–50 | 86 |
| 1vi [42] |  |  | 0.5 | 48–50 | 66 |
2.2. Physicochemical Studies
2.2.1. Determination of Lipophilicity
| No. | Ar1 | Ar2 | Clog P | RM | Rf # | m.p. (°C) | Yields (%) |
|---|---|---|---|---|---|---|---|
| 2i |  |  | 5.90 | 0.091 | 0.7 | 165–166 | 49 |
| 2ii |  |  | 5.05 | 0.073 | 0.6 | 219–220 | 96 |
| 2iii |  |  | 6.37 | −0.061 | 0.9 | 233–235 | 43 |
| 2iv |  |  | 5.26 | −0.149 | 0.8 | 270–272 | 17 |
| 2v |  |  | 6.04 | Nd | 0.9 | 285–286 | 30 |
| 2vi |  |  | 4.07 | −0.685 | 0.7 | 247–248 | 32 |
2.2.2. Theoretical Calculation of Physicochemical Properties
| No. | E(HOMO) (eV) | E(LUMO) (eV) | ΔE(HOMO-LUMO) (eV) | SM2 (eV) | Elpot_MIN (eV) | Elpot_MAX (eV) | Dipole (D) |
|---|---|---|---|---|---|---|---|
| 2i | −7.95 | 2.42 | 10.37 | 2.12 | −2.16 | 3.12 | 6.06 |
| 2ii | −7.12 | 2.77 | 9.89 | 2.02 | −2.30 | 2.86 | 4.33 |
| 2iii | −7.78 | 2.45 | 10.23 | 0.75 | −2.70 | 3.16 | 6.37 |
| 2iv | −7.21 | 2.41 | 9.62 | 2.53 | −2.26 | 2.95 | 4.13 |
| 2v | −7.29 | 2.31 | 9.6 | 2.43 | −2.13 | 3.01 | 4.99 |
| 2vi | −7.50 | -1.91 | 5.59 | 9.85 | −2.12 | 1.63 | 4.14 |
| No | CMR | MR (Ar1) | MR (Ar2) | Surface Area (Å2) | Volume (Å3) |
|---|---|---|---|---|---|
| 2i | 9.723 | 5.250 | 2.395 | 381.77 | 376.32 |
| 2ii | 9.902 | 4.860 | 2.586 | 351.86 | 343.56 |
| 2iii | 11.266 | 6.491 | 2.395 | 430.45 | 421.74 |
| 2iv | 9.328 | 4.274 | 2.586 | 345.60 | 341.68 |
| 2v | 10.404 | 4.274 | 3.077 | 363.97 | 359.95 |
| 2vi | 8.611 | 3.614 | 2.586 | 317.63 | 314.83 |
2.3. Biological Evaluation of Antioxidant and Anti-Inflammatory Activity
| Comp. | RA % (100 μΜ) | RA % (1000 μΜ) | CPE % (0.01 mmol/kg Body Weight) | ||
|---|---|---|---|---|---|
| 20 min | 60 min | 20 min | 60 min | ||
| 1i | 43 | 2 | 5 | na | 41 c |
| 1ii | 31 | 13 | 38 | 42 | 69 d |
| 1iii | na | na | na | na | 86 d |
| 1iv | na | na | na | na | 40 d |
| 1v | na | na | na | na | 44 d |
| 1vi | na | na | na | na | 42 c |
| NDGA | 81 | 83 | 96 | 96 | − |
| Indomethacin | 53 d | ||||
| Comp. | RA % (100 μΜ) | RA % (1000 μΜ) | O2•− % (100 μΜ) | LP IC50 (μΜ) | ||
|---|---|---|---|---|---|---|
| 20 min | 60 min | 20 min | 60 min | |||
| 2i | na | na | na | na | 57 | 40 |
| 2ii * | 86 | 88 | 99 | 96 | na | na |
| 2iii | na | na | 4 | 5 | 57 | 100 |
| 2iv | na | na | 8 | 10 | 76 | 72 |
| 2v | na | na | na | na | na | 150 |
| 2vi | na | na | na | na | 86 | na |
| NDGA | 81 | 83 | 95.6 | − | − | − |
| Tolmetin | 3 | 3 | 4 | 9 | − | − |
| Caffeic acid | − | − | − | − | 86.1 | 6 |
| Comp. | LOX IC50 (μΜ)/% (100 μM) | COX-2 % (100 μM) | TP IC50 (μΜ) | CPE % (0.01 mmol/kg Body Weight) or ED50 mmol/kg Body Weight |
|---|---|---|---|---|
| 2i | 1000 μΜ | 67.2 | 60 | 0.061 c |
| 2ii | na | 47 | na | 89% d |
| 2iii | na | na | 9 | 0.0525(89%) c |
| 2iv | 10% | na | 65 | 0.064 (75%) c |
| 2v | 32% | 81 | 8 | 0.063 (78%) c |
| NDGA | 43 μΜ | nt | nt | nt |
| Tolmetin | 190 μΜ | nt | nt | 76% |
| Indomethacin | − | 95 | nt | 53% d |
| Salicylic acid | − | - | 100 | nt |
| IL-6 pg/mL | Comp. 2i (10 μΜ) % Inhibition | Comp. 2v (10 μΜ) % Inhibition | IMA (10 μΜ) % Inhibition |
|---|---|---|---|
| 1500 | 99 | 100 | 100 |
| 750.0 | 98 | 99 | na |
| 375.0 | 99 | 99 | na |
| 187.5 | 99 | na | na |
| 93.7 | na | na | na |
| 46.8 | na | na | na |
| 23.4 | na | na | na |

3. Experimental Section
3.1. Materials and Instruments
3.2. Chemistry General Procedure
3.2.1. Chemistry General Procedure for Chalcones
