
Synthetic Routes to N-9 Alkylated 8-Oxoguanines; Weak Inhibitors of the Human DNA Glycosylase OGG1

Abstract
:
1. Introduction
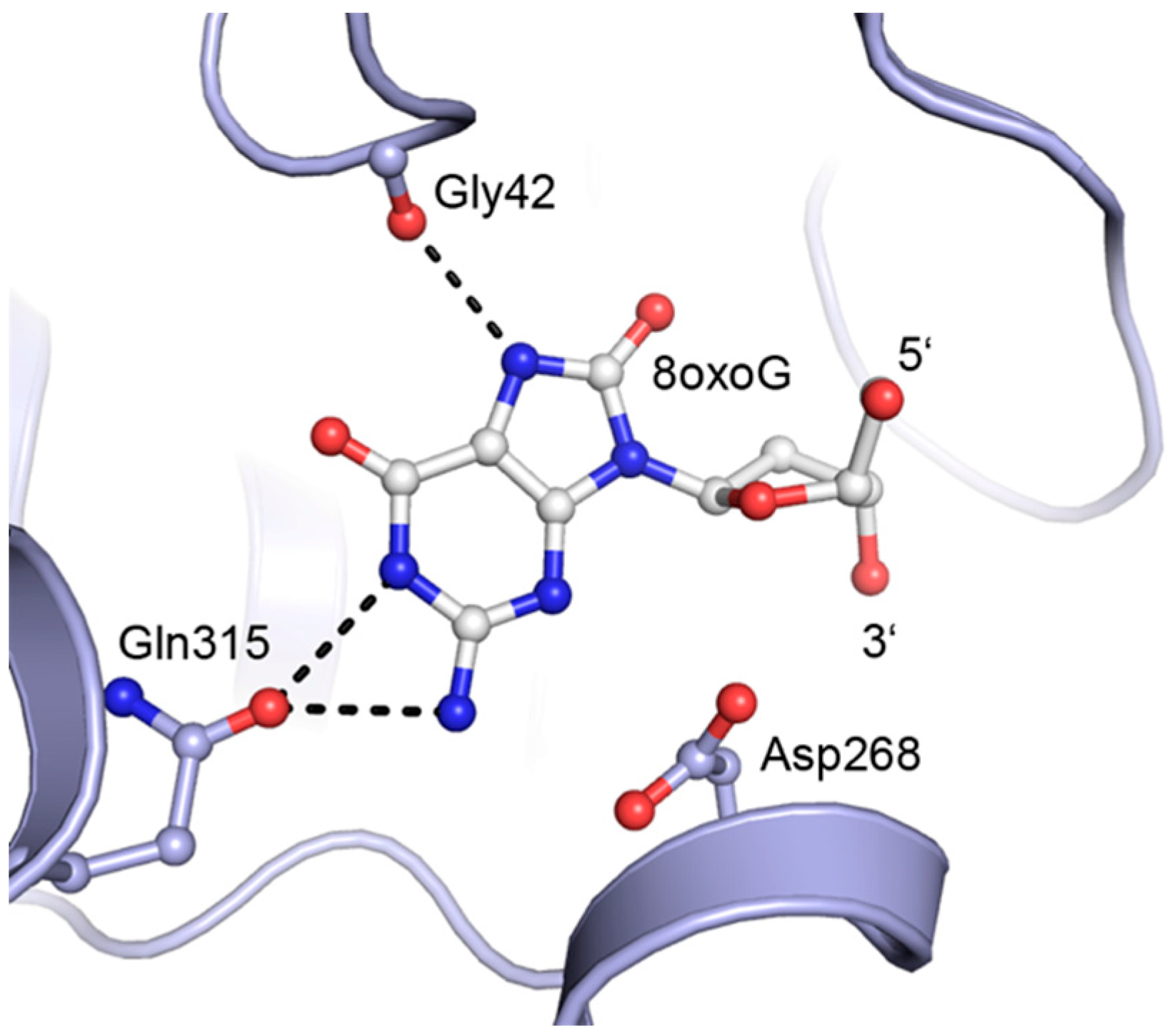
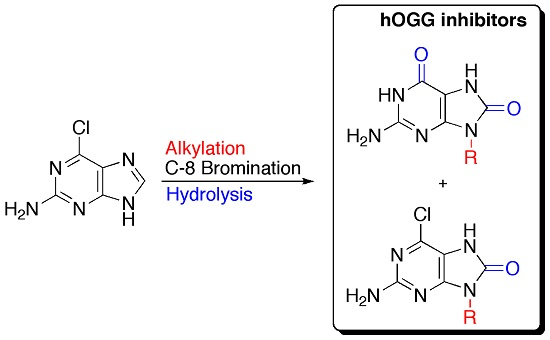
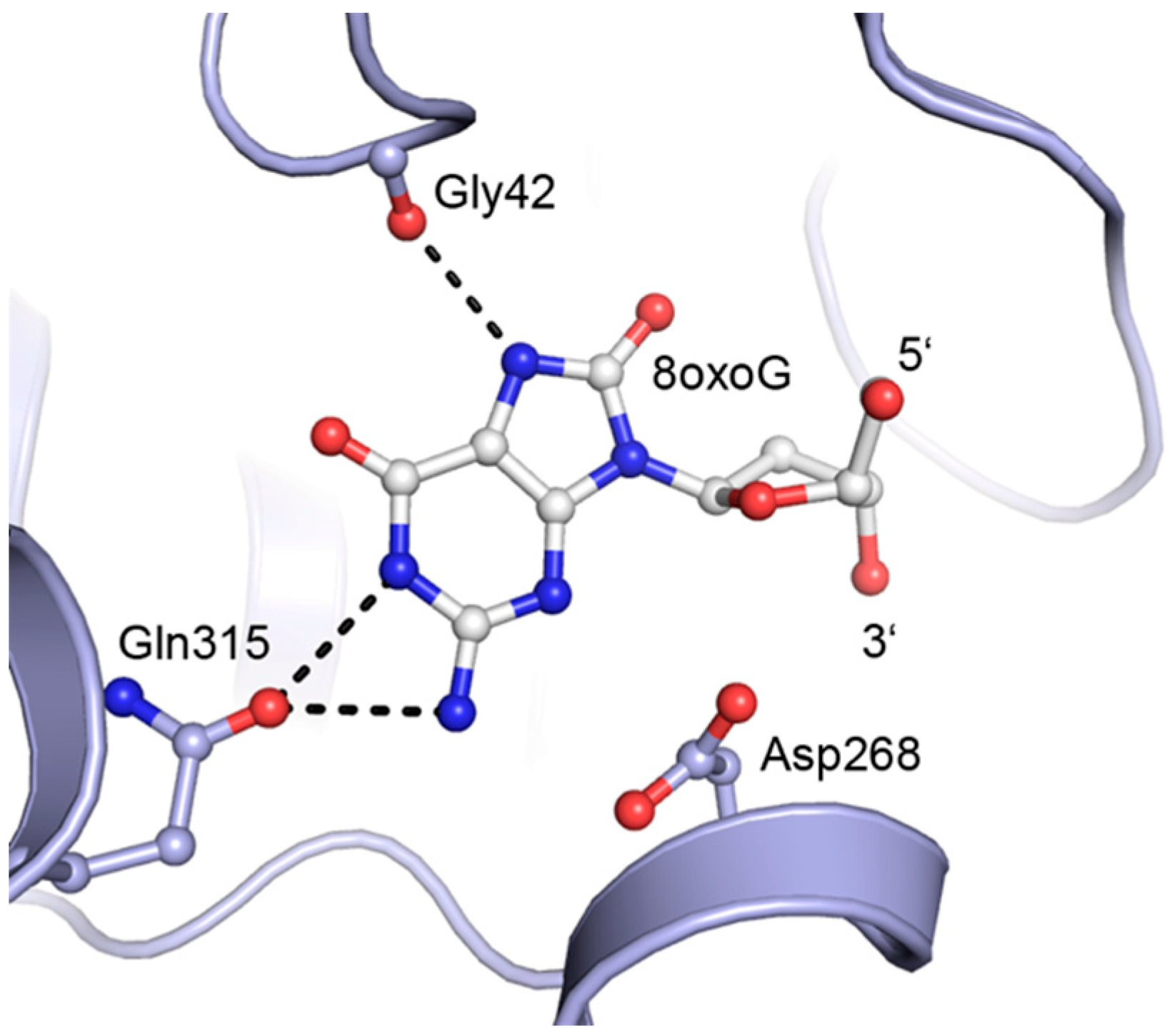
2. Results and Discussion
2.1. Chemistry


{kind=link}
{kind=link}
{kind=link}
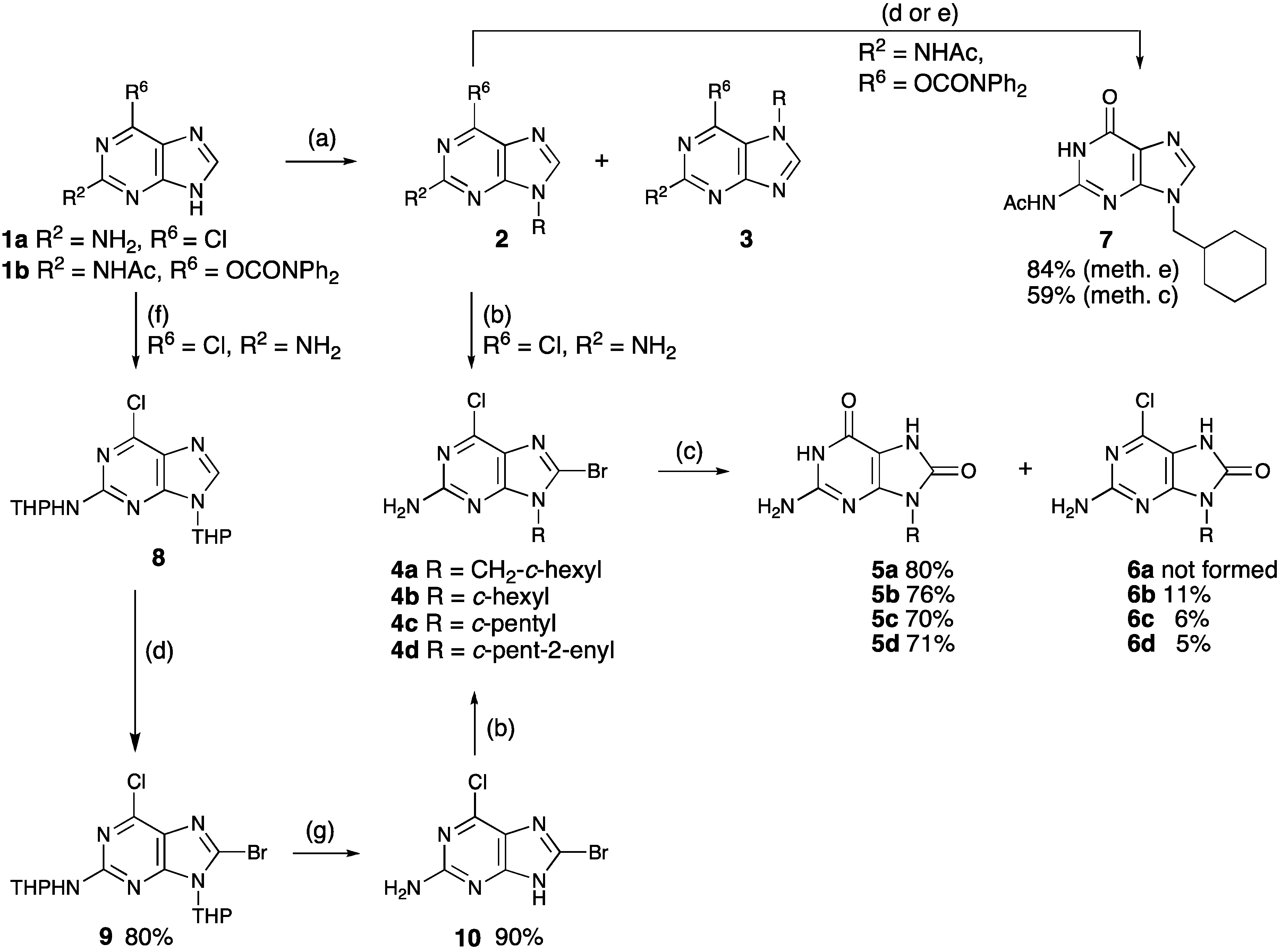{kind=link}
| Entry | R2 | R6 | R | Reagents and Conditions | Ratio 2:3:1 a | Yield (%) 2 b | Yield (%) 3 b |
|---|---|---|---|---|---|---|---|
| 1 | Cl | NH2 | CH2-c-hexyl | RBr, K2CO3, DMF, rt, 72 h | 80:20:0 | 67, 2a | 10, 3a |
| 2 | Cl | NH2 | CH2-c-hexyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | 93:7:0 | 76, 2a | 5, 3a |
| 3 | OCONPh2 | NHAc | CH2-c-hexyl | RBr, K2CO3, DMF, rt, 72 h | 81:19:0 | 45, 2e | 7, 3e |
| 4 | OCONPh2 | NHAc | CH2-c-hexyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | 82:18:0 | 70, 2e | 3, 3e |
| 5 | Cl | NH2 | c-hexyl | RI, K2CO3, DMF, rt, 72 h | 15:0:85 | – c | – |
| 6 | Cl | NH2 | c-hexyl | ROTs, K2CO3, DMF, rt, 72 h | – d | 33, 2b | – c |
| 7 | Cl | NH2 | c-hexyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | 8:4:88 | – c | – c |
| 8 | Cl | NH2 | c-hexyl | ROH, DIAD, PPh3, THF, ultrasound, 14 h | 27:0:73 | 20, 2b | – |
| 9 | Cl | NH2 | c-hexyl | ROH, DIAD, PPh3, DMF, 150 °C, μW, 2 h | 41:8:51 | – c | – c |
| 10 | OCONPh2 | NHAc | c-hexyl | ROTs, K2CO3, THF, rt, 72 h | – d | 30, 2f | – c |
| 11 | OCONPh2 | NHAc | c-hexyl | ROTs, K2CO3, DMF, 80 °C, 72 h | – d,e | – c | – c |
| 12 | OCONPh2 | NHAc | c-hexyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | – d | 22, 2f | – c |
| 13 | Cl | NH2 | c-pentyl | RBr, K2CO3, DMF, rt, 72 h | 86:14:0 | 71, 2c | 5, 3c |
| 14 | Cl | NH2 | c-pentyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | 91:9:0 | 72, 2c | 6, 3c |
| 15 | OCONPh2 | NHAc | c-pentyl | RBr, K2CO3, DMF, rt, 72 h | 76:15:09 | 52, 2g | – c |
| 16 | OCONPh2 | NHAc | c-pentyl | ROH, DIAD, PPh3, THF, 70 °C, 14 h | 90:10:0 | 58, 2g | – c |
| 17 | Cl | NH2 | c-pent-2-enyl | RBr, K2CO3, DMF, rt, 24 h | 23:16:61 | 18, 2d | – c |
| 18 | Cl | NH2 | c-pent-2-enyl | ROH, DIAD, PPh3, THF, 70 °C, 42 h | 55:18:27 | 40, 2d | – c |
