
Unconventional Bifunctional Lewis-Brønsted Acid Activation Mode in Bicyclic Guanidine-Catalyzed Conjugate Addition Reactions

Abstract
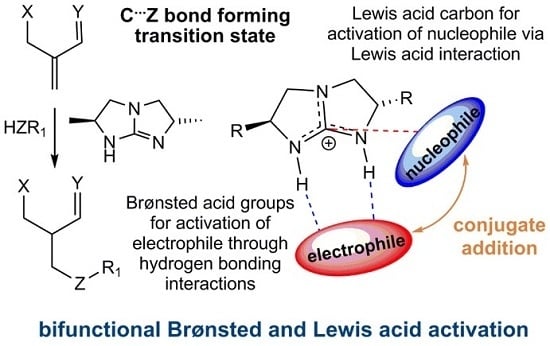
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
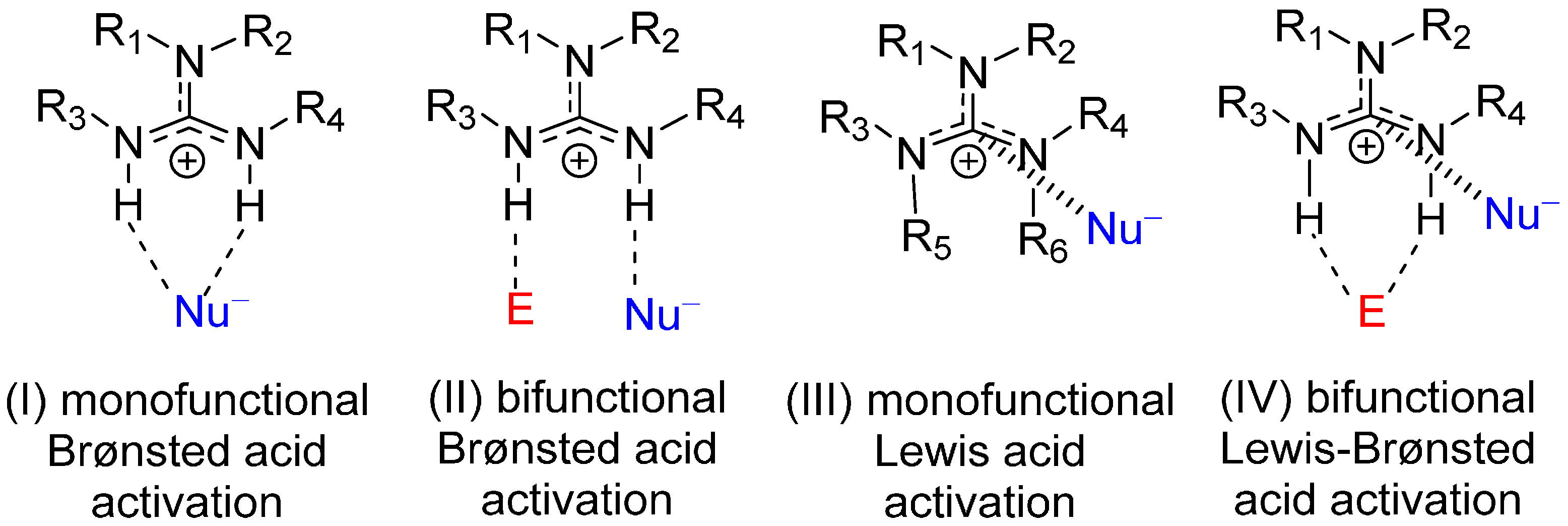
1. Introduction
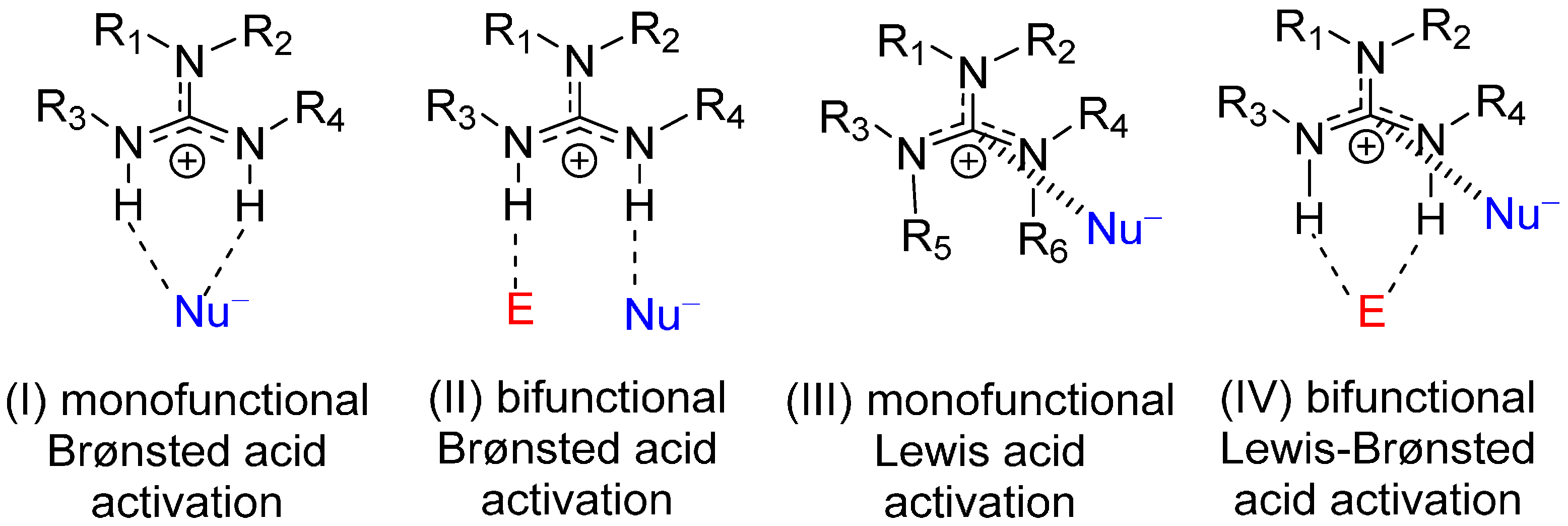

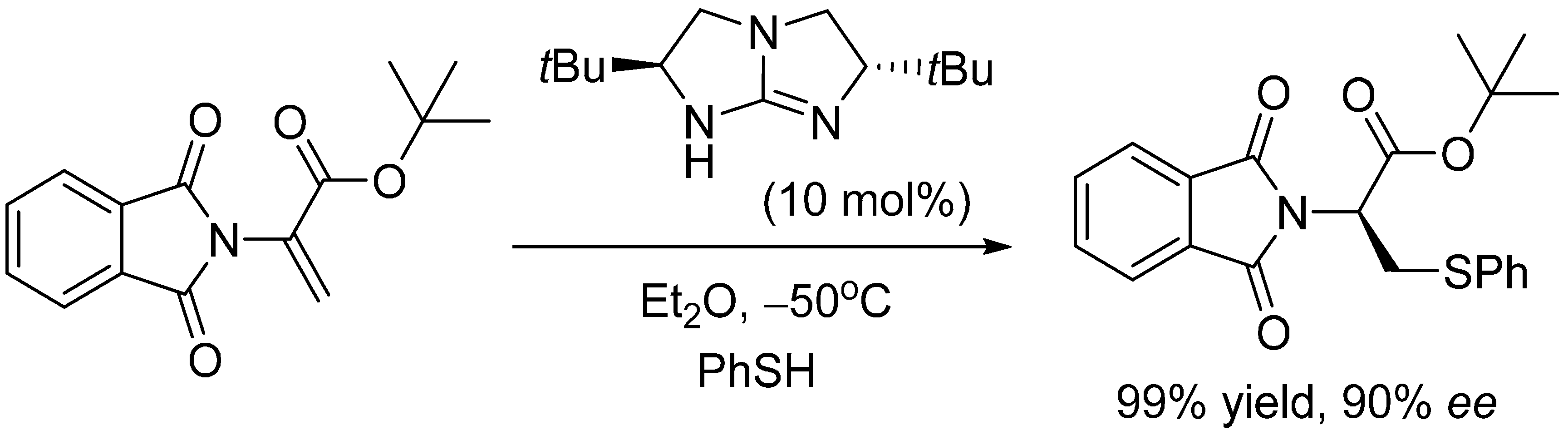
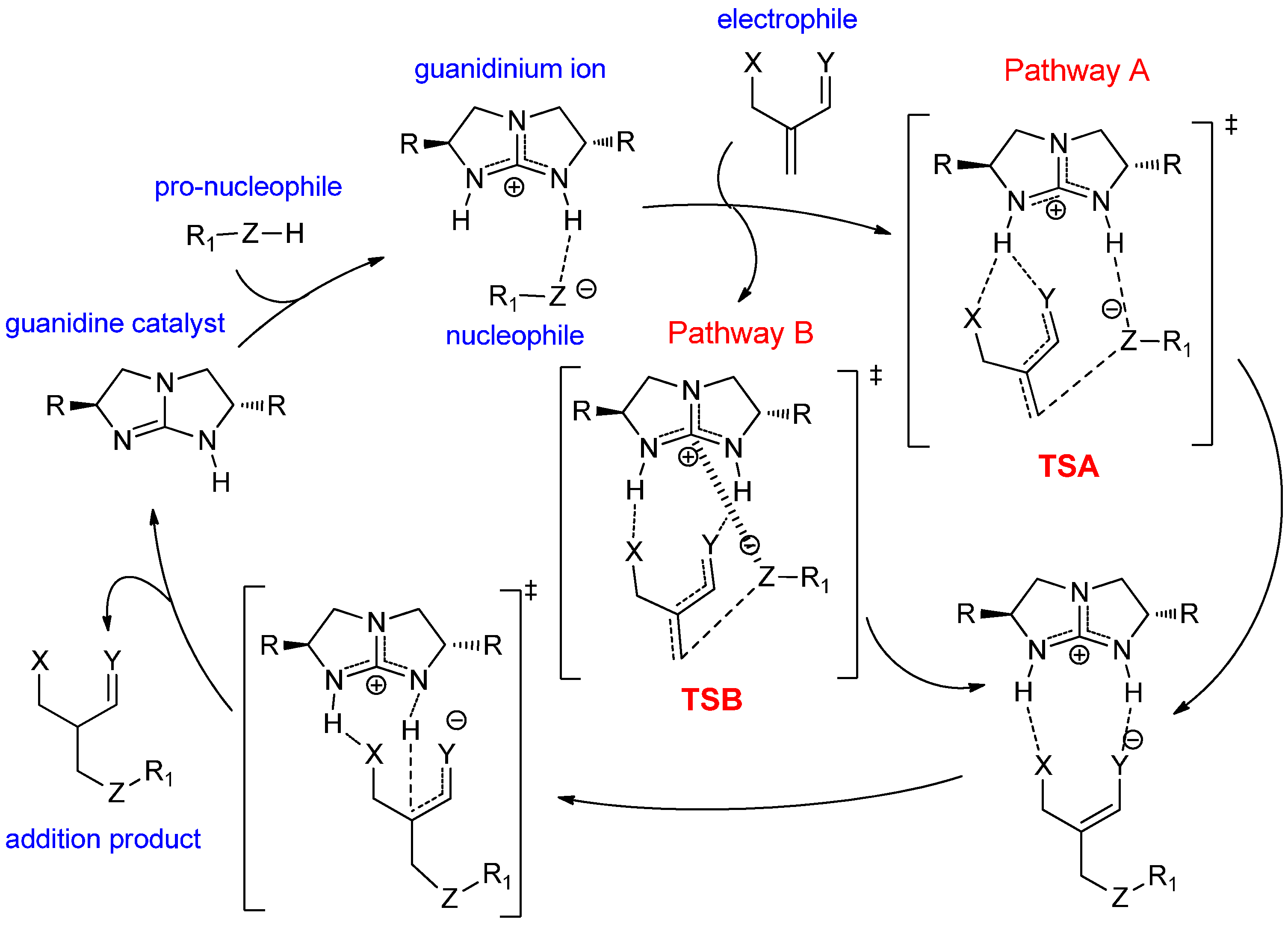
2. Results and Discussion
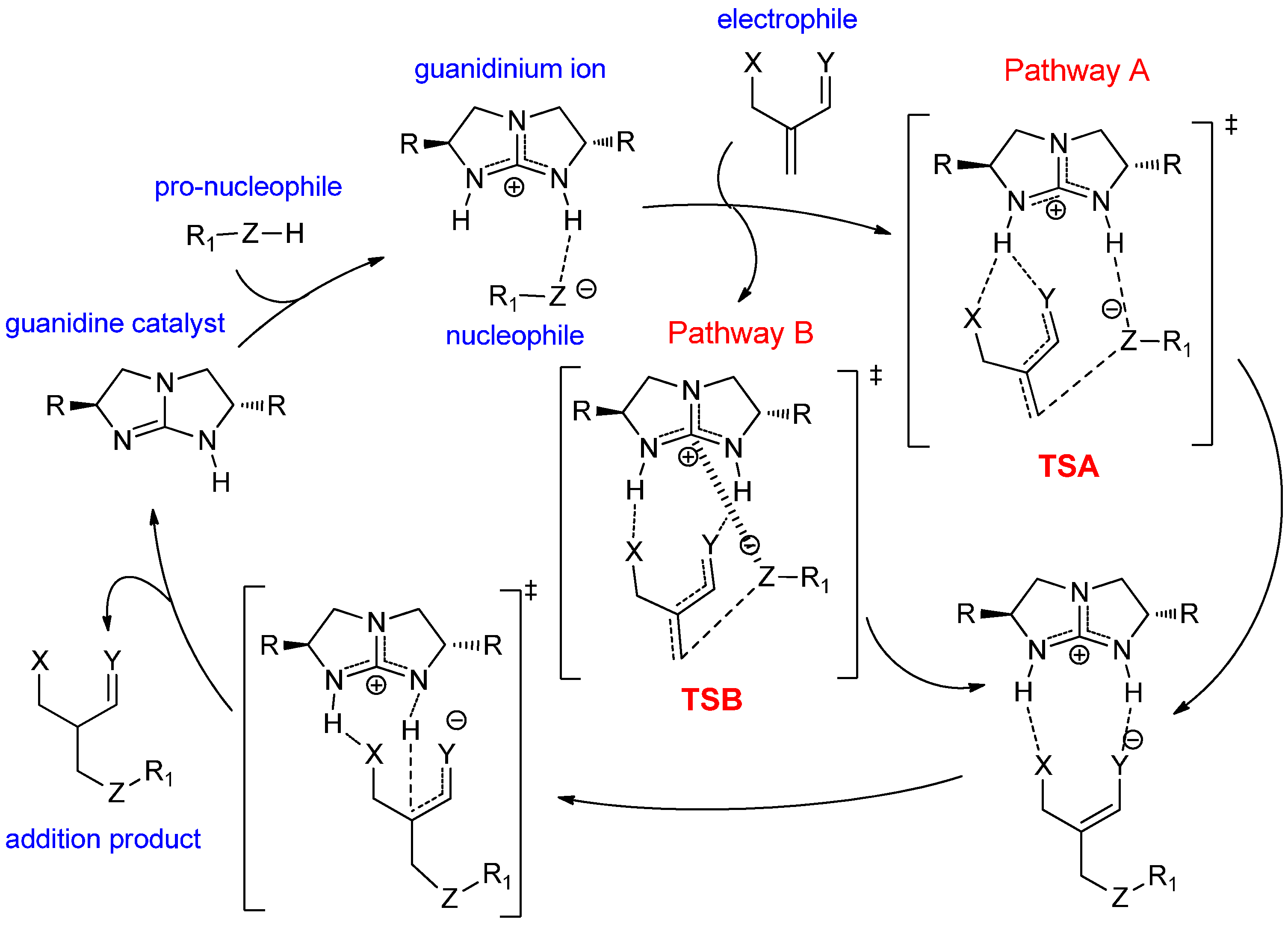
2.1. Model Systems for Conjugate Nucleophilic Addition
- (1)
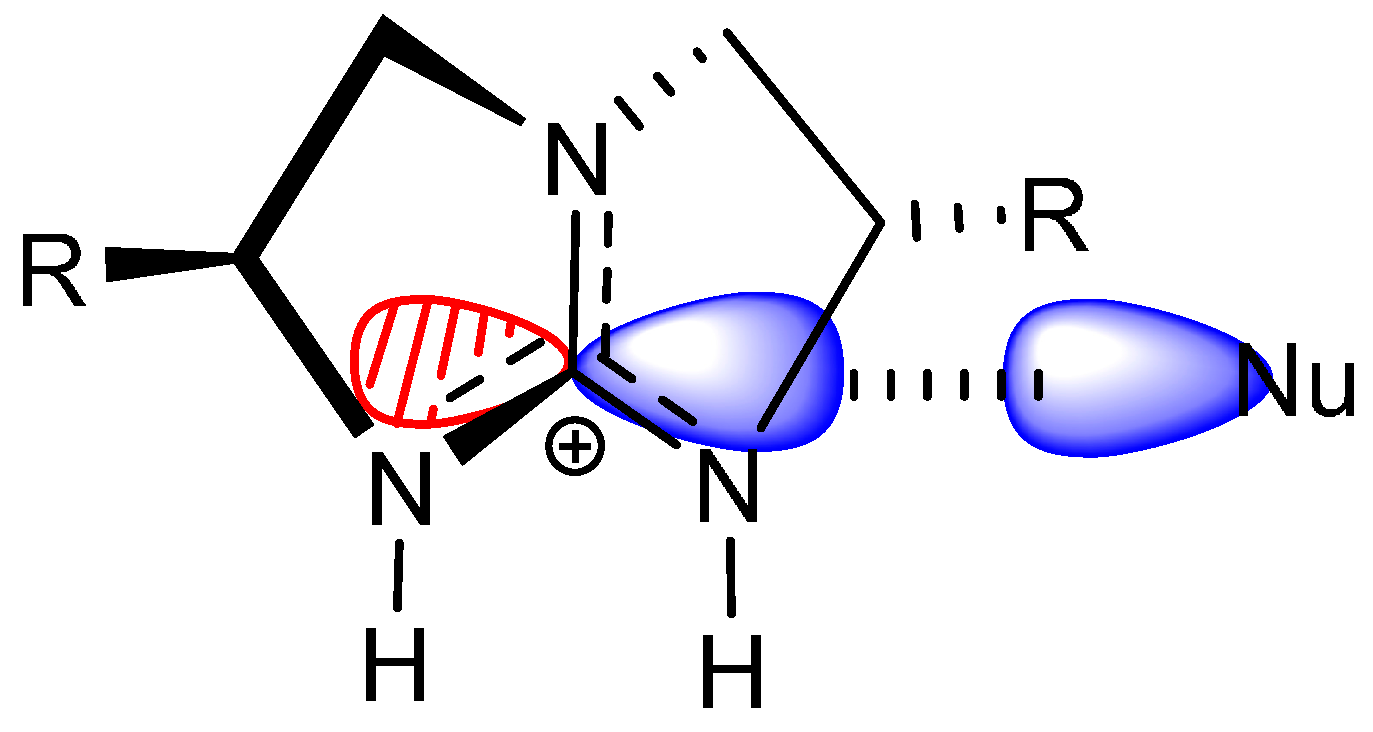
- The electrophile has two Brønsted basic sites (i.e., hydrogen bond acceptors X and Y, see Scheme 4) for dual hydrogen bonding interaction with the two guanidinium N−H protons.
- (2)
- The nucleophile, preferably in an anionic form, should have significant Lewis basicity to interact favorably with the guanidinium central electrophilic carbon, via the “Lewis acid” interaction (Scheme 2).
- (3)
- The electrophile must have sufficient flexibility to enable the nucleophile to access the Lewis acidic site and the unsaturated carbon for conjugation addition simultaneously.
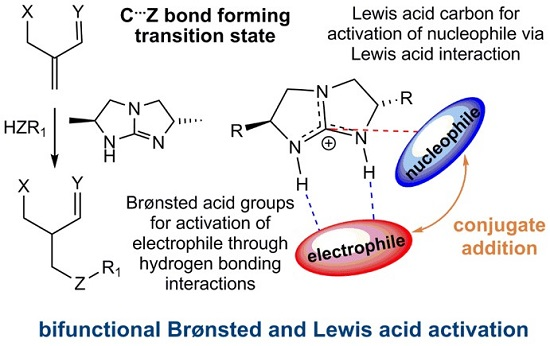
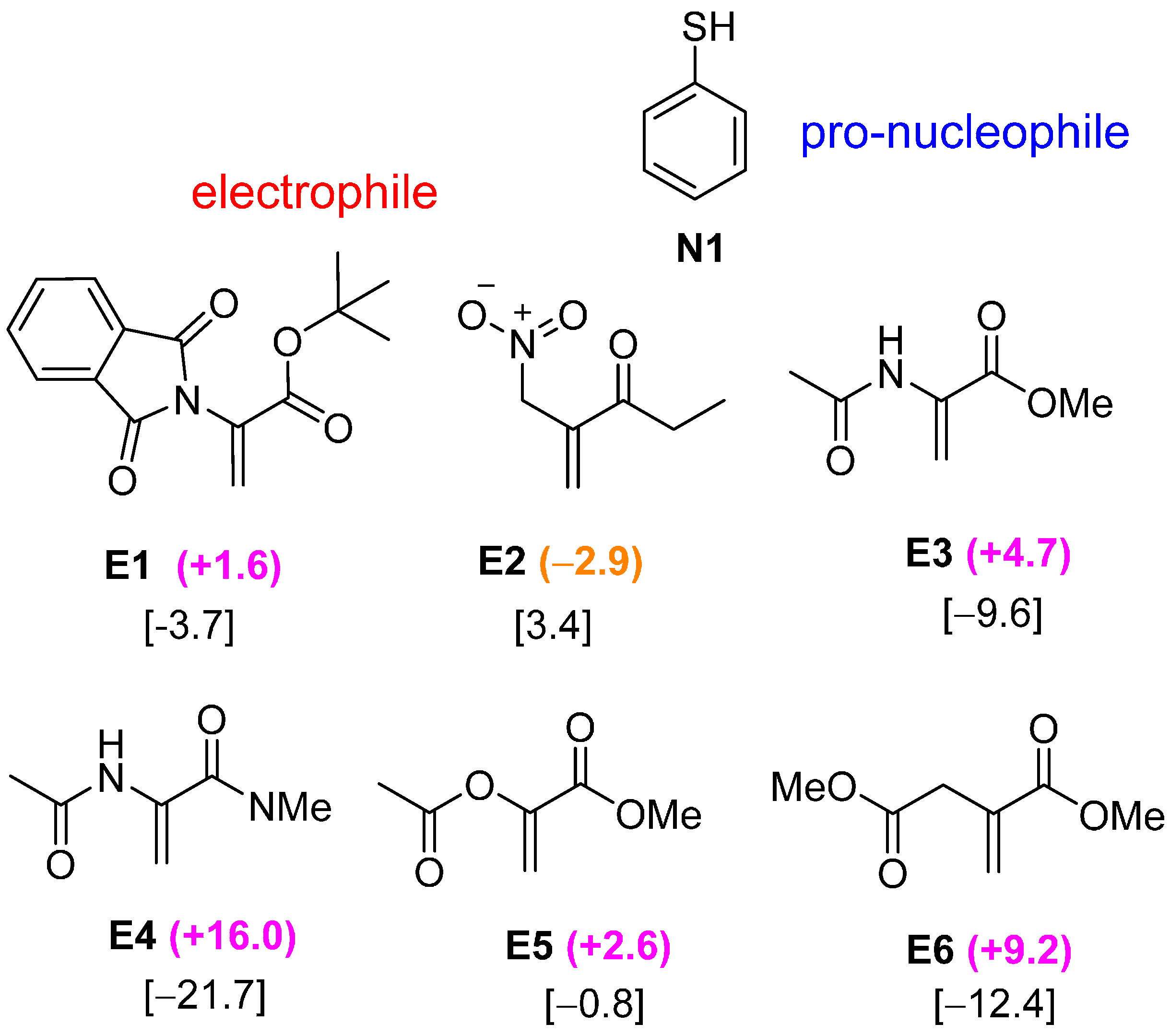
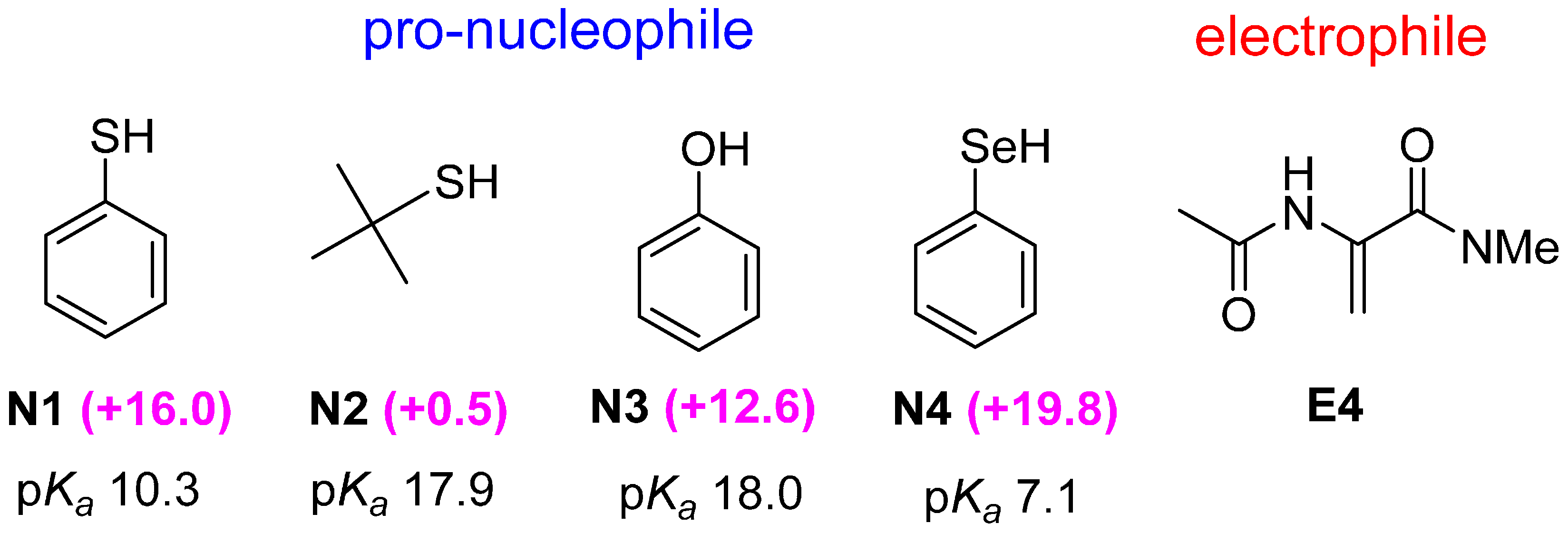
