
Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs

Abstract
:1. Introduction
2. Results and Discussion
2.1. Pharmacophore Model Studies
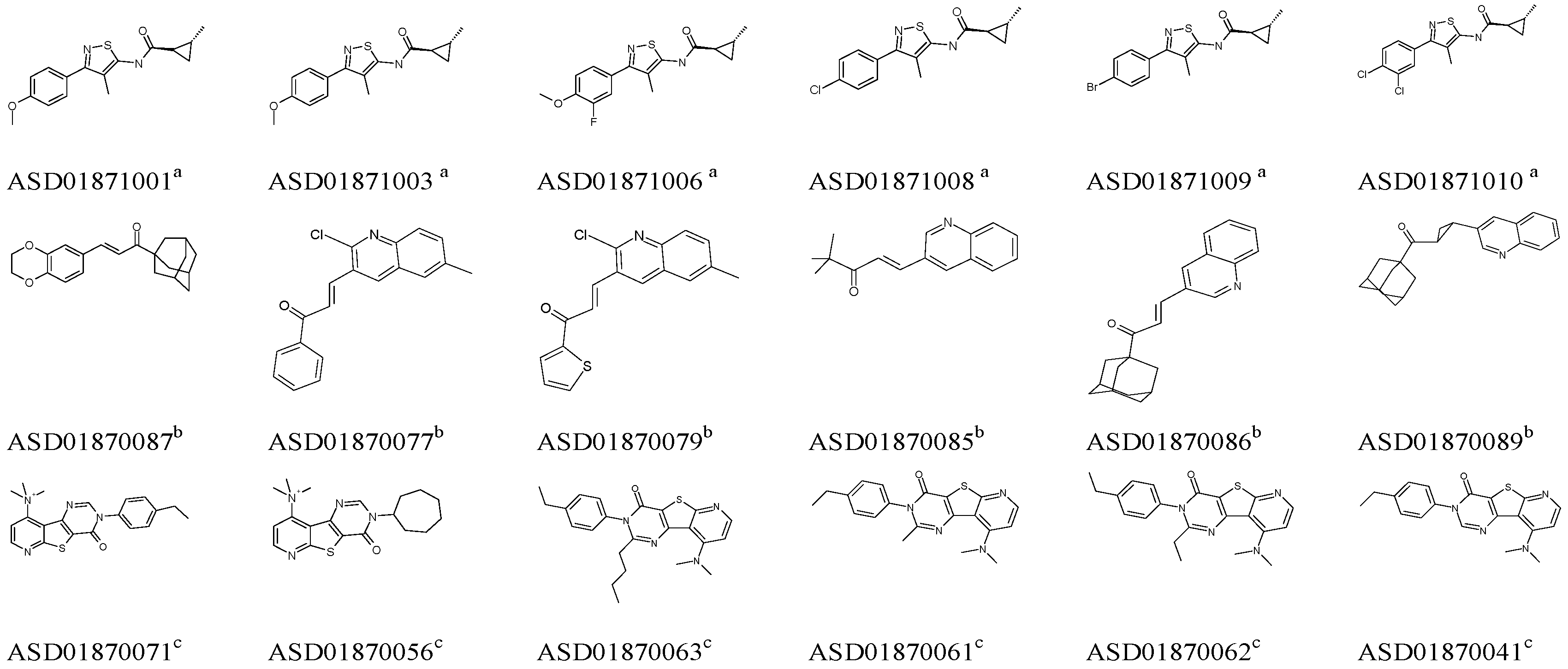

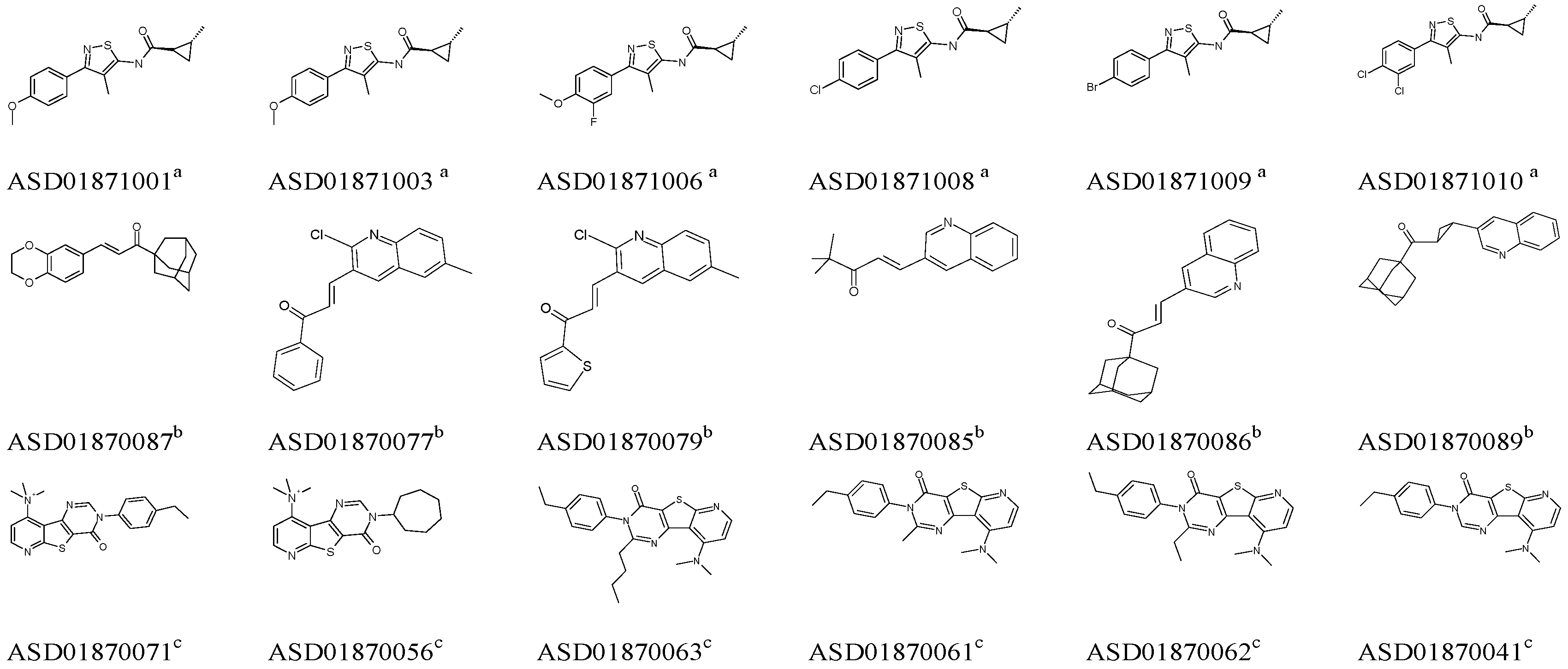{kind=link}
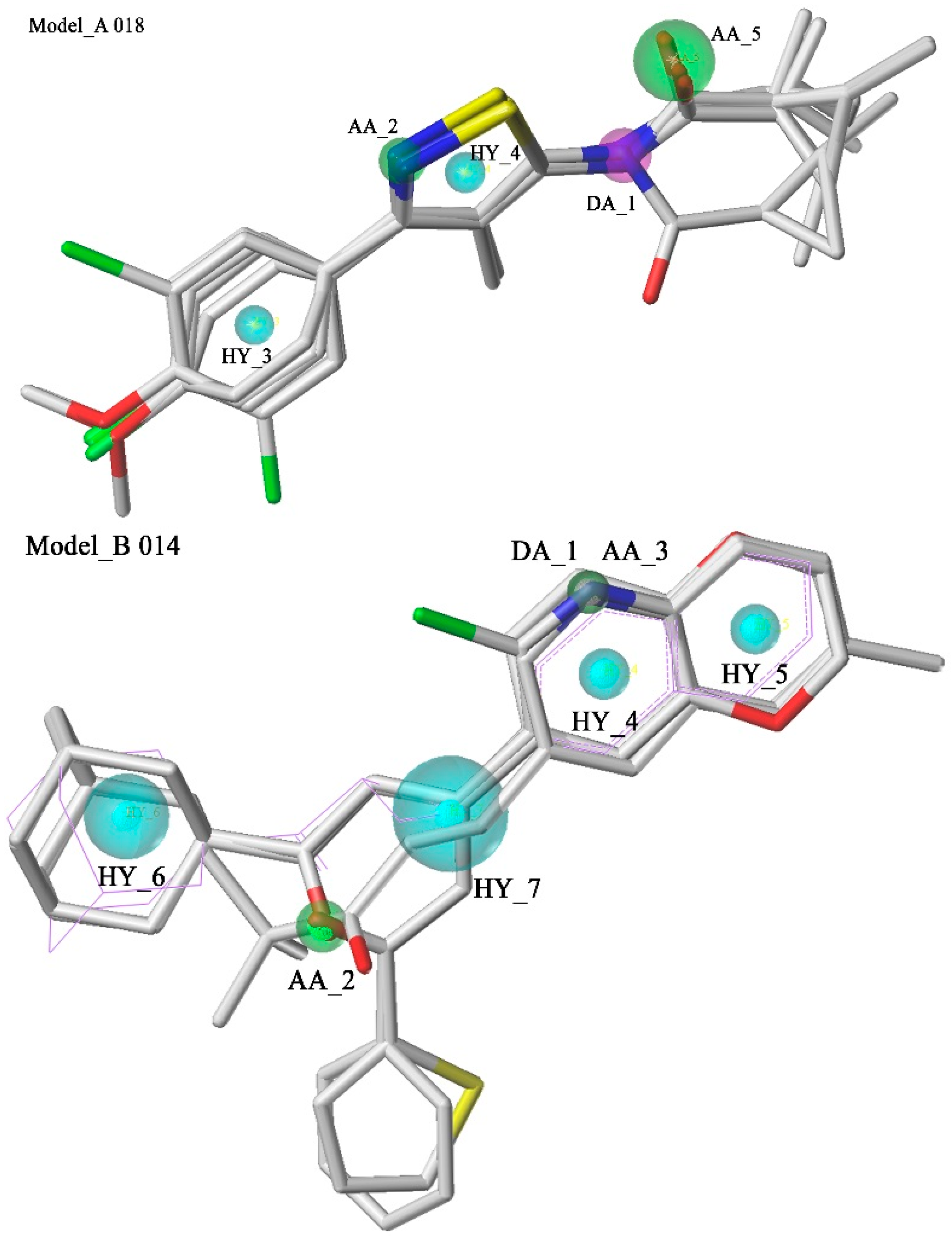{kind=link}
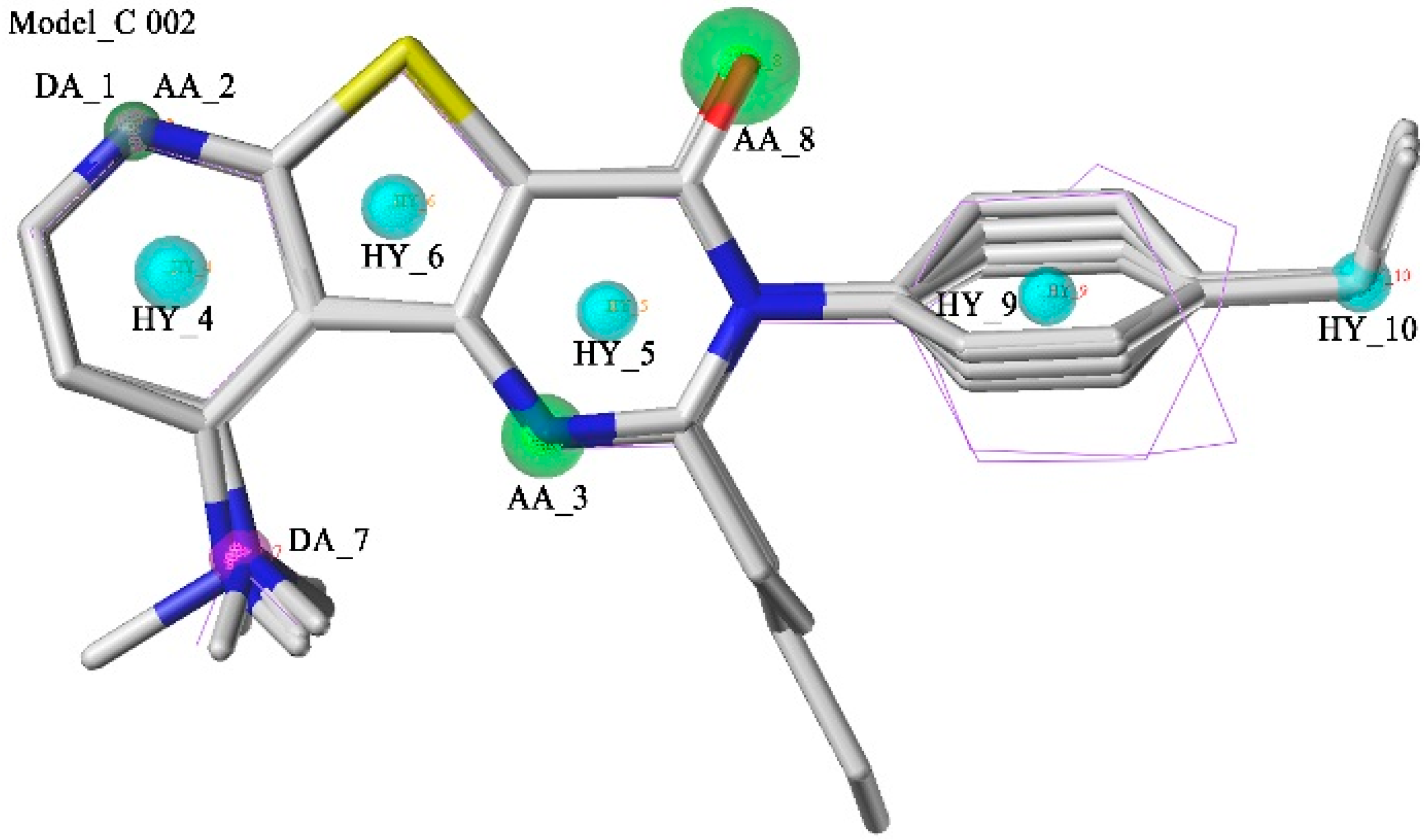{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Specificity | Energy | N_Hits | Ha a | Ht b | Ht-Ha c | A% d | N e | CAI f |
|---|---|---|---|---|---|---|---|---|---|
| Model_A018 | 4.33 | 2.89 | 6 | 12 | 29 | 17 | 16.22% | 1.66 | 0.27 |
| Model_A014 | 3.97 | 3.57 | 6 | 58 | 154 | 96 | 78.38% | 1.51 | 1.18 |
| Model_A010 | 3.96 | 3.80 | 6 | 20 | 124 | 104 | 27.02% | 0.65 | 0.17 |
| Model_A009 | 3.97 | 3.97 | 6 | 57 | 141 | 84 | 77.02% | 1.62 | 1.25 |
| Model_A002 | 3.97 | 4.06 | 6 | 57 | 148 | 91 | 77.02% | 1.54 | 1.19 |
| Model_B014 | 3.81 | 1.76 | 5 | 11 | 13 | 2 | 14.87% | 3.39 | 0.50 |
| Model_B013 | 3.81 | 2.20 | 6 | 10 | 13 | 3 | 13.52% | 3.08 | 0.42 |
| Model_B008 | 3.81 | 1.01 | 6 | 14 | 31 | 17 | 18.92% | 1.81 | 0.34 |
| Model_B015 | 3.66 | 3.33 | 6 | 11 | 20 | 9 | 14.87% | 2.20 | 0.33 |
| Model_B006 | 3.81 | 2.01 | 4 | 14 | 48 | 34 | 18.92% | 1.17 | 0.22 |
| Model_C002 | 4.02 | 13.66 | 4 | 39 | 41 | 2 | 52.70% | 3.81 | 2.00 |
| Model_C001 | 4.02 | 19.73 | 4 | 39 | 42 | 3 | 52.70% | 3.71 | 1.96 |
| Model_C003 | 4.02 | 13.87 | 4 | 39 | 44 | 5 | 52.70% | 3.55 | 1.87 |
| Model_C004 | 4.02 | 12.78 | 4 | 39 | 45 | 6 | 52.70% | 3.47 | 1.83 |