| 19 | Cl | NH2 | c-pent-2-enyl | ROAc, Pd(PPh3)4,NaH, DMSO, f 50 °C, 48 h | 75:25:0 | 53, 2d | 18, 3d |
| Entry | Starting Material a | Reagents and Conditions | Yield (%) 4 a,b |
|---|---|---|---|
| 1 | 2a | Br2, H2O | 79%, 4a |
| 2 | 10 | RBr, K2CO3, DMF | 34%, 4a |
| 3 | 10 | ROH, DIAD, PPh3, THF, 70 °C | 56%, 4a |
| 4 | 2b | Br2, H2O | 70%, 4b |
| 5 | 2c | Br2, H2O | 81%, 4c |
| 6 | 2d | 1. LDA, 2. CCl2BrCCl2Br, THF, −78 °C | 32%, 4d |
| 7 | 10 | ROH, DEAD, PPh3, THF, 70 °C | 42%, 4d |
| 8 | 10 | ROAc, Pd(PPh3)4, NaH, DMF, 50 °C | 29%, 4d |
2.2. Biology

| Compound | X | R | % Activity |
|---|---|---|---|
| 5a | OH a | CH2-c-hexyl | 89 ± 5 |
| 5b | OH a | c-hexyl | 92 ± 2 |
| 6b | Cl | c-hexyl | 70 ± 11 |
| 5c | OH | c-pentyl | 101 ± 12 |
| 6c | Cl | c-pentyl | 72 ± 9 |
| 5d | OH | c-pent-2-enyl | 92 ± 7 |
| 6d | Cl | c-pent-2-enyl | 84 ± 3 |
| Compound | X | R | % Activity |
|---|---|---|---|
| 5a | OH a | CH2-c-hexyl | 96 ± 3 |
| 5b | OH a | c-hexyl | 123 ± 20 |
| 6b | Cl | c-hexyl | 73 ± 37 |
| 5c | OH | c-pentyl | 102 ± 16 |
| 6c | Cl | c-pentyl | 108 ± 18 |
| 5d | OH | c-pent-2-enyl | 104 ± 21 |
| 6d | Cl | c-pent-2-enyl | 89 ± 13 |
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. 2-Amino-6-chloro-9-(cyclohexylmethyl)-9H-purine (2a) and 2-Amino-6-chloro-7-(cyclohexylmethyl)-7H-purine (3a)
3.2.2. 2-Amino-6-chloro-9-(cyclohexyl)-9H-purine (2b)
3.2.3. 2-Amino-6-chloro-9-(cyclopentyl)-9H-purine (2c) and 2-Amino-6-chloro-7-(cyclopentyl)-7H-purine (3c)
3.2.4. 2-Amino-6-chloro-9-(cyclopent-2-enyl)-9H-purine (2d) and 2-Amino-6-chloro-7-(cyclopent-2-enyl)-7H-purine (3d)
3.2.5. 2-Acetamido-9-(cyclohexylmethyl)-9H-purin-6-yl diphenylcarbamate (2e) and 2-Acetamido-7-(cyclohexylmethyl)-7H-purin-6-yl diphenylcarbamate (3e)
3.2.6. 2-Acetamido-9-(cyclohexyl)-9H-purin-6-yl diphenylcarbamate (2f)
3.2.7. 2-Acetamido-9-(cyclopentyl)-9H-purin-6-yl diphenylcarbamate (2g)
3.2.8. 2-Amino-8-bromo-6-chloro-9-(cyclohexylmethyl)-9H-purine (4a)
3.2.9. 2-Amino-8-bromo-6-chloro-9-(cyclohexyl)-9H-purine (4b)
3.2.10. 2-Amino-8-bromo-6-chloro-9-(cyclopentyl)-9H-purine (4c)
3.2.11. 2-Amino-8-bromo-6-chloro-9-(cyclopent-2-enyl)-9H-purine (4d)
3.2.12. 9-(Cyclohexylmethyl)-8-oxoguanine (5a)
3.2.13. 9-(Cyclohexyl)-8-oxoguanine (5b) and 2-Amino-6-chloro-9-cyclohexyl-7H-purin-8(9H)-one (6b)