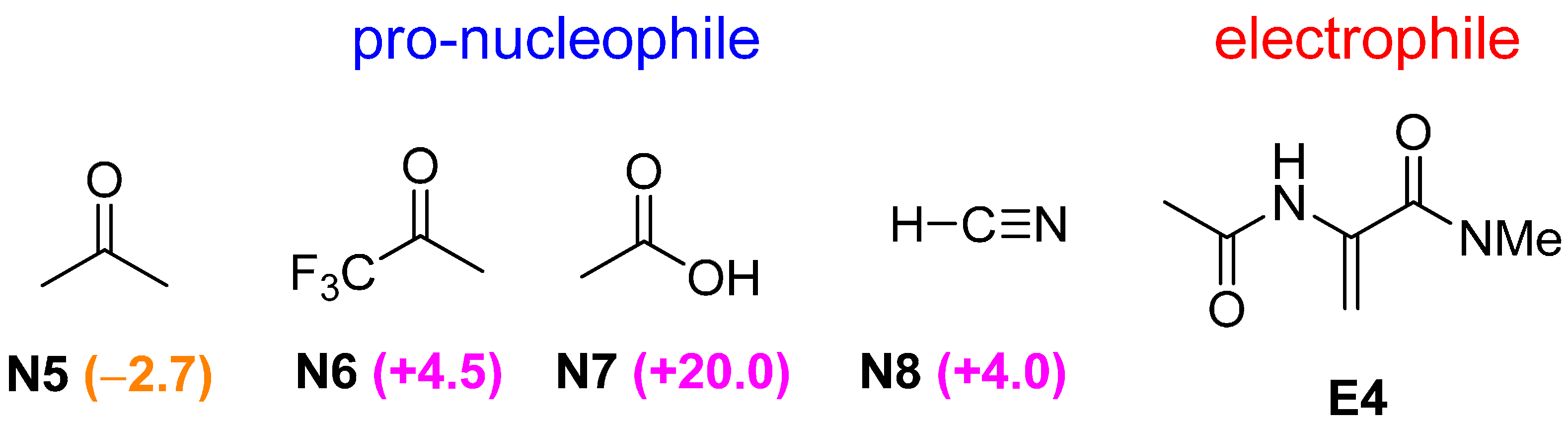
2.2. Optimal Brønsted Basic Functional Groups
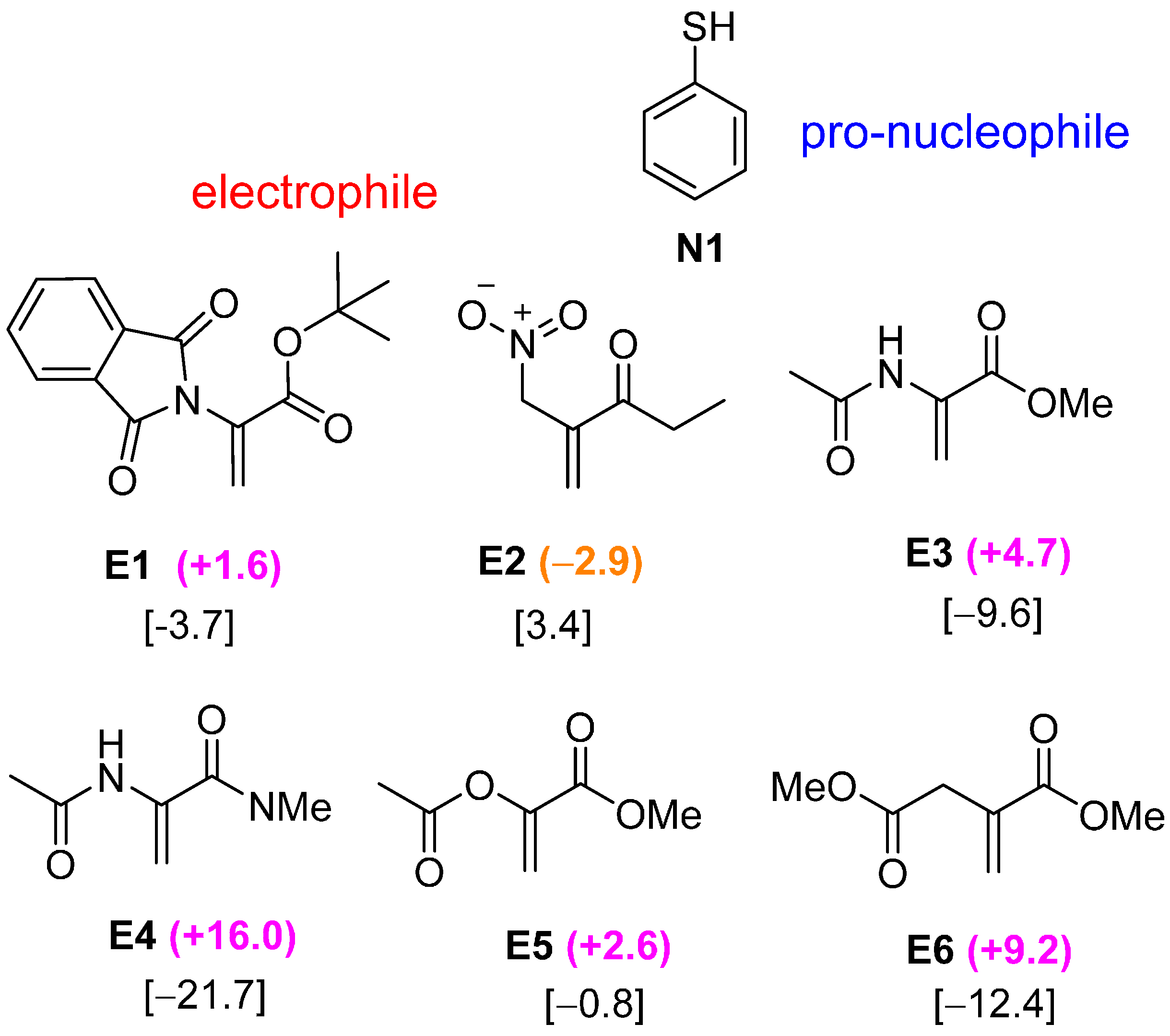
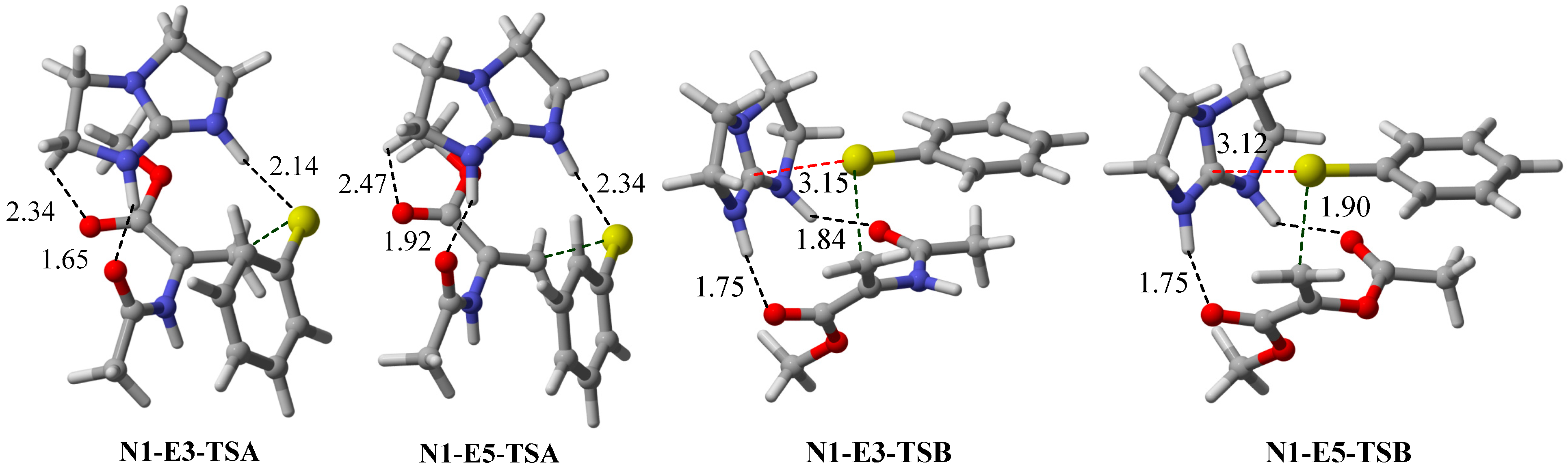
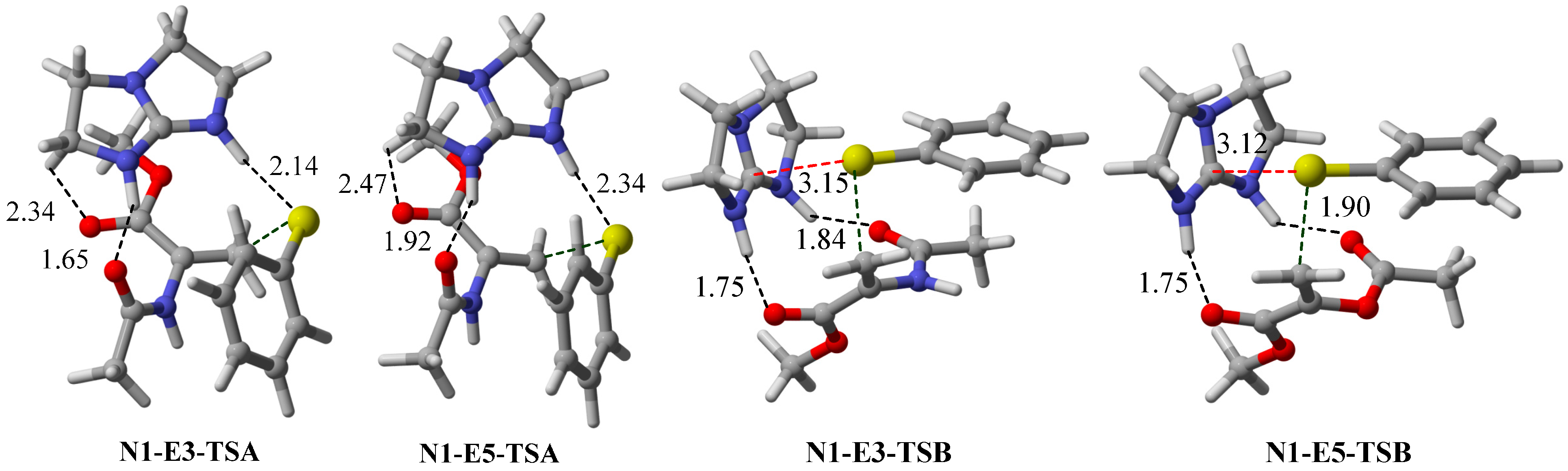
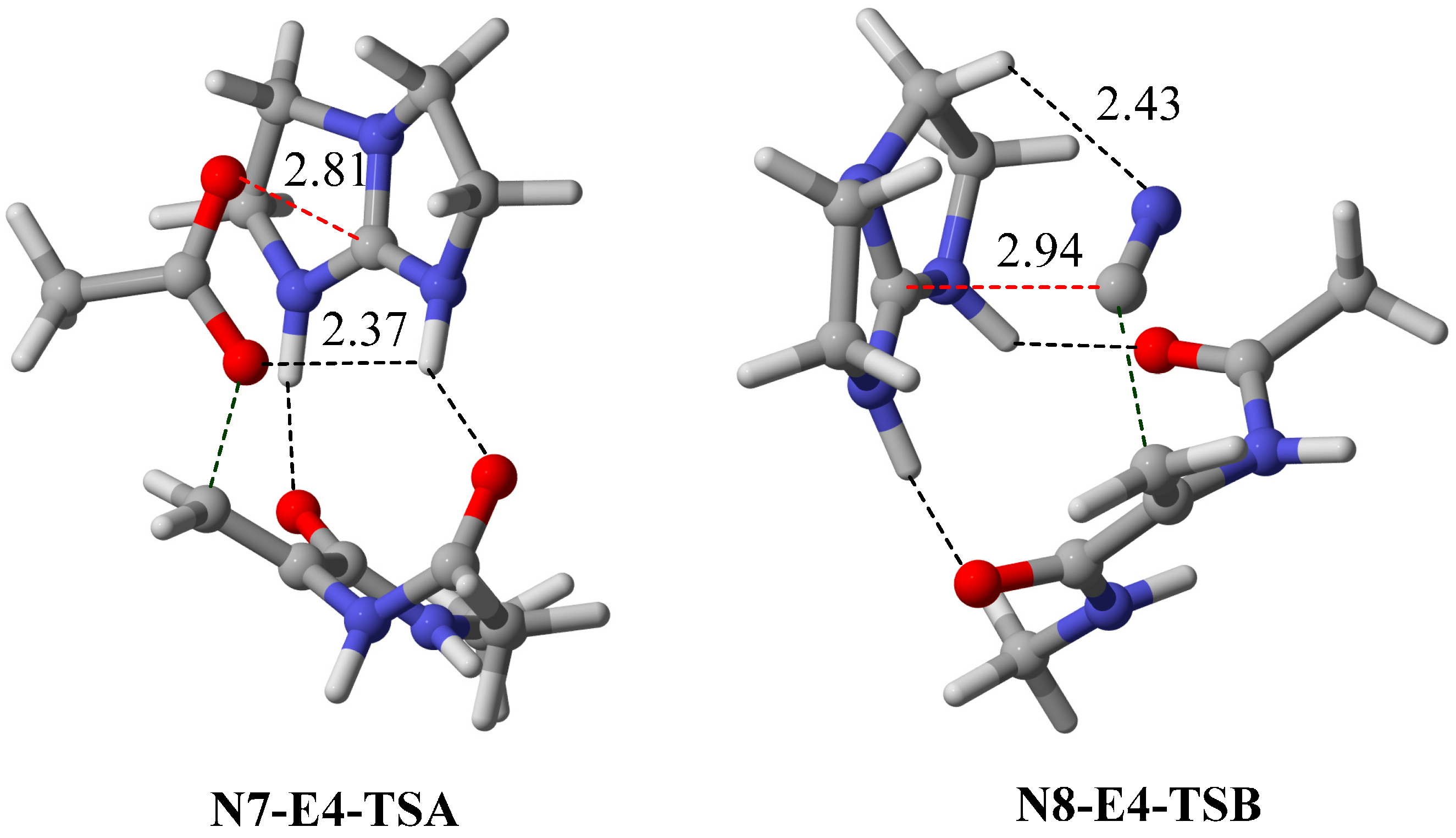
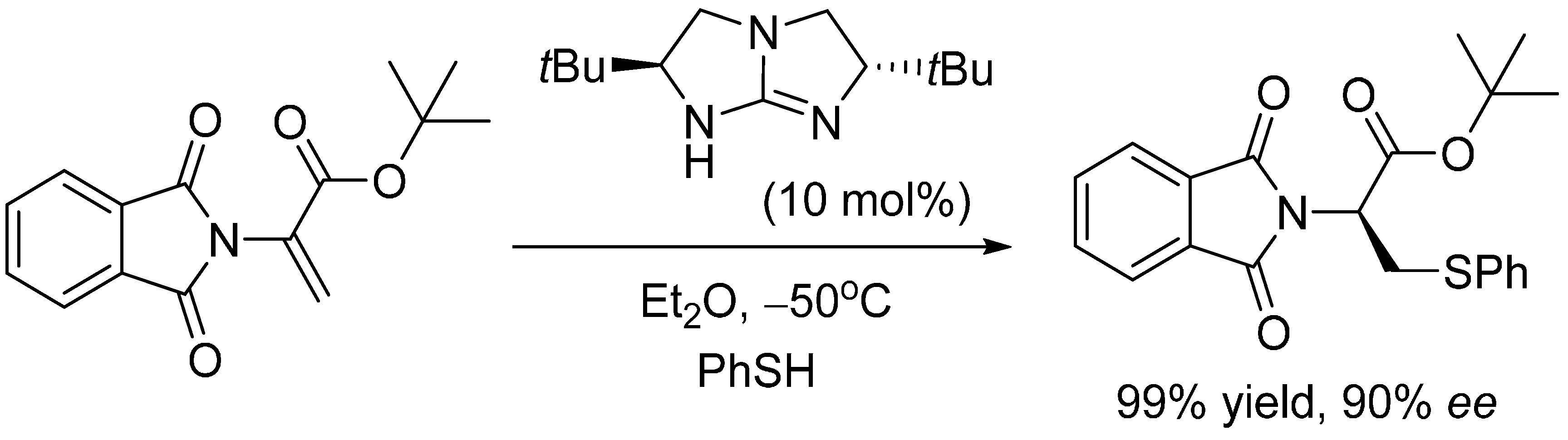
2.3. Optimal Pro-Nucleophiles
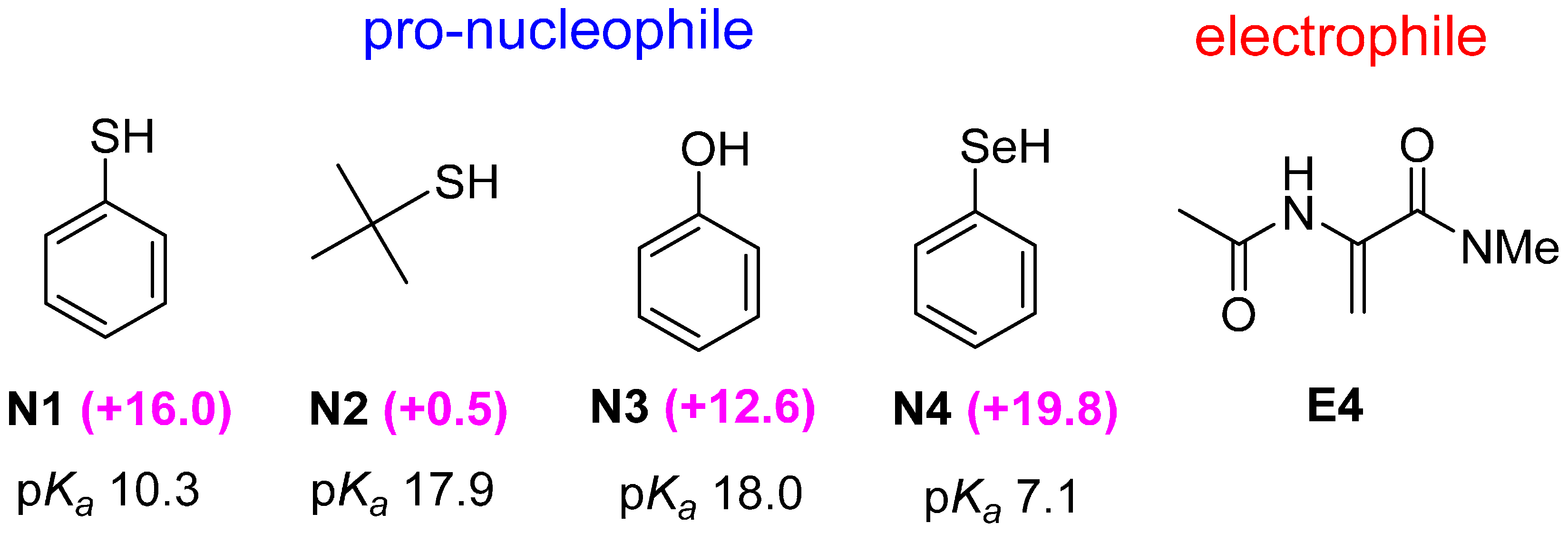
2.4. Bifunctional Lewis-Brønsted Acid Activation in Michael and Hetero-Michael Reactions


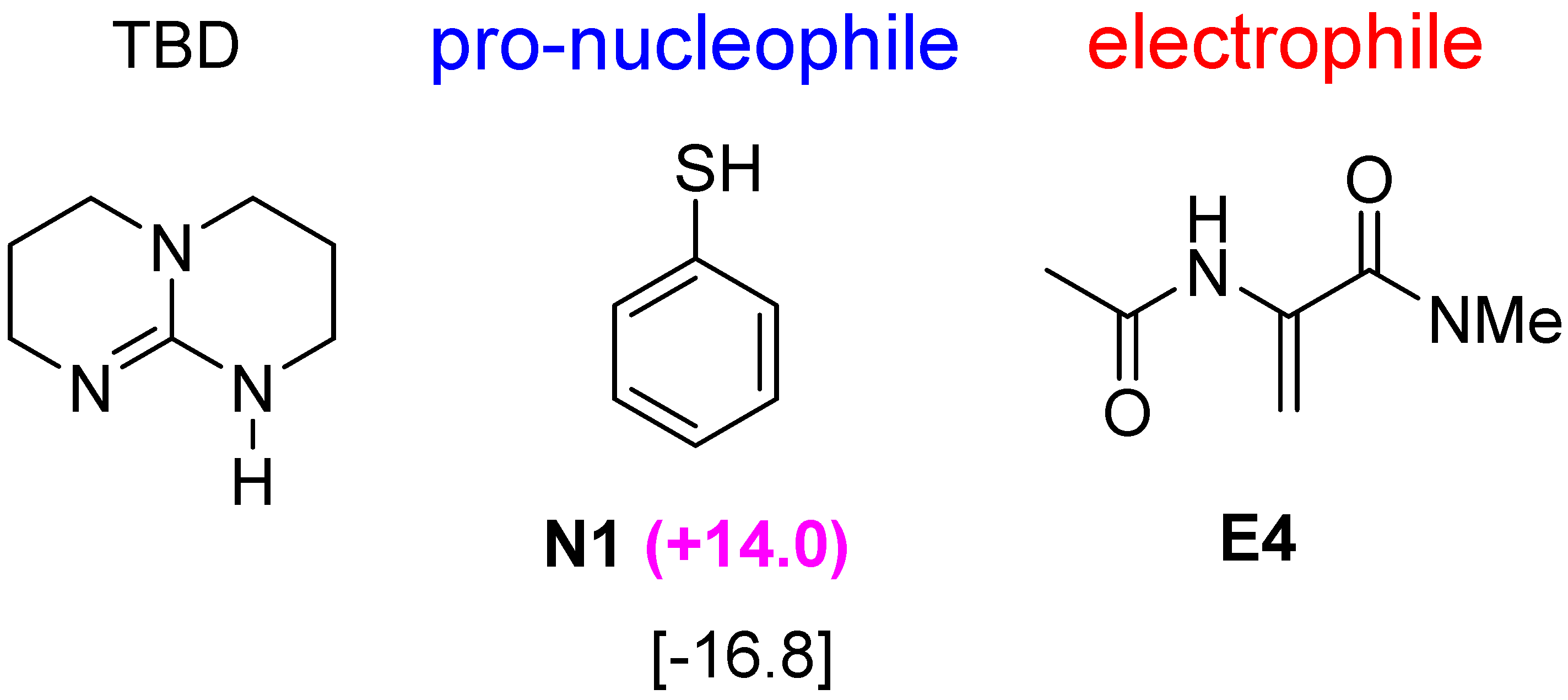
2.5. Other Bicyclic Guanidine Catalyst
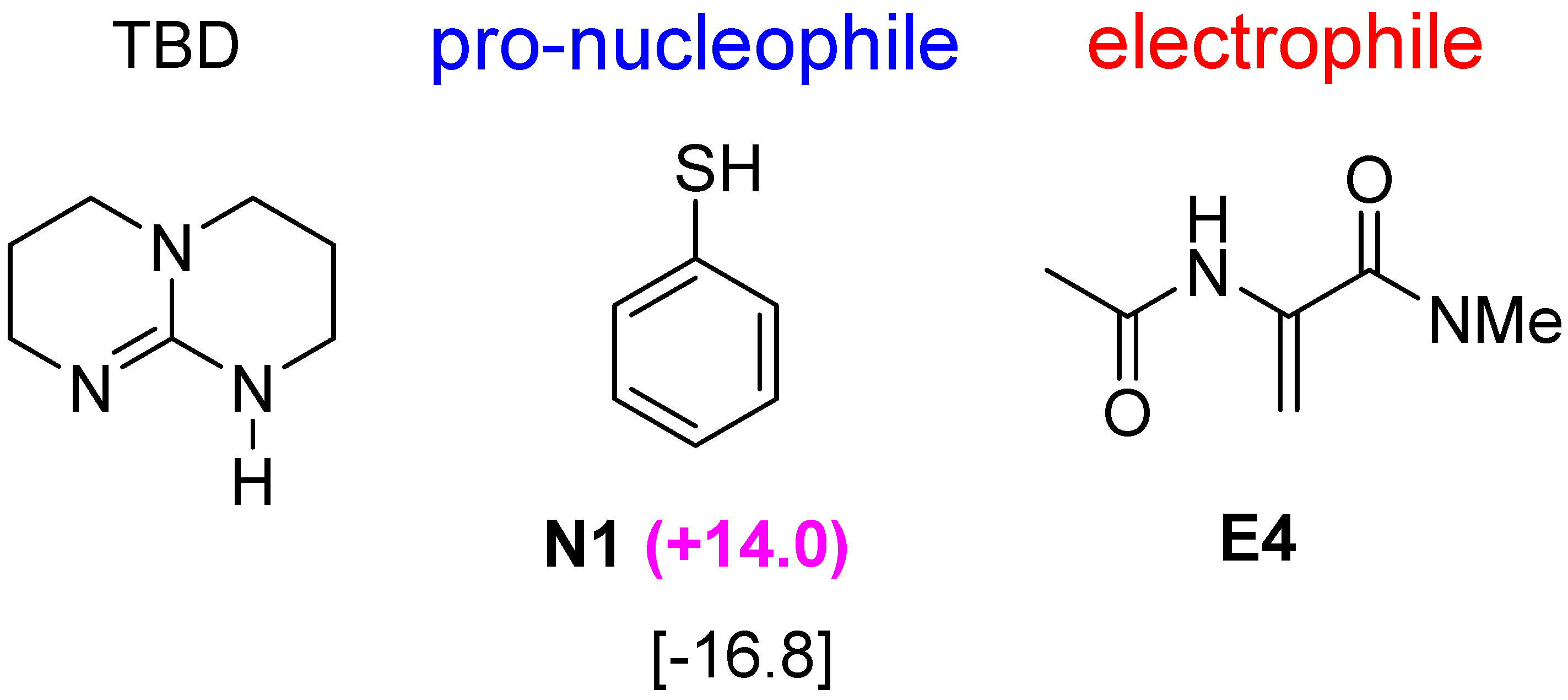
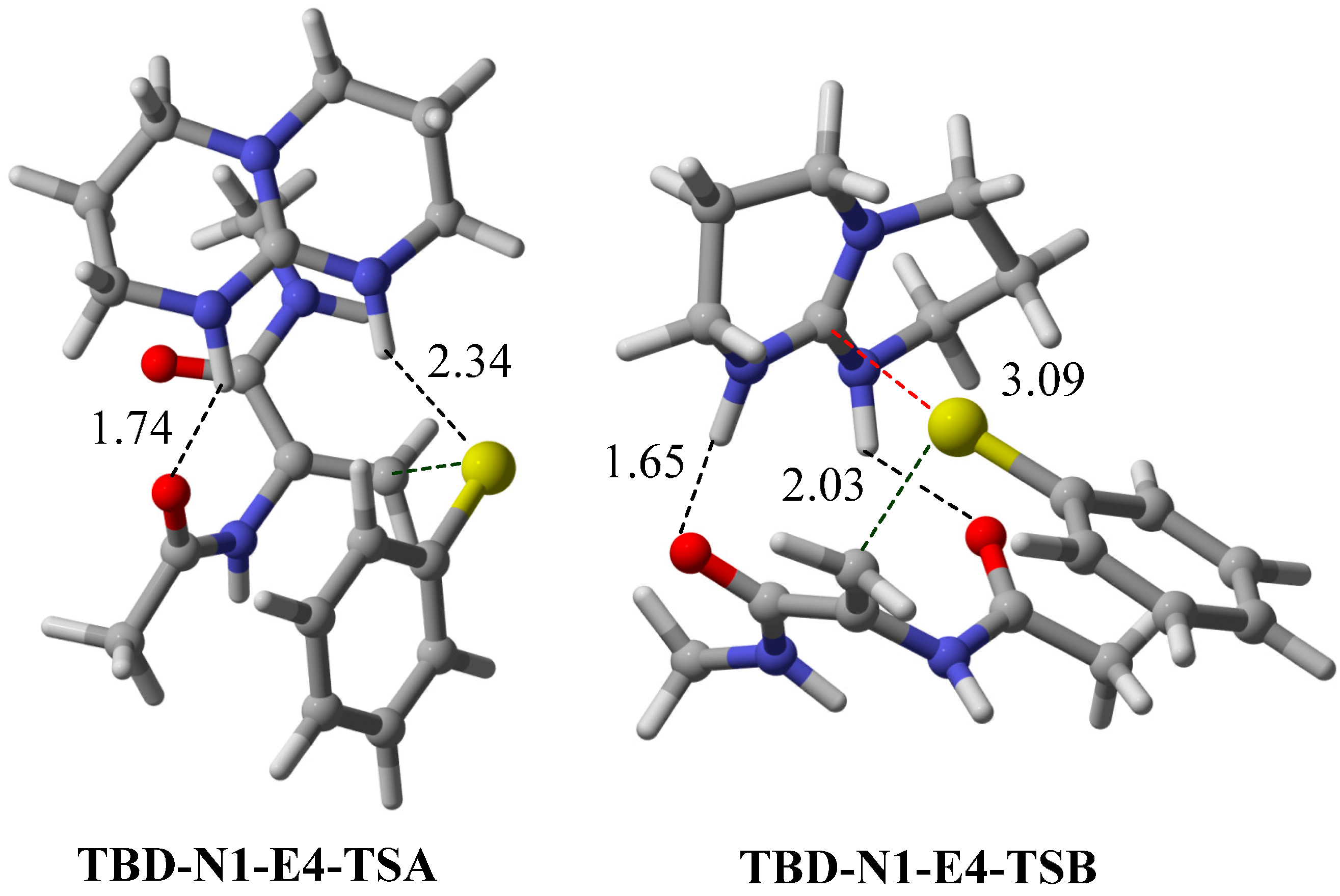
3. Computational Methods
Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fu, X.; Tan, C.-H. Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar] [CrossRef] [PubMed]
- Selig, P. Guanidine organocatalysis. Synthesis-Stuttgart 2013, 45, 703–718. [Google Scholar] [CrossRef]
- Leow, D.; Tan, C.H. Chiral guanidine catalyzed enantioselective reactions. Chem. Asian J. 2009, 4, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kumamoto, T. Guanidine in organic synthesis. Synthesis 2006, 5, 737–752. [Google Scholar] [CrossRef]