3.2.2. General Method for the Synthesis of Pyrrolyl Derivatives
3.3. Physicochemical Studies
3.4. Biological Assays
3.4.1. Biological Assays in Vitro
Interaction with the Stable Free Radical 2,2-Diphenyl-1-picrylhydrazyl
Superoxide Radical Scavenging Activity
Inhibition of Heme-Dependent Lipid Peroxidation of Linoleic Acid
Inhibition of Soybean Lipoxygenase
Inhibition of Cycloxygenase-2 (COX-2)
Inhibition of Interleukin-6 (IL-6) (Interleukin-6 (Mouse))
Inhibition of the Proteolytic Activity of Trypsin
3.4.2. Biological Assay in Vivo
Inhibition of Rat Paw Edema Induced by Carrageenan
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References and Notes
- Salzano, S.; Checconi, P.; Hanschmann, E.M.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef] [PubMed]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord. Drug Targets 2011, 10, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Michaux, C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2003, 38, 645–659. [Google Scholar] [CrossRef]
- Botting, R.M. Mechanism of action of acetaminophen: Is there a cycloxygenase 3? Clin. Infect. Dis. 2000, 31, S202–S210. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Atukorala, I.; Hunter, D.J. Valdecoxib: The rise and fall of a COX-2 inhibitor. Expert Opin. Pharmacother. 2013, 14, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Marwali, M.R.; Mehta, J.L. COX-2 inhibitors and cardiovascular risk. Inferences based on biology and clinical studies. Thromb. Haemost. 2006, 96, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W.; Tomlinson, A.; Willoughby, D.A. Differential effects of inhibitors of cyclooxygenase (cyclooxygenase 1 and cyclooxygenase 2) in acute inflammation. Eur. J. Pharmacol. 1998, 355, 211–217. [Google Scholar] [CrossRef]
- Leone, S.; Ottani, A.; Bertolini, A. Dual acting anti-inflammatory drugs. Curr. Top. Med. Chem. 2007, 7, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Aïd, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, A.J.; Crews, B.C.; Daniel, C.M.; Blobaum, A.L.; Kingsley, P.J.; Ghebreselasie, K.; Marnett, L.J. Cyclooxygenase-1-selective inhibitors based on the (E)-2′-des-methyl-sulindac sulfide scaffold. J. Med. Chem. 2012, 55, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Asiri, A.M.; Alamry, K.A.; El-Daly, S.A.; Zayed, M.A. Eco-friendly synthesis and in vitro antibacterial activities of some novel chalcones. Bioorg. Khim. 2013, 39, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Łącka, I.; Konieczny, M.T.; Bułakowska, A.; Rzymowski, T.; Milewski, S. Antifungal action of the oxathiolone-fused chalcone derivative. Mycoses 2011, 54, 407–414. [Google Scholar] [CrossRef] [PubMed]
- De León, E.J.; Alcaraz, M.J.; Dominguez, J.N.; Charris, J.; Terencio, M.C. 1-(2,3,4-Trimethoxyphenyl)-3-(3-(2-chloroquinolinyl))-2-propen-1-one, a chalcone with analgesic, anti-inflammatory and immunomodulatory properties. Inflamm. Res. 2003, 52, 246–257. [Google Scholar] [PubMed]