3.2.14. 9-(Cyclopentyl)-8-oxoguanine (5c) and 2-Amino-6-chloro-9-cyclopentyl-7H-purin-8(9H)-one (6c)
3.2.15. 9-(Cyclopent-2-enyl)-8-oxoguanine (5d) and 2-Amino-6-chloro-9-(cyclopent-2-enyl)-7H-purin-8(9H)-one (6d)
3.2.16. N-[9-(Cyclohexylmethyl)-6-oxo-6,9-dihydro-1H-purin-2-yl]acetamide (7)
3.2.17. 8-Bromo-6-chloro-N,9-bis(tetrahydro-2H-pyran-2-yl)-9H-purin-2-amine (9)
3.2.18. 2-Amino-8-bromo-6-chloro-1H-purine (10)
3.3. DNA Glycosylase Activity Assay
4. Conclusions
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chabner, B.A.; Roberts, T.G., Jr. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Middleton, M.R. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat. Rev. 2005, 31, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin. Cancer Res. 2010, 16, 4527–4531. [Google Scholar] [CrossRef] [PubMed]
- Pallis, A.G.; Karamouzis, M.W. DNA repair pathways and their implication in cancer treatment. Cancer Metast. Rev. 2010, 29, 677–685. [Google Scholar] [CrossRef] [PubMed]
- See for instance: Dalhus, B.; Lærdahl, J.K.; Backe, P.H.; Bjørås, M. DNA base repair–recognition and initiation of catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078. [Google Scholar]
- Bruner, S.D.; Norman, D.P.G.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [PubMed]
- Siah, H.-S.M.; Gundersen, L.-L.; Gørbitz, C.H. NMR and X-ray structural studies on 3-benzyl-8-bromoadenine. J. Heterocycl. Chem. 2011, 48, 1375–1378. [Google Scholar] [CrossRef]
- Siah, H.-S.M.; Gundersen, L.-L. Synthetic strategies to 9-substituted 8-oxoadenines. Synth. Commun. 2013, 43, 1469–1476. [Google Scholar] [CrossRef]
- Perini, F.; Tieckelmann, H. Conversion of ureidomalonates and 5-carbalkoxyhydantoins to 5-ureido-4,6-pyrimidinediones. J. Org. Chem. 1970, 35, 812–816. [Google Scholar] [CrossRef]
- Brown, R.; Joseph, M.; Leigh, T.; Swain, M.L. Synthesis and reactions of 7,8-dihydro-8-methylpterin and 9-methylguanine 7-oxide. J. Chem. Soc. Perkin 1977, 1, 1003–1009. [Google Scholar] [CrossRef]
- Müller, H.; Carell, T.A. Carbocyclic analog of the oxidatively generated DNA lesion spiroiminodihydantoin. Eur. J. Org. Chem. 2007, 1438–1445. [Google Scholar] [CrossRef]