- Coles, M.P. Bicyclic-guanidines, -guanidinates and -guanidinium salts: Wide ranging applications from a simple family of molecules. Chem. Commun. 2009, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Grogan, E.J. Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, W.Y.; Han, K.L.; He, G.Z.; Li, C. Density functional study on the mechanism of bicyclic guanidine-catalyzed Strecker reaction. J. Org. Chem. 2003, 68, 8786–8789. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Lim, X.; Chen, T.; Tan, G.K.; Tan, C.-H.; Huang, K.W. Selective formation of bicyclic guanidinium chloride complexes: Implication of the bifunctionality of guanidine. Tetrahedron Lett. 2009, 50, 1560–1562. [Google Scholar] [CrossRef]
- Xue, H.; Jiang, D.; Jiang, H.; Kee, C.H.; Hirao, H.; Nishimura, T.; Wong, M.W.; Tan, C.H. Mechanistic insights of chiral bicyclic guanidine-catalyzed enantioselective reactions from microscopic and macroscopic perspectives. J. Org. Chem. 2015, 80, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Pan, Y.; Zhao, Y.; Ma, T.; Lee, R.; Yang, Y.; Huang, K.W.; Wong, M.W.; Tan, C.H. Synthesis of a chiral quaternary carbon center bearing a fluorine atom: Enantioselective and diastereoselective guanidine-catalyzed addition of fluorocarbon nucleophiles. Angew. Chem. Int. Ed. 2009, 48, 3627–3631. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.W.; Ng, A.M.E. Asymmetric michael addition using bifunctional bicyclic guanidine organocatalyst: A theoretical perspective. Aust. J. Chem. 2014, 67, 1100–1109. [Google Scholar] [CrossRef]