| Model_C010 | 4.02 | 14.99 | 4 | 40 | 46 | 6 | 54.05% | 3.48 | 1.88 |
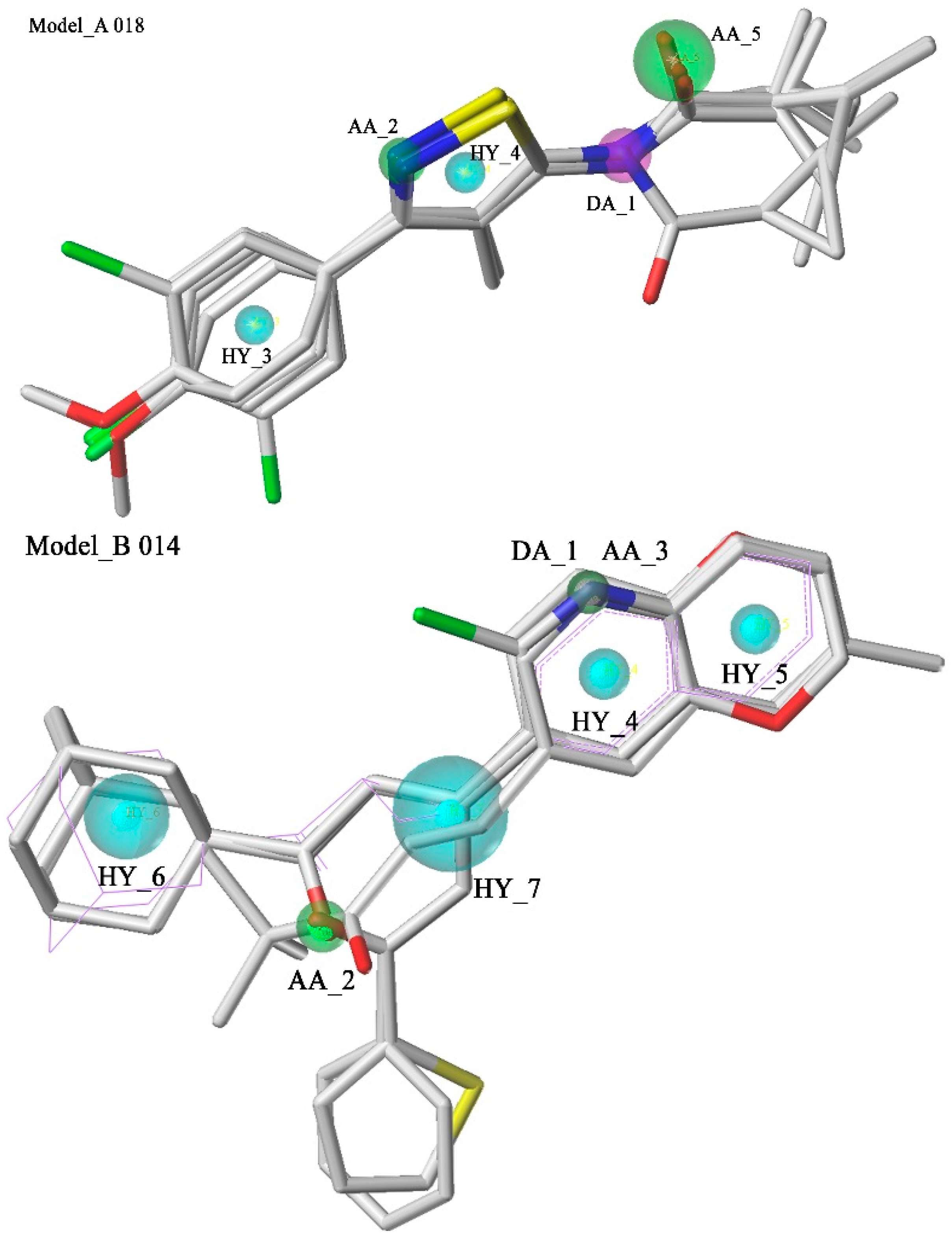
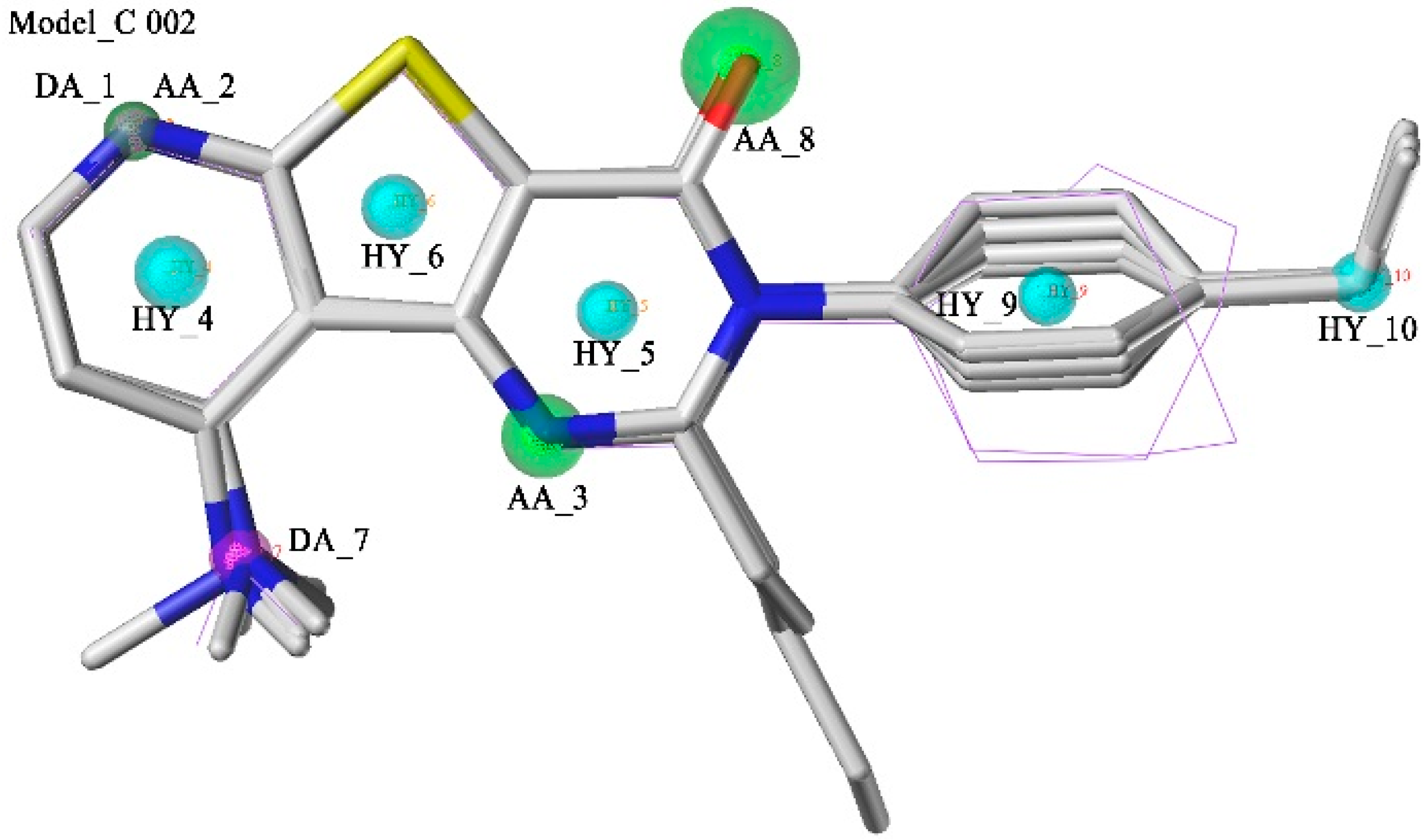
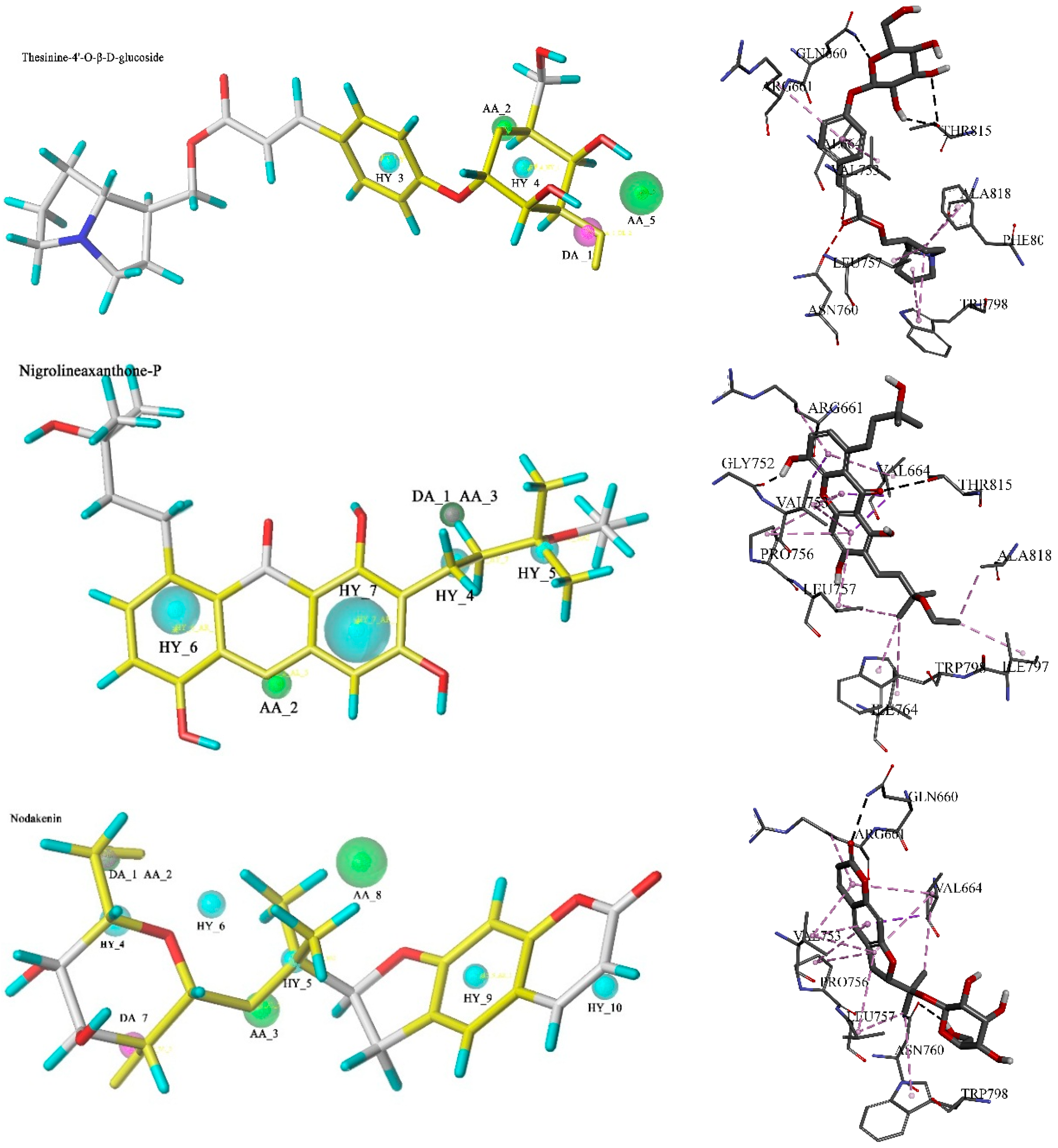
2.2. Database Searching
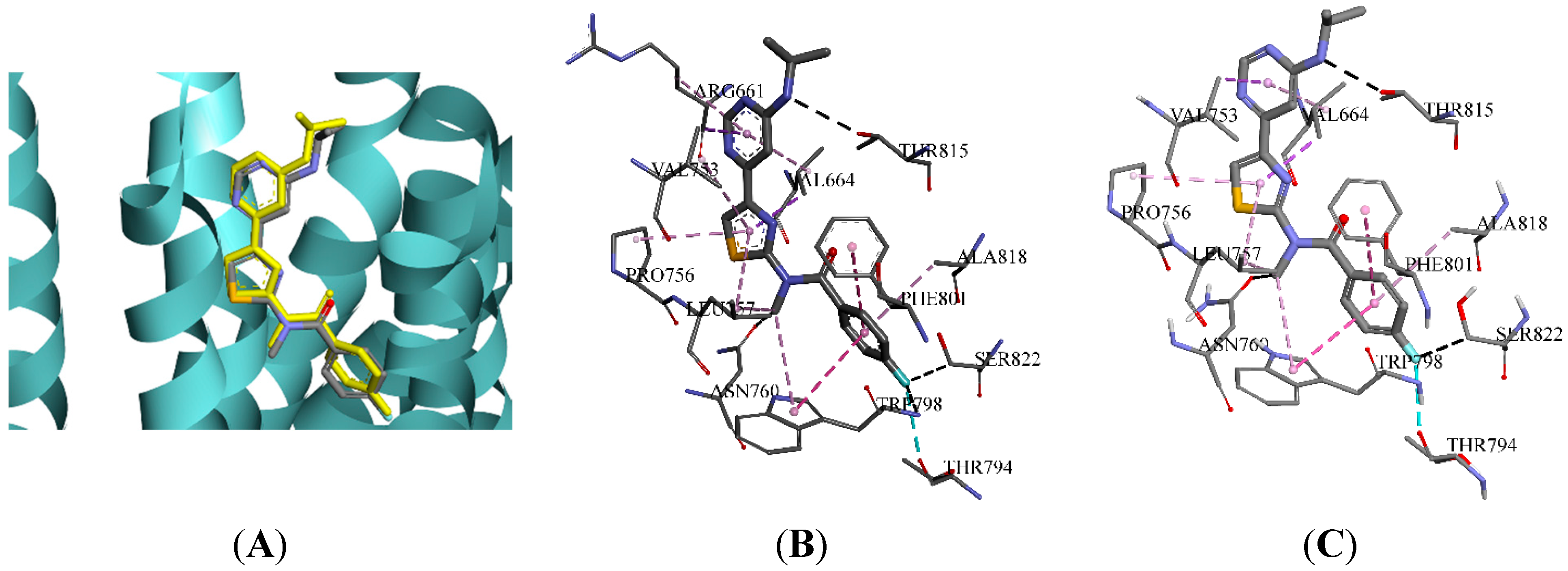
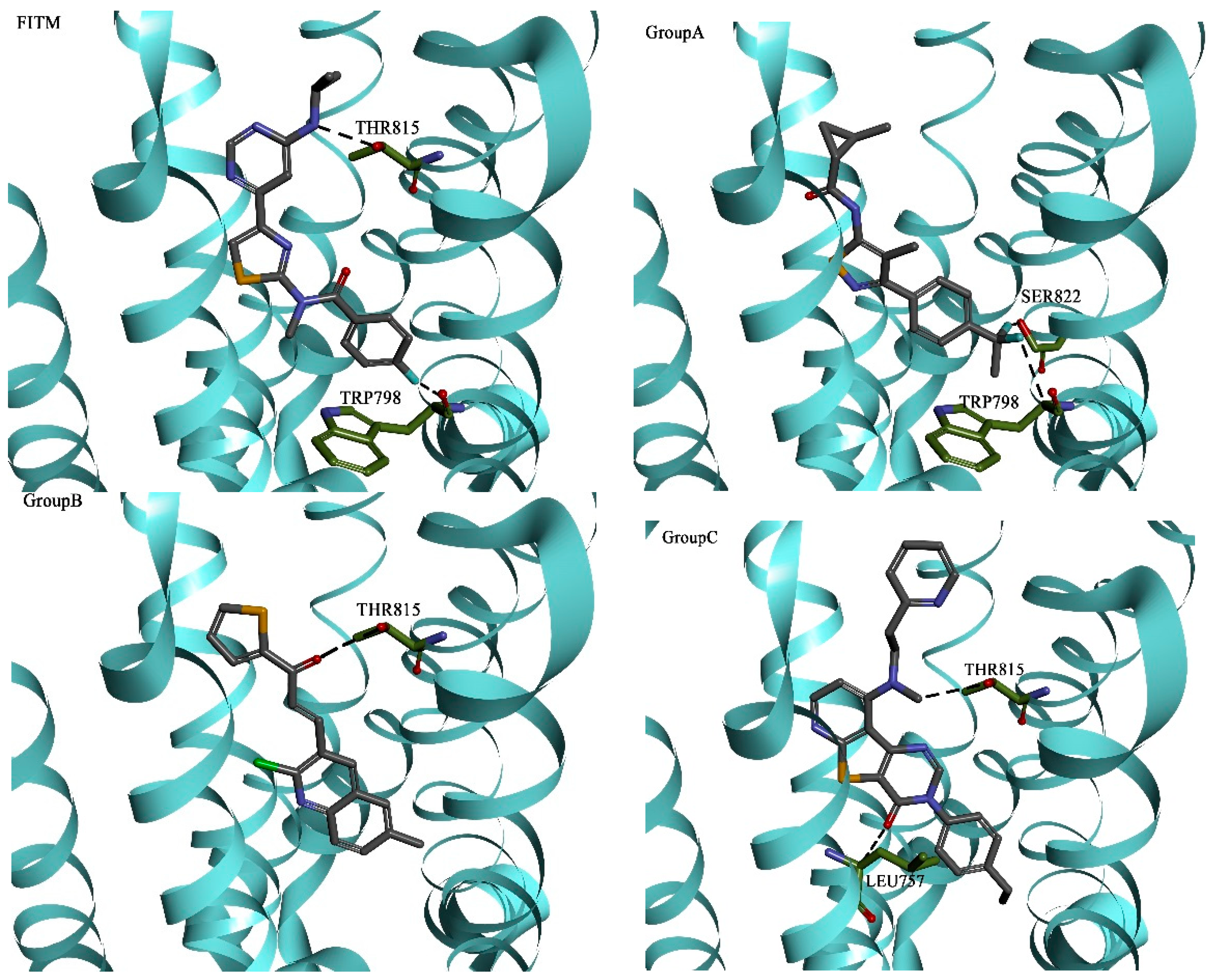
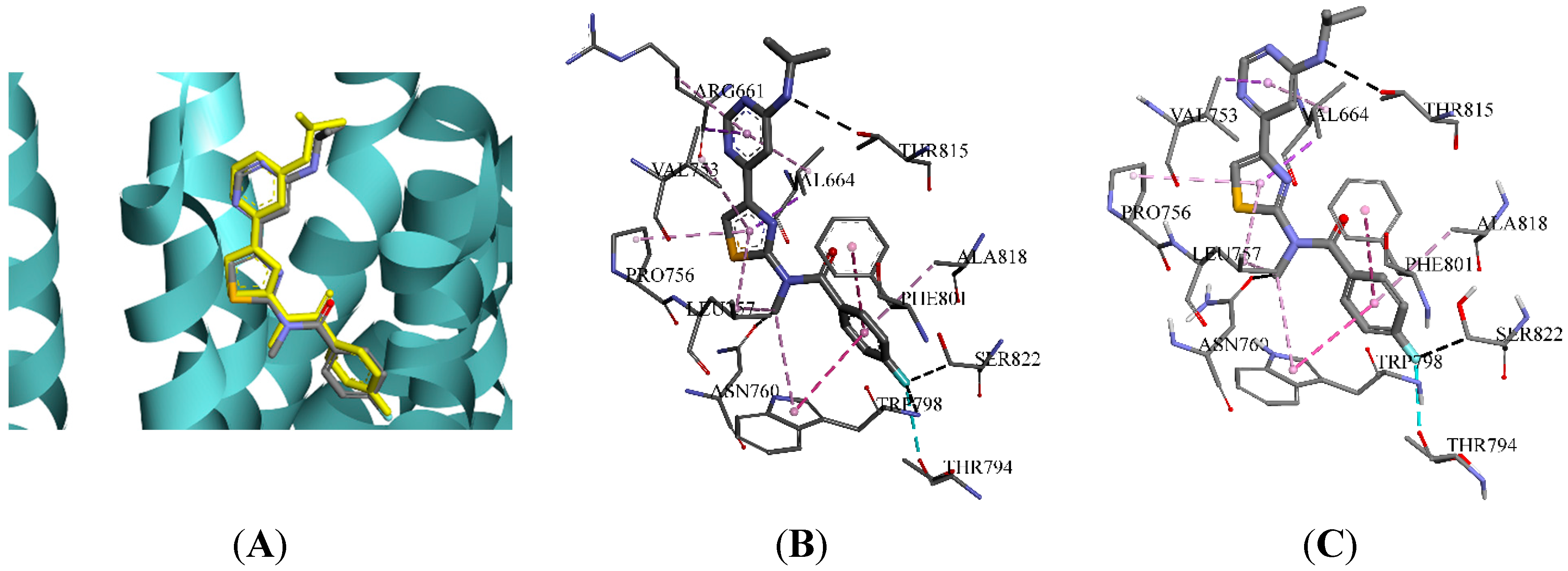
2.3. Molecular Docking and Database Search
| Indexes | 4OR2_A | 4OR2_B | |
|---|---|---|---|
| LibDock | RMSD | 0.56 Å | 3.89 Å |
| LibDock Score | 143.73 | 146.22 | |
| CDOCKER | RMSD | 1.18 Å | 1.12 Å |
| -CDOCKER_ENERGY | 40.80 | 43.90 | |
| -CDOCKER_INTERACTION_ENERGY | 55.74 | 58.46 | |
| Flexible Docking | RMSD | 1.17 Å | 1.21 Å |
| -CDOCKER_ENERGY | 44.15 | 38.56 | |
| -CDOCKER_INTERACTION_ENERGY | 58.36 | 55.79 | |
| LibDock Score | 127.83 | 139.35 | |
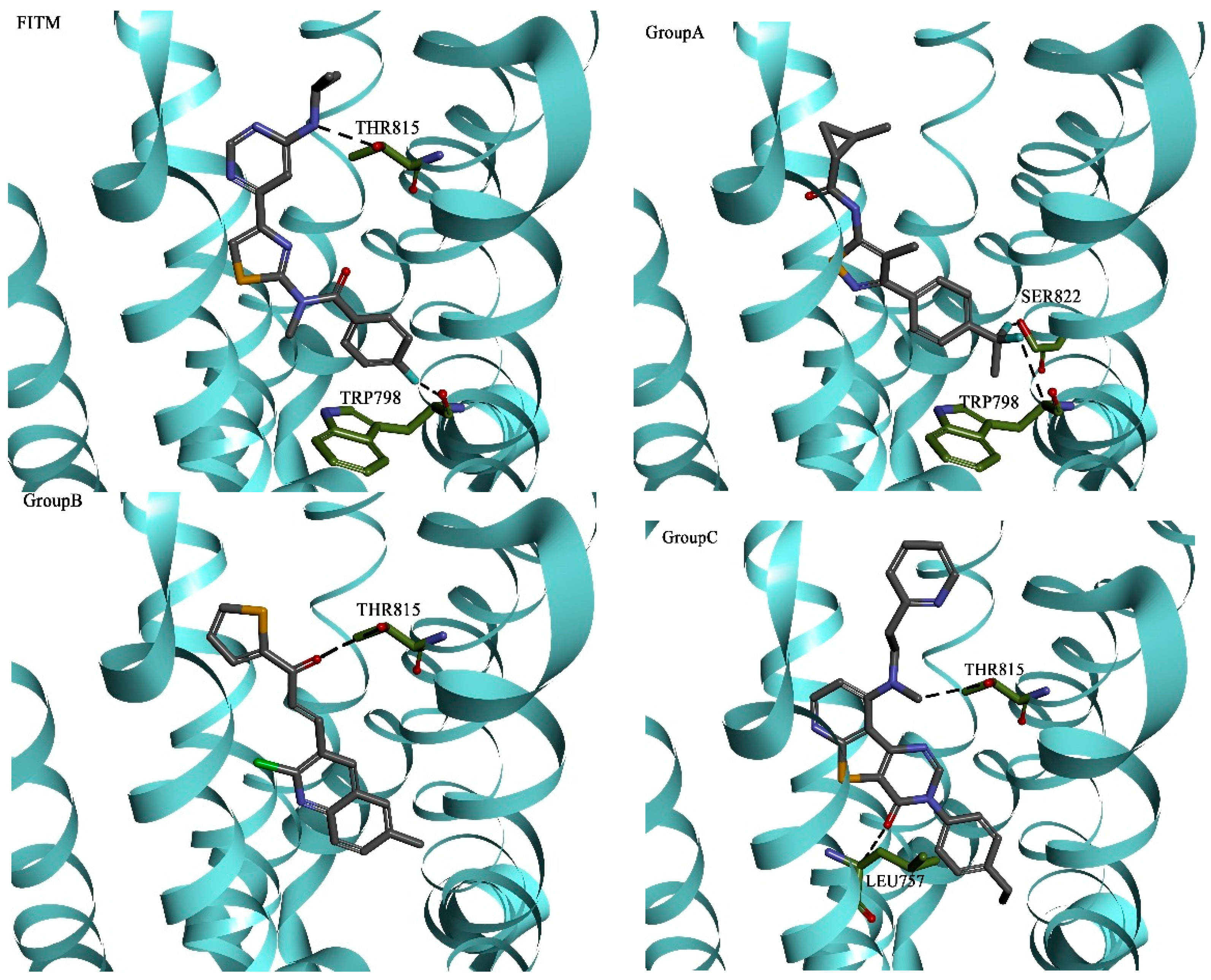
| Hydrogen Bonding Interactions | Hydrophobic Interactions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group A | TRP798 | LEU757 | ARG661 | THR815 | THR794 | VAL664 | LEU757 | PHE801 | VAL753 | PRO756 |
| Group B | LEU757 | TRP798 | GLY665 | ARG661 | THR815 | VAL664 | VAL753 | LEU757 | ALA818 | PRO756 |
| Group C | ARG661 | GLY752 | THR815 | LEU757 | ASN760 | VAL664 | VAL753 | PRO756 | LEU757 | ARG661 |
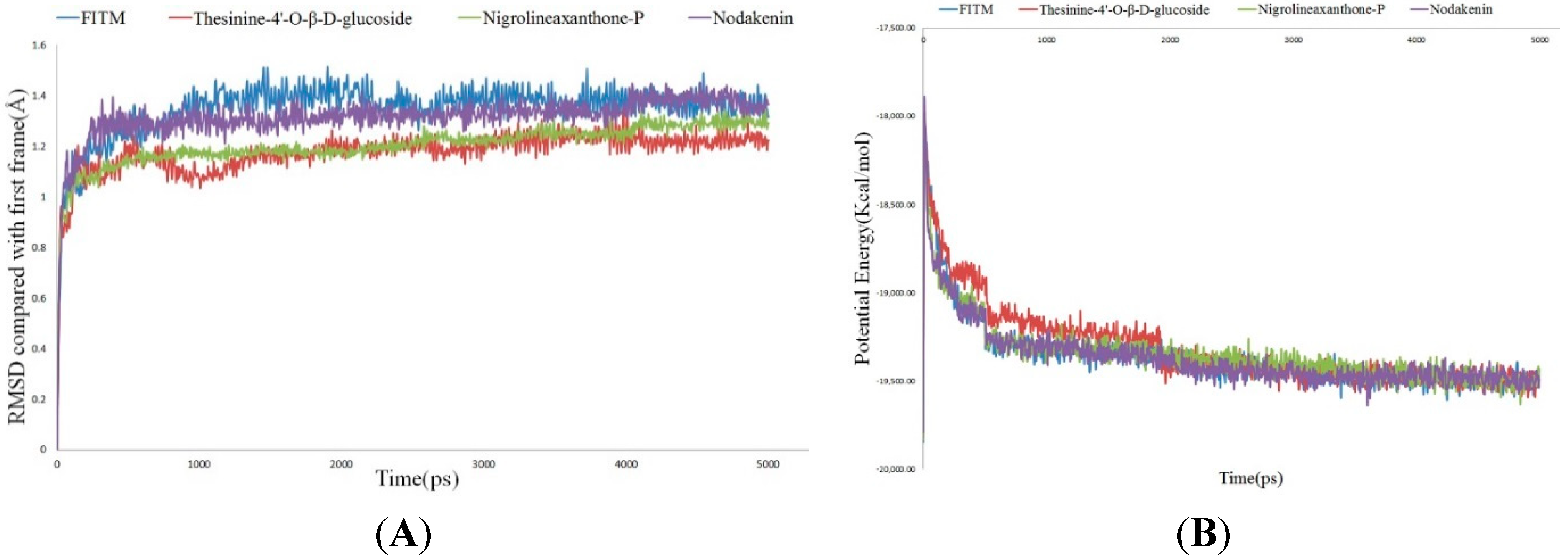
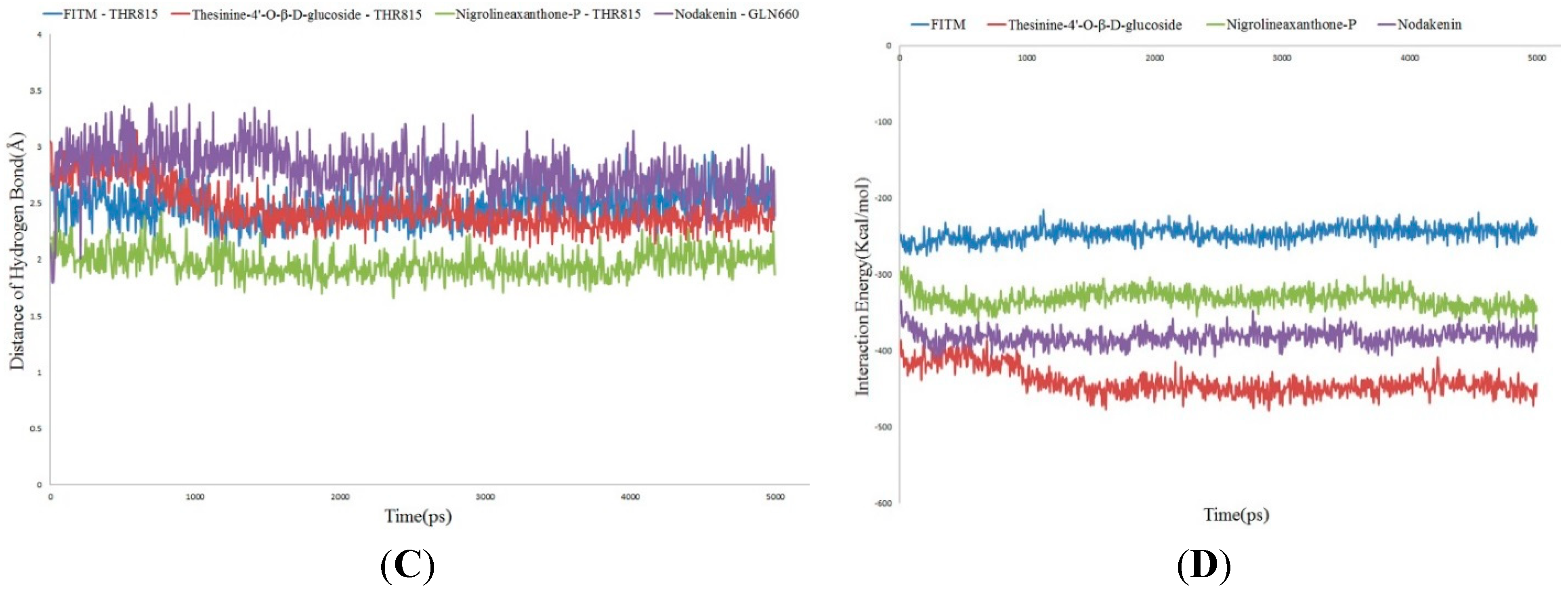
2.4. Molecular Dynamics Simulation
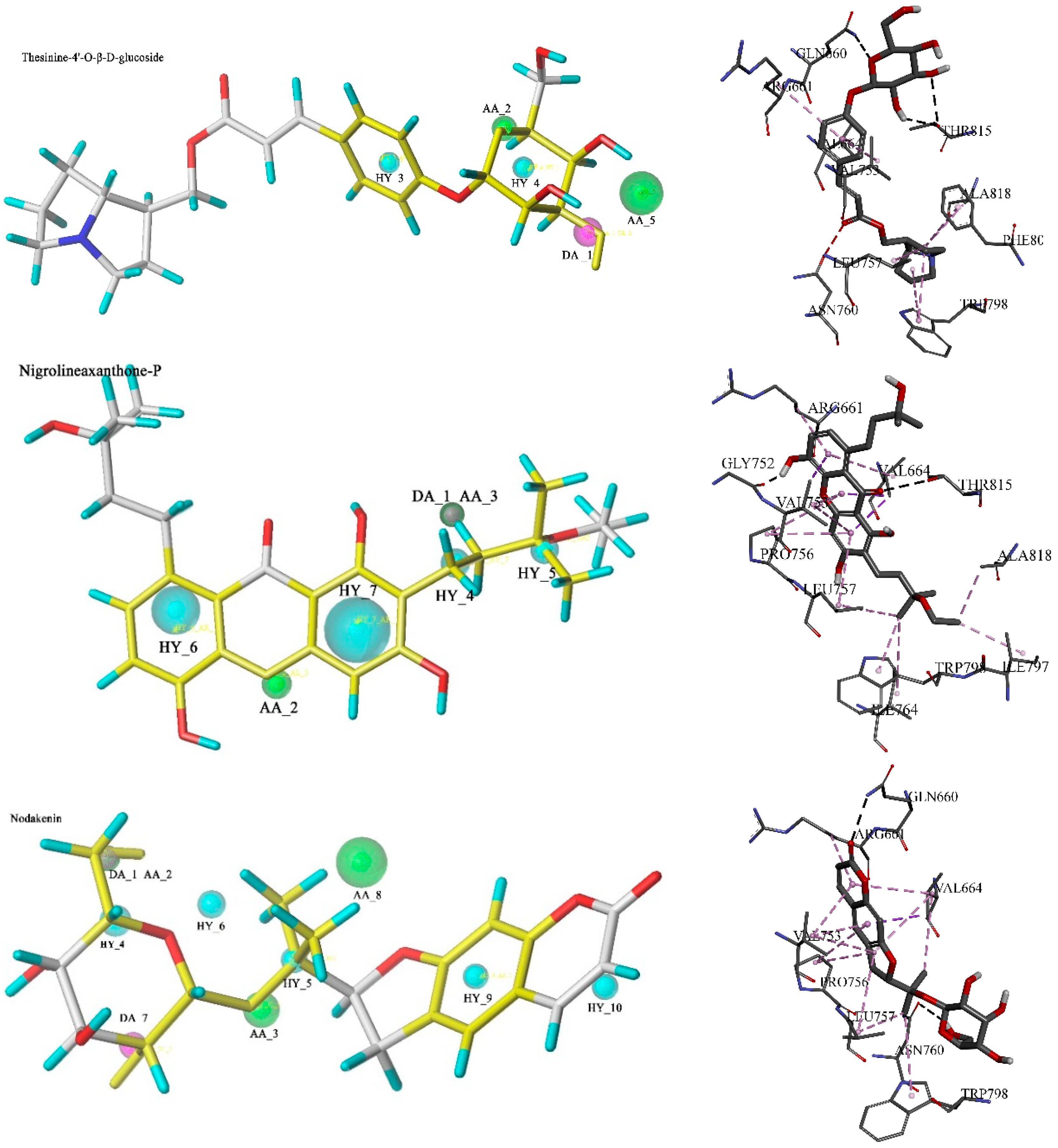
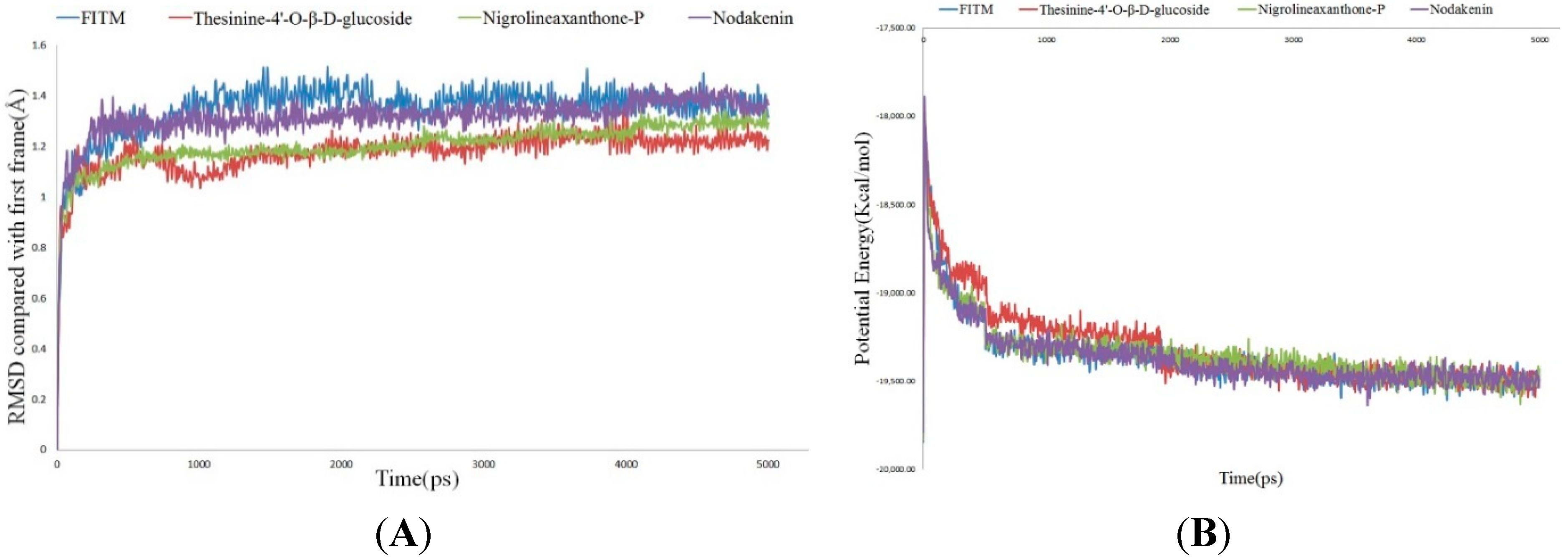
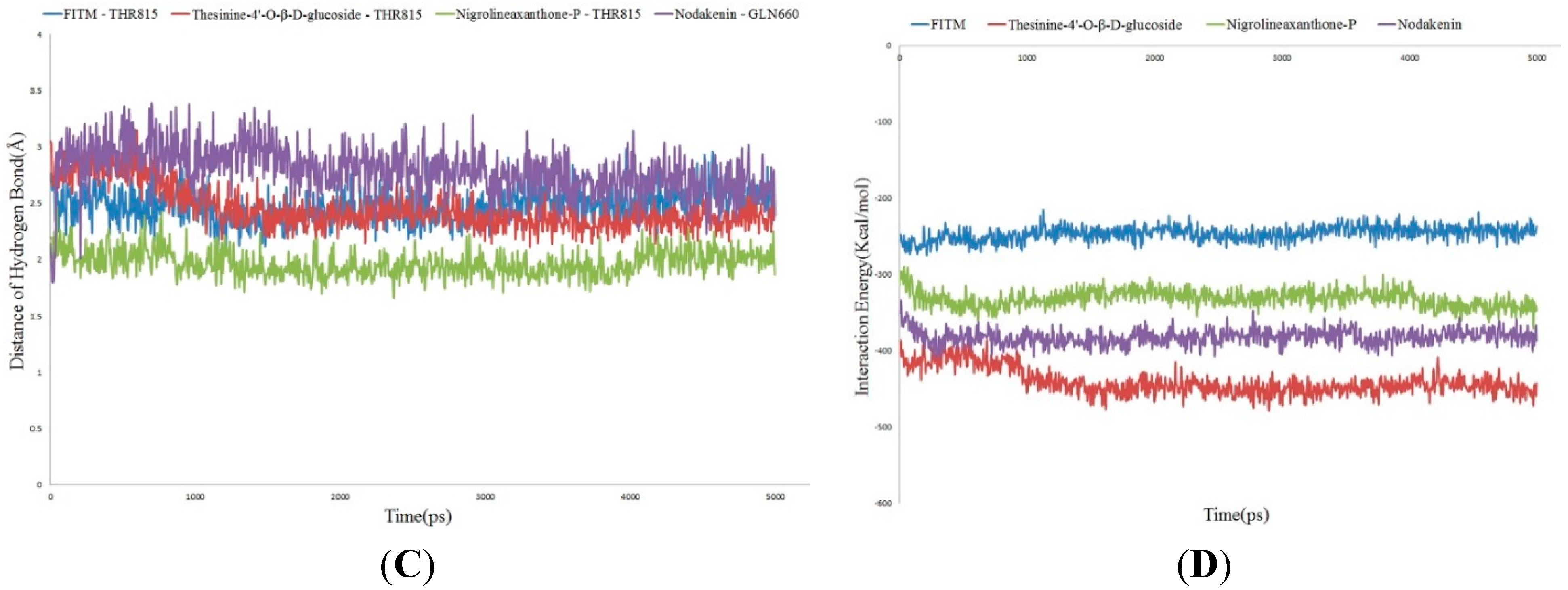