- Bukhari, S.N.; Jantan, I.; Jasamai, M. Anti-inflammatory trends of 1,3-diphenyl-2-propen-1-one derivatives. Mini Rev. Med. Chem. 2013, 13, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Srinivasulu, V.; Nayak, V.L.; Sathish, M.; Shankaraiah, N.; Bagul, C.; Reddy, N.V.; Rangaraj, N.; Nagesh, N. Design and synthesis of C3-pyrazole/chalcone-linked beta-carboline hybrids: Antitopoisomerase I, DNA-Interactive and apoptosis-inducing anticancer agents. ChemMedChem 2014, 9, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.S.; Loh, W.S.; Ooi, C.W.; Quah, C.K.; Fun, H.K. Structural correlation of some heterocyclic chalcone analogues and evaluation of their antioxidant potential. Molecules 2013, 18, 1196–2011. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Song, R.; Li, Y.; Zhang, S.; Liu, Z.J.; Fu, J.; Zhu, H.L. Design, synthesis, and biological evaluation of chalcone oxime derivatives as potential immunosuppressive agents. Bioorg. Med. Chem. Lett. 2012, 22, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Okunrobo, L.O.; Usifoh, C.O.; Uwaya, J.O. Anti-inflammatory and gastroprotective properties of some chalcones. Acta Pol. Pharm. 2006, 63, 195–199. [Google Scholar] [PubMed]
- Sharma, H.; Patil, S.; Sanchez, T.W.; Neamati, N.; Schinazi, R.F.; Buolamwini, J.K. Synthesis, biological evaluation and 3D-QSAR studies of 3-keto salicylic acid chalcones and related amides as novel HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2011, 19, 2030–2045. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Christensen, S.B.; Cruciani, G.; Kharazmi, A.; Liljefors, T. Antileishmanial chalcones: Statistical design, synthesis and three-dimensional structure–activity relationship analysis. J. Med. Chem. 1998, 41, 4819–4832. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, D.; Kaiser, M.; White, K.L.; Norval, S.; Riley, J.; Wyatt, P.G.; Charman, S.A.; Read, K.D.; Yeates, C.; Gilbert, I.H. Structure-activity relationship studies of pyrrolone antimalarial agents. ChemMedChem 2013, 8, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.; Kamel, R.; Fatahala, S.S. Synthesis and biological evaluation of some thio containing pyrrolo[2,3-d]pyrimidine derivatives for their anti-inflammatory and anti-microbial activities. Eur. J. Med. Chem. 2010, 45, 2994–3004. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Choi, M.J.; Shin, J.S.; Kim, M.; Choi, H.E.; Kang, S.M.; Jin, J.H.; Lee, K.T.; Lee, J.Y. Synthesis, biological evaluation, and docking analysis of a novel family of 1-methyl-1H-pyrrole-2,5-diones as highly potent and selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, N.; Raghunathan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno[4,3-b]pyrroles and indolizino[6,7-b]indoles as potent antimicrobial and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, H.; Zhu, Q.; Jiang, S.; Liao, Y. Structure-based design, synthesis and biological evaluation of new N-carboxyphenylpyrrole derivatives as HIV fusion inhibitors targeting gp41. Bioorg. Med. Chem. Lett. 2010, 20, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xiao, Y.; Wang, F.; Xu, Y.; Qian, X.; Zhang, R.; Cui, J.; Liu, J. Novel acenaphtho[1,2-b]pyrrole-carboxylic acid family: synthesis, cytotoxicity, DNA-binding and cell cycle evaluation. Bioorg. Med. Chem. 2009, 17, 7615–7621. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Valsasina, B.; Ballinari, D.; Bertrand, J.; Brasca, M.G.; Caldarelli, M.; Cappella, P.; Fiorentini, F.; Gianellini, L.M.; Scolaro, A.; et al. 5-(2-amino-pyrimidin-4-yl)-1H-pyrrole and 2-(2-amino-pyrimidin-4-yl)-1,5,6,7-tetrahydro-pyrrolo[3,2-c]pyridin-4-one derivatives as new classes of selective and orally available Polo-like kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Weagle, G.; Gupta, A.; Bérubé, G.; Chapados, C. Evaluation of in vivo biological activities of tetrapyrrole