- Robins, M.J.; Hatfield, P.W.; Balzarini, J.; de Clercq, E. Nucleic acid related compounds. 47. Synthesis and biological activities of pyrimidine and purine “acyclic” nucleoside analogs. J. Med. Chem. 1984, 27, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.A.; Cottam, H.B.; Smee, D.F.; Robins, R.K.; Kini, G.D. Alkylpurines as immunopotentiating agents. Synthesis and antiviral activity of certain alkylguanines. J. Med. Chem. 1993, 36, 3431–3437. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.-H.; Wang, J.-Q.; Fei, X.; Hutchins, G.D. Synthesis of 8-methoxypenciclovir and 8-methoxyganciclovir through methyl triflate, a new potential approach to label penciclovir and ganciclovir with carbon-11. Synthesis 2003, 2785–2794. [Google Scholar]
- DeClue, M.S.; Monnard, P.-A.; Bailey, J.A.; Maurer, S.E.; Collins, G.E.; Ziock, H.-J.; Rasmussen, S.; Boncella, J.M. Nucleobase mediated, photocatalytic vesicle formation from an ester precursor. J. Am. Chem. Soc. 2009, 131, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Kaiya, T.; Ohta, M.; Kohda, K. Electrophilic amination of imidazole moieties of 9-ethylguanine and 1-methylbenzimidazole derivatives and reactivities of N-aminated products. Tetrahedron 1993, 49, 8795–8804. [Google Scholar] [CrossRef]
- Werbovetz, K.A.; Macdonald, T.L. On the mechanism of arylamine-DNA adduct formation. Bioorg. Med. Chem. Lett. 1994, 4, 2323–2326. [Google Scholar] [CrossRef]
- Kaiya, T.; Fujiwara, T.; Kohda, K. Syntheses and properties of 1-methyl-3-phenylaminobenzimidazolium salts, models of DNA adducts of N7-arylaminodeoxyguanosinium salt. Chem. Res. Toxicol. 2000, 13, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Ferenc, G.; Padar, P.; Szolomajer, J.; Kovacs, L. N-alkylated guanine derivatives. Curr. Org. Chem. 2009, 13, 1085–1135. [Google Scholar] [CrossRef]
- Kjellberg, J.; Liljenberg, M.; Johansson, N.G. Regioselective alkylation of 6-(β-methoxyethoxy)guanine to give the 9-alkylguanine derivative. Tetrahedron Lett. 1986, 27, 877–880. [Google Scholar] [CrossRef]
- Kjellberg, J.; Hagberg, C.E.; Malm, A.; Noren, J.O.; Johansson, N.G. Studies on the Alkylation of Guanine. 2. The synthesis of acyclic guanosine analogs via the precursor 7-methyl-10-oxo-9,10-dihydropyrimido[1,2-a]purine. Acta Chem. Scand. B 1986, 40, 310–312. [Google Scholar] [CrossRef]