3. Experimental Section
3.1. Data Collection and Preparation
3.2. Pharmacophore Model Studies
3.2.1. GALAHAD Pharmacophore Hypotheses Generation
3.2.2. Validation of the Pharmacophore Model
3.3. Database Search
3.4. Molecular Docking Studies
3.4.1. Define Binding Site
3.4.2. Docking Strategy
3.5. Molecular Dynamics Simulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in gpcr allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Flight, M.H. Drug discovery structure-led design. Nature 2013, 502, S50–S52. [Google Scholar] [CrossRef] [PubMed]
- Engers, D.W.; Lindsley, C.W. Allosteric modulation of class c gpcrs: A novel approach for the treatment of cns disorders. Drug Discov. Today Technol. 2013, 10, e269–e276. [Google Scholar] [CrossRef] [PubMed]
- Durand, D.; Pampillo, M.; Caruso, C.; Lasaga, M. Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology 2008, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Molck, C.; Harpsoe, K.; Gloriam, D.E.; Mathiesen, J.M.; Nielsen, S.M.; Braeuner Osborne, H. Mglur5: Exploration of orthosteric and allosteric ligand binding pockets and their applications to drug discovery. Neurochem. Res. 2014, 39, 1862–1875. [Google Scholar] [CrossRef] [PubMed]
- Chun, L.; Zhang, W.H.; Liu, J.F. Structure and ligand recognition of class c gpcrs. Acta Pharmacol. Sin. 2012, 33, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Li, G.B.; Yang, L.L.; Feng, S.; Zhou, J.P.; Huang, Q.; Xie, H.Z.; Li, L.L.; Yang, S.Y. Discovery of novel mglur1 