ethanolamides as novel anticancer agents. J. Photochem. Photobiol. B. 2010, 100, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Su, M.; Wang, J.; Deng, G.; Deng, S.; Li, Z.; Tang, C.; Li, J.; Li, J.; Zhao, L.; Jiang, H.; Liu, H. Design, synthesis and biological evaluation of hetero-aromatic moieties substituted pyrrole-2-carbonitrile derivatives as dipeptidyl peptidase IV inhibitors. Eur. J. Med. Chem. 2014, 75, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Hasuoka, A.; Arikawa, Y.; Kurasawa, O.; Hirase, K.; Inatomi, N.; Hori, Y.; Sato, F.; Tarui, N.; Imanishi, A.; et al. Discovery, synthesis, and biological evaluation of novel pyrrole derivatives as highly selective potassium-competitive acid blockers. Bioorg. Med. Chem. 2012, 20, 3925–3938. [Google Scholar] [CrossRef] [PubMed]
- Bratton, L.D.; Auerbach, B.; Choi, C.; Dillon, L.; Hanselman, J.C.; Larsen, S.D.; Lu, G.; Olsen, K.; Pfefferkorn, J.A.; Robertson, A.; et al. Discovery of pyrrole-based hepatoselective ligands as potent inhibitors of HMG-CoA reductase. Bioorg. Med. Chem. 2007, 15, 5576–5589. [Google Scholar] [CrossRef] [PubMed]
- Dannhardt, G.; Kiefer, W.; Krämer, G.; Maehrlein, S.; Nowe, U.; Fiebich, B. The pyrrole moiety as a template for COX-1/COX-2 inhibitors. Eur. J. Med. Chem. 2000, 35, 499–510. [Google Scholar] [CrossRef]
- Kouskoura, M.; Hadjipavlou-Litina, D.; Giakoumakou, M. Synthesis and AntiInflammatory Activity of Chalcones and Related Mannich Bases. Med. Chem. 2008, 4, 586–596. [Google Scholar]
- Artico, M.; Di Santo, R.; Costi, R.; Massa, S.; Retico, A.; Artico, M.; Apuzzo, G.; Simonetti, G.; Strippoli, V. Antifungal agents. 9. 3-Aryl-4-[α-(1H-imidazol-1-yl)arylmethyl]pyrroles: A new class of potent anti-Candida agents. J. Med. Chem. 1995, 38, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E. Supersensitization of Photographic Emulsions. US Patent 2852385 A, 16 September 1958. [Google Scholar]
- Chiaradia, L.D.; Mascarello, A.; Purificação, M.; Vernal, J.; Cordeiro, M.N.; Zenteno, M.E.; Villarino, A.; Nunes, R.J.; Yunes, R.A.; Terenzi, H. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg. Med. Chem. Lett. 2008, 18, 6227–6230. [Google Scholar] [CrossRef] [PubMed]
- Stroba, A.; Schaeffer, F.; Hindie, V.; Lopez-Garcia, L.; Adrian, I.; Fröhner, W.; Hartman, R.W.; Biondi, R.M.; Engel, M. 3,5-Diphenylpent-2-enoic acids as allosteric activators of the protein kinase PDK1: Structure-activity relationships and thermodynamic characterization of binding as paradigms for PIF-binding pocket-targeting compound. J. Med. Chem. 2009, 52, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Cocconcelli, G.; Diodato, E.; Caricasole, A.; Gaviraghi, G.; Genesio, E.; Ghiron, C.; Magnoni, L.; Pecchioli, E.; Plazzi, P.V.; Terstappen, G.C. Aryl azoles with neuroprotective activity-parallel synthesis and attempts at target identification. Bioorg. Med. Chem. 2008, 16, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Rekker, R. Hydrophobic Fragmental Constant; Elsevier Scientific Co.: Amsterdam, The Netherlands, 1977; Volume 1, p. 19. [Google Scholar]
- Hansch, C.; Leo, A.J. Exploring QSAR Fundamentals and Applications in Chemistry and Biolorgy; Heller, S.R., Ed.; ACS Professional Reference Book: Washington, DC, USA, 1995; Volume I, pp. 279–280. [Google Scholar]
- BioByte Corporation, C-QSAR database, 201 W Fourth str. Suite # 204, Claremont, CA 91711–4707, USA.