- Kjellberg, J.; Johansson, N.G. Regioselective alkylation of guanine via diacyloxyglyoxal-N2-acetylguanine adduct to obtain 7-alkylguanine derivatives. Studies on alkylation of guanine I. J. Heterocycl. Chem. 1986, 23, 625–627. [Google Scholar] [CrossRef]
- Kjellberg, J.; Johansson, N.G. Studies on the alkylation of derivatives of guanines. Nucleosides Nucleotides 1989, 8, 225–256. [Google Scholar] [CrossRef]
- Geen, G.R.; Kincey, P.M.; Choudary, B.M. Regiospecific Michael additions with 2-aminopurines. Tetrahedron Lett. 1992, 33, 4609–4612. [Google Scholar] [CrossRef]
- Bisacchi, G.S.; Singh, J.; Godfrey, J.D., Jr.; Kissick, T.P.; Mitt, T.; Malley, M.F.; di Marco, J.D.; Gougoutas, J.Z.; Mueller, R.H.; Zahler, R. Regioselective coupling of tetraalkylammonium salts of 6-iodo-2-aminopurine to a cyclobutyl triflate: Efficient preparation of homochiral BMS-180,194, a potent antiviral carbocyclic nucleoside. J. Org. Chem. 1995, 60, 2902–2905. [Google Scholar] [CrossRef]
- Geen, G.R.; Kincey, P.M.; Spoors, P.G. Regioselective alkylation of guanines using 2-acetoxytetrahydrofurans. Tetrahedron Lett. 2001, 42, 1781–1784. [Google Scholar] [CrossRef]
- Zou, R.; Robbins, M.J. High-yield regioselective synthesis of 9-glycosyl guanine nucleosides and analogues via coupling with 2-N-acetyl-6-O-diphenylcarbamoylguanine. Can. J. Chem. 1987, 65, 1436–1437. [Google Scholar] [CrossRef]
- Dalpozzo, R.; de Nino, A.; Maiuolo, L.; Procopio, A.; de Munno, G.; Sindona, G. 9-Vinylguanine: an easy access to aza-analogs of 2′,3′-dideoxyguanosine. Tetrahedron 2001, 57, 4035–4038. [Google Scholar] [CrossRef]
- Hocek, M.; Holy, A. A facile synthesis of 6-cyanopurine bases. Collect. Czech. Chem. Commun. 1995, 60, 1386–1389. [Google Scholar] [CrossRef]
- Langli, G.; Gundersen, L.-L.; Rise, F. Regiochemistry in Stille couplings of 2,6-dihalopurines. Tetrahedron 1996, 52, 5625–5638. [Google Scholar] [CrossRef]
- Brændvang, M.; Gundersen, L.-L. Synthesis, biological activity and SAR of antimycobacterial 2- and 8-substituted 6-(2-furyl)-9-(p-methoxybenzyl)purines. Bioorg. Med. Chem. 2007, 15, 7144–7165. [Google Scholar] [CrossRef] [PubMed]