antagonists: A multistep virtual screening approach based on an svm model and a pharmacophore hypothesis significantly increases the hit rate and enrichment factor. Bioorg. Med. Chem. Lett. 2011, 21, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Noeske, T.; Jirgensons, A.; Starchenkovs, I.; Renner, S.; Jaunzeme, L.; Trifanova, D.; Hechenberger, M.; Bauer, T.; Kauss, V.; Parsons, C.G.; et al. Virtual screening for selective allosteric mglur1 antagonists and structure-activity relationship investigations for cournarine derivatives. ChemMedChem 2007, 2, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Vanejevs, M.; Jatzke, C.; Renner, S.; Mueller, S.; Hechenberger, M.; Bauer, T.; Klochkova, A.; Pyatkin, I.; Kazyulkin, D.; Aksenova, E.; et al. Positive and negative modulation of group i metabotropic glutamate receptors. J. Med. Chem. 2008, 51, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Omer, A.; Prasad, C.S. Designing allosteric modulators for active conformational state of m-glutamate g-protein coupled receptors. Bioinformation 2012, 8, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The g-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Niswender, C.M.; Katritch, V.; Meiler, J.; Cherezov, V.; et al. Structure of a class c gpcr metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 2014, 344, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Dore, A.S.; Okrasa, K.; Patel, J.C.; Serrano Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of class