- Cho, B.; Tan, C.-H.; Wong, M.W. Sequential catalytic role of bifunctional bicyclic guanidine in asymmetric phospha-Michael reaction. Org. Biomol. Chem. 2011, 9, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Salvio, R. The guanidinium unit in the catalysis of phosphoryl transfer reactions: From molecular spacers to nanostructured supports. Chem. Eur. J. 2015, 21, 10960–10971. [Google Scholar] [CrossRef] [PubMed]
- Baldini, L.; Cacciapaglia, R.; Casnati, A.; Mandolini, L.; Salvio, R.; Sansone, F.; Ungaro, R. Upper rim guanidinocalix[4]arenes as artificial phosphodiesterases. J. Org. Chem. 2012, 77, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Corona-Martinez, D.O.; Taran, O.; Yatsimirsky, A.K. Mechanism of general acid-base catalysis in transesterification of an RNA model phosphodiester studied with strongly basic catalyst. Org. Biomol. Chem. 2010, 8, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Perreault, D.M.; Cabell, L.A.; Anslyn, E.V. Using guanidinium groups for the recognition of RNA and as catalysts for the hydrolysis of RNA. Bioorg. Med. Chem. 1997, 5, 1209–1220. [Google Scholar] [CrossRef]
- Cho, B.; Tan, C.-H.; Wong, M.W. Origin of asymmetric induction in bicyclic guanidine-catalyzed thio-Michael reaction: A bifunctional mode of Lewis and Brønsted acid activations. J. Org. Chem. 2012, 77, 6553–6562. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.; LePerchec, P.; Senet, J.P. Reaction of epoxides with chlorocarbonylated compounds catalyzed by hexaalkylguanidinium chloride. J. Org. Chem. 1994, 59, 4925–4930. [Google Scholar] [CrossRef]
- Li, H.; Wu, J.; Brunel, S.; Monnet, C.; Baudry, R.; le Perchec, P. Polymerization of lactides and lactones by metal-free initiators. Ind. Eng. Chem. Res. 2005, 44, 8641–8643. [Google Scholar] [CrossRef]
- Foulon, F.; Fixari, B.; Picq, D.; LePerchec, P. Catalytic decomposition of alkyl chloroformates by hexabutylguanidinium chloride. Tetrahedron Lett. 1997, 38, 3387–3339. [Google Scholar] [CrossRef]
- Leow, D.; Lin, S.; Chittimalla, S.K.; Fu, X.; Tan, C.H. Enantioselective protonation catalyzed by a chiral bicyclic guanidine derivative. Angew. Chem. Int. Ed. 2008, 47, 5641–5645. [Google Scholar] [CrossRef] [PubMed]