- Toyota, A.; Katagiri, N.; Kaneko, C. Mitsunobu reactions for the synthesis of carbocyclic analogues of nucleosides: Examination of the regioselectivity. Synth. Commun. 1993, 23, 1295–1305. [Google Scholar] [CrossRef]
- Toyota, A.; Katagiri, N.; Kaneko, C. Synthesis of nucleosides and related compounds. 31. The alkylation of 2-amino-6-chloropurine with alcohols by Mitsunobu reaction for a synthesis of carbocyclic guanosine analogs. Heterocycles 1993, 36, 1625–1630. [Google Scholar] [CrossRef]
- Maruyama, T.; Kozai, S.; Uchida, M. Synthesis of N-aryl uracils and hypoxanthines and their biological properties. Nucleosides Nucleotides 1999, 18, 661–671. [Google Scholar] [CrossRef]
- Lu, W.; Sengupta, S.; Peterson, J.L.; Akhmedow, N.G.; Shi, X. Mitsunobu coupling of nucleobases and alcohols: An efficient, practical synthesis for novel nonsugar carbon nucleosides. J. Org. Chem. 2007, 72, 5012–5015. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Madsen, R.; Guile, S.G.; Brown, B. Palladium-catalyzed enantioselective synthesis of carbanucleosides. J. Am. Chem. Soc. 2000, 122, 5947–5956. [Google Scholar] [CrossRef]
- Choo, H.; Chong, Y.; Chu, C.K. Solid phase synthesis of carbocyclic l-2′-deoxynucleosides. Org. Lett. 2001, 3, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Guillarme, S.; Legoupy, S.; Aubertin, A.-M.; Olicard, C.; Bourgougnon, N.; Huet, F. Rapid access to acyclic nucleosides via conjugate addition. Tetrahedron 2003, 59, 2177–2184. [Google Scholar] [CrossRef]
- Velcicky, J.; Lanver, A.; Lex, J.; Prokop, A.; Wieder, T.; Schmalz, H.-G. Transition-metal-mediated synthesis of novel carbocyclic nucleoside analogues with antitumoral activity. Chem. Eur. J. 2004, 10, 5087–5110. [Google Scholar] [CrossRef] [PubMed]
- Pautus, S.; Sehr, P.; Lewis, J.; Fortune, A.; Wolkerstorfer, A.; Szolar, O.; Guilligay, D.; Lunardi, T.; Decout, J.-L.; Cusack, S. New 7-methylguanine derivatives targeting the influenza polymerase PB2 Cap-binding domain. J. Med. Chem. 2013, 56, 8915–8930. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Chambers, V.C. Small-ring compounds. VIII. Some nucleophilic displacement reactions of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl p-toluenesulfonates and halides. J. Am. Chem. Soc. 1951, 73, 5034–5040. [Google Scholar] [CrossRef]