- Flohe, L.; Beckman, R.; Giertz, H.; Loschen, G. Oxygen-centered free radicals as mediators of inflammation. In Oxidative Stress; Sies, H., Ed.; Academic Press: London, UK, 1985; pp. 403–435. [Google Scholar]
- Niki, E. Do antioxidants impair signaling by reactive oxygen species and lipid oxidation products? FEBS Lett. 2012, 586, 3767–3770. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.P.; Cecchini, R.; Halliwell, B. Oxidative damage to lipids and alpha 1-antiproteinase by phenylbutazone in the presence of haem proteins: Protection by ascorbic acid. Biochem. Pharmacol. 1992, 44, 981–984. [Google Scholar] [CrossRef]
- Crooks, S.W.; Stockley, R.A. Leukotriene B4. Int. J. Biochem. Cell. Biol. 1998, 30, 173–178. [Google Scholar] [CrossRef]
- Peperidou, A.; Kapoukranidou, D.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Multitarget Molecular Hybrids of Cinnamic Acids. Molecules 2014, 19, 20197–20226. [Google Scholar] [CrossRef] [PubMed]
- Demopoulos, V.J.; Rekka, E. Isomeric benzoylpyrroloacetic acids: Some structural aspects for aldose reductase inhibitory and anti-inflammatory activities. J. Pharm. Sci. 1995, 84, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Antioxidant and anti-inflammatory activity of aryl-acetic and hydroxamic acids as novel lipoxygenase inhibitors. Med. Chem. 2006, 2, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Kulmacz, R.J.; Lands, W.E.M. Requirements for hydroperoxide by the cyclooxygenase and peroxidase activities of prostaglandin H synthase. Prostaglandins 1983, 25, 531–540. [Google Scholar]
- Michaelidou, A.; Hadjipavlou-Litina, D.; Matsini, I.; Tsitsogianni, E. Heterocyclic aryl(phenyl)acetic acid and aryl acetohydroxamic acids as antiinflammatory-antioxidant agents and inhibitors of lipoxygenase and serine proteases. Med. Chem. 2007, 3, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1i–1vi and 2i–2vi are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konstantinidou, M.; Gkermani, A.; Hadjipavlou-Litina, D. Synthesis and Pharmacochemistry of New Pleiotropic Pyrrolyl Derivatives. Molecules 2015, 20, 16354-16374. https://doi.org/10.3390/molecules200916354
Konstantinidou M, Gkermani A, Hadjipavlou-Litina D. Synthesis and Pharmacochemistry of New Pleiotropic Pyrrolyl Derivatives. Molecules. 2015; 20(9):16354-16374. https://doi.org/10.3390/molecules200916354
Chicago/Turabian StyleKonstantinidou, Markella, Alice Gkermani, and Dimitra Hadjipavlou-Litina. 2015. "Synthesis and Pharmacochemistry of New Pleiotropic Pyrrolyl Derivatives" Molecules 20, no. 9: 16354-16374. https://doi.org/10.3390/molecules200916354
APA StyleKonstantinidou, M., Gkermani, A., & Hadjipavlou-Litina, D. (2015). Synthesis and Pharmacochemistry of New Pleiotropic Pyrrolyl Derivatives. Molecules, 20(9), 16354-16374. https://doi.org/10.3390/molecules200916354