- Zhachkina, A.; Liu, M.; Sun, X.; Amegayibor, F.; Lee, J.K. Gas-phase thermochemical properties of the damaged base O(6)-methylguanine versus adenine and guanine. J. Org. Chem. 2009, 74, 7429–7440. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G. Equilibrium acidities in dimethyl-sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Nising, C.F.; Brase, S. The oxa-Michael reaction: From recent developments to applications in natural product synthesis. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.C.; Joshi, N.N. Catalytic, enantioselective Michael addition reactions. ARKIVOC 2002, 7, 167–196. [Google Scholar] [CrossRef]
- Flemer, S., Jr. Selenol protecting groups in organic chemistry: Special emphasis on selenocysteine se-protection in solid phase peptide synthesis. Molecules 2011, 16, 3232–3251. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Beutner, G.L.; Wynn, T.; Eastgate, M.D. Lewis base activation of Lewis acids: Catalytic, enantioselective addition of silyl ketene acetals to aldehydes. J. Am. Chem. Soc. 2005, 127, 3744–3789. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Wilson, T.W. N-silyl oxyketene imines are underused yet highly versatile reagents for catalytic asymmetric synthesis. Nat. Chem. 2010, 2, 937–994. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Feng, X. Asymmetric Strecker reaction. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef] [PubMed]