- For a review of Pd-catalyzed N-allylation of purines, see for instance: Gundersen, L.-L. Metal-mediated C–C and C–N bond formation in the synthesis of bioactive purines. Targets Heterocycl. Syst. 2008, 12, 85–119. [Google Scholar]
- Kobayashi, S.; Kawamoto, T.; Uehara, S.; Fukuyama, T.; Ryu, I. Black-light-induced radical/ionic hydroxymethylation of alkyl iodides with atmospheric CO in the presence of tetrabutylammonium borohydride. Org. Lett. 2010, 12, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Gamadeku, T.; Gundersen, L.-L. Synthesis of 8-bromo-N-benzylpurines via 8-lithiated purines; Scopes and Limitations. Synth. Commun. 2010, 40, 2723–2735. [Google Scholar] [CrossRef]
- Marzouk, V.H.R.; Hennum, M.; Gundersen, L.-L. Efficient synthesis of cytotoxic pyrido[1,2-e]purines from purines employing direct C-allylation and RCM-oxidation as key-steps. Tetrahedron Lett. 2013, 54, 3437–3439. [Google Scholar] [CrossRef]
- Ikehara, M.; Tada, H.; Muneyama, K. Synthesis of 8-hydroxypurine nucleosides. Chem. Pharm. Bull. 1965, 13, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-Y.; Lin, Y.; de Jonghe, S.; Gao, L.-J.; Vanderhoydonck, B.; Froeyen, M.; Rozenski, J.; Herman, J.; Louat, T.; van Belle, K.; et al. Discovery of 7-N-piperazinylthiazolo[5,4-d]pyrimidine analogues as a novel class of immunosuppressive agents with in vivo biological activity. J. Med. Chem. 2011, 54, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, K.; Muramoto, N.; Misaki, T.; Sugimura, T. DMEAD: A new dialkyl azodicarboxylate for the Mitsunobu reaction. Tetrahedron 2009, 65, 6109–6114. [Google Scholar] [CrossRef]
- Fromme, J.C.; Bruner, S.D.; Yang, W.; Karplus, M.; Verdine, G.L. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Biol. 2003, 10, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Winstein, S.; Grunwald, E.; Buckles, R.E.; Hanso, C. The role of neighboring groups in replacement reactions. XI. Some reactivities involving neighboring groups. J. Am. Chem. Soc. 1948, 70, 816–821. [Google Scholar] [CrossRef]