c gpcr metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lakkaraju, S.K.; Hanscom, M.; Zhao, Z.; Wu, J.; Stoica, B.; MacKerell, A.D., Jr.; Faden, A.I.; Xue, F. Acyl-2-aminobenzimidazoles: A novel class of neuroprotective agents targeting mglur5. Bioorg. Med. Chem. 2015, 23, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, S.K.; Mbatia, H.; Hanscom, M.; Zhao, Z.; Wu, J.; Stoica, B.; MacKerell, A.D., Jr.; Faden, A.I.; Xue, F. Cyclopropyl-containing positive allosteric modulators of metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem. Lett. 2015, 25, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- King, V.F.; Garcia, M.L.; Himmel, D.; Reuben, J.P.; Lam, Y.K.; Pan, J.X.; Han, G.Q.; Kaczorowski, G.J. Interaction of tetrandrine with slowly inactivating calcium channels. Characterization of calcium channel modulation by an alkaloid of Chinese medicinal herb origin. J. Biol. Chem. 1988, 263, 2238–2244. [Google Scholar] [CrossRef]
- Alexeev, M.; Grosenbaugh, D.K.; Mott, D.D.; Fisher, J.L. The natural products magnolol and honokiol are positive allosteric modulators of both synaptic and extra-synaptic gaba(a) receptors. Neuropharmacology 2012, 62, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.; Chung, K.; Ng, K.; Yu, Y.; Ziea, E.T.; Ng, B.F. A systematic review on the efficacy, safety and types of chinese herbal medicine for depression. J. Psychiatr. Res. 2014, 57, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zuo, P. Neural regeneration: Role