- Chuma, A.; Horn, H.W.; Swope, W.C.; Pratt, R.C.; Zhang, L.; Lohmeijer, B.G.; Wade, C.G.; Waymouth, R.M.; Hedrick, J.L.; Rice, J.E. The reaction mechanism for the organocatalytic ring-opening polymerization of L-lactide using a guanidine-based catalyst: Hydrogen-bonded or covalently bound? J. Am. Chem. Soc. 2008, 130, 6749–6754. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Goodman, J.M. The mechanism of TBD-catalyzed ring-opening polymerization of cyclic esters. J. Org. Chem. 2007, 72, 9656–9662. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Xu, J.; Tan, C.H. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) catalyzed Michael reactions. Tetrahedron Lett. 2005, 46, 6875–6878. [Google Scholar] [CrossRef]
- Simoni, D.; Rondanin, R.; Morini, M.; Baruchello, R.; Invidiata, F.P. 1,5,7-triazabicyclo [4.4.0]dec-1-ene (TBD), 7-methyl-TBD (MTBD) and the polymer-supported TBD (P-TBD): Three efficient catalysts for the nitroaldol (Henry) reaction and for the addition of dialkyl phosphites to unsaturated systems. Tetrahedron Lett. 2000, 41, 1607–1610. [Google Scholar] [CrossRef]
- Hammar, P.; Ghobril, C.; Antheaume, C.; Wagner, A.; Baati, A.; Himo, F. Theoretical mechanistic study of the TBD-catalyzed intramolecular aldol reaction of ketoaldehydes. J. Org. Chem. 2010, 75, 4728–4736. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Yang, H.; Wong, M.W. Oxyanion hole stabilization by CH∙∙∙O interaction in a transition state—A three-point interaction model for cinchona alkaloid-catalyzed asymmetric methanolysis of meso-cyclic anhydrides. J. Am. Chem. Soc. 2013, 135, 5808–5818. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wong, M.W. β-amino acid-catalyzed asymmetric Michael additions: Design of organocatalysts with catalytic acid/base dyad inspired by serine proteases. J. Org. Chem. 2011, 76, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, B.; Wong, M.W. Unconventional Bifunctional Lewis-Brønsted Acid Activation Mode in Bicyclic Guanidine-Catalyzed Conjugate Addition Reactions. Molecules 2015, 20, 15108-15121. https://doi.org/10.3390/molecules200815108
Cho B, Wong MW. Unconventional Bifunctional Lewis-Brønsted Acid Activation Mode in Bicyclic Guanidine-Catalyzed Conjugate Addition Reactions. Molecules. 2015; 20(8):15108-15121. https://doi.org/10.3390/molecules200815108
Chicago/Turabian StyleCho, Bokun, and Ming Wah Wong. 2015. "Unconventional Bifunctional Lewis-Brønsted Acid Activation Mode in Bicyclic Guanidine-Catalyzed Conjugate Addition Reactions" Molecules 20, no. 8: 15108-15121. https://doi.org/10.3390/molecules200815108