- Forkel, N.V.; Henderson, D.A.; Fuchter, M.J. Lanthanide replacement in organic synthesis: Luche-type reduction of α,β-unsaturated ketones in the presence of calcium triflate. Green Chem. 2012, 14, 2129–2132. [Google Scholar] [CrossRef]
- Jacquet, O.; Bergholz, T.; Magnier-Bouvier, C.; Mellah, M.; Guillot, R.; Fiaud, J.-C. Palladium-catalyzed and samarium-promoted coupling of stereochemically-biased allylic acetates with carbonyl compounds. Tetrahedron 2010, 66, 222–226. [Google Scholar] [CrossRef]
- Kim, K.; McComas, W. Chemoselective high-throughput purification mediated by solid-supported reagents: Its application to the first 6,9-disubstituted purine library synthesis. Comb. Chem. High Throughput Screen. 2000, 3, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Legraverend, M.; Ludwig, O.; Bisagni, E.; Leclerc, S.; Meijer, L.; Giocanti, N.; Sadri, R.; Favaudon, V. Synthesis and in vitro evaluation of novel 2,6,9-trisubstituted purines acting as cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. 1999, 7, 1281–1293. [Google Scholar] [CrossRef]
- Lukin, K.A.; Yang, C.X.; Bellettini, J.R.; Narayanan, B.A. New purine derivatives as efficient preparation of nucleoside analogs via alkylation. Nucleosides Nucleotides Nucleic Acids 2000, 19, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Gültekin, Z. Palladium-catalyzed synthesis of 9-(2-cyclopentenyl)guanine. Asian J. Chem. 2006, 18, 1462–1466. [Google Scholar]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahajan, T.R.; Ytre-Arne, M.E.; Strøm-Andersen, P.; Dalhus, B.; Gundersen, L.-L. Synthetic Routes to N-9 Alkylated 8-Oxoguanines; Weak Inhibitors of the Human DNA Glycosylase OGG1. Molecules 2015, 20, 15944-15965. https://doi.org/10.3390/molecules200915944
Mahajan TR, Ytre-Arne ME, Strøm-Andersen P, Dalhus B, Gundersen L-L. Synthetic Routes to N-9 Alkylated 8-Oxoguanines; Weak Inhibitors of the Human DNA Glycosylase OGG1. Molecules. 2015; 20(9):15944-15965. https://doi.org/10.3390/molecules200915944
Chicago/Turabian StyleMahajan, Tushar R., Mari Eknes Ytre-Arne, Pernille Strøm-Andersen, Bjørn Dalhus, and Lise-Lotte Gundersen. 2015. "Synthetic Routes to N-9 Alkylated 8-Oxoguanines; Weak Inhibitors of the Human DNA Glycosylase OGG1" Molecules 20, no. 9: 15944-15965. https://doi.org/10.3390/molecules200915944