of traditional chinese medicine in neurological diseases treatment. J. Pharmacol. Sci. 2012, 120, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gomez Santacana, X.; Rovira, X.; Dalton, J.A.; Goudet, C.; Pin, J.P.; Gorostiza, P.; Giraldo, J.; Llebaria, A. A double effect molecular switch leads to a novel potent negative allosteric modulator of metabotropic glutamate receptor 5. MedChemComm 2014, 5, 1548–1554. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, Y.; Shen, K.; Wang, X.; Li, F.; He, Y. The discovery of potentially selective human neuronal nitric oxide synthase (nnos) inhibitors: A combination of pharmacophore modelling, comfa, virtual screening and molecular docking studies. Int. J. Mol. Sci. 2014, 15, 8553–8569. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, Z.; He, Y.; Xiang, Y.; Zhang, Y.; Qiao, Y. A combination of pharmacophore modeling, molecular docking and virtual screening for inos inhibitors from chinese herbs. Biomed. Mater. Eng. 2014, 24, 1315–1322. [Google Scholar] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, J.; Suzuki, G.; Kimura, T.; Nagatomi, Y.; Ito, S.; Kawamoto, H.; Ozaki, S.; Ohta, H. Identification of a novel transmembrane domain involved in the negative modulation of mglur1 using a newly discovered allosteric mglur1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4h-chromen-4-one. Neuropharmacology 2009, 57, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, P.; Kratochwil, N.; Knoflach, F.; Zenner, M.-T.; Kew, J.N.C.; Kratzeisen, C.; Maerki, H.P.; Adam, G.; Mutel, V. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J. Biol. Chem. 2003, 278, 8340–8347. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.P.; Engers, D.W.; Venable, D.F.; Niswender, C.M.; Lindsley, C.W.; Conn, P.J.; Emmitte, K.A.; Rodriguez, A.L. A novel class of succinimide-derived negative allosteric modulators of metabotropic glutamate receptor subtype 1 provides insight into a disconnect in activity between the rat and human receptors. ACS Chem. Neurosci. 2014, 5, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhu, L.; Cao, Y.; Wu, G.; Liu, X.; Chen, Y.; Wang, Q.; Shi, T.; Zhao, Y.; Wang, Y.; et al. Asd: A comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011, 39, D663–D669. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.J.; Backer, R.T.; Barth, V.N.; Garbison, K.E.; Gruber, J.M.; Heinz, B.A.; Iyengar, S.; Hollinshead, S.P.; Kingston, A.; Kuklish, S.L.; et al. 3-Phenyl-5-isothiazole carboxamides with potent mglur1 antagonist activity. Bioorg. Med. Chem. Lett. 2012, 22, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Noeske, T.; Trifanova, D.; Kauss, V.; Renner, S.; Parsons, C.G.; Schneider, G.; Weil, T. Synergism of virtual screening and medicinal chemistry: Identification and optimization of allosteric antagonists of metabotropic glutamate receptor 1. Bioorg. Med. Chem. 2009, 17, 5708–5715. [Google Scholar] [CrossRef] [PubMed]
- Nataraja Sekhar, Y.; Ravikumar, M.; Ravi Shashi Nayana, M.; Mallena, S.C.; Kishore Kumar, M. 3D-QSAR studies of triazafluorenone inhibitors of metabotropic glutamate receptor subtype 1. Eur. J. Med. Chem. 2008, 43, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Smellie, A.; Teig, S.L.; Towbin, P. Poling: Promoting conformational variation. J. Comput. Chem. 1995, 16, 171–187. [Google Scholar] [CrossRef]
- Holland, J.H. Genetic algorithms. Sci. Am. 1992, 267, 66. [Google Scholar] [CrossRef]
- Richmond, N.J.; Willett, P.; Clark, R.D. Alignment of three-dimensional molecules using an image recognition algorithm. J. Mol. Graph. Model. 2004, 23, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, S.J.; Gillet, V.J.; Taylor, R.; Wilton, D.J. Generation of multiple pharmacophore hypotheses using multiobjective optimisation techniques. J. Comput. Aided Mol. Des. 2004, 18, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Xiang, Y.; Ren, Z.; Qiao, Y. Pharmacophore model generation of thrombin inhibitors. J. Softw. Eng. Appl. 2012, 5, 84–87. [Google Scholar] [CrossRef]
- He, Y.; Jiang, L.; Yang, Z.; Qiao, Y.; Zhang, Y. A combination of pharmacophore modeling, molecular docking, and virtual screening for p2y12receptor antagonists from chinese herbs. Can. J. Chem. 2015, 93, 311–316. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a g-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Zhang, X.; Chen, X.; He, Y.; Qiao, L.; Zhang, Y.; Li, G.; Xiang, Y. Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs. Molecules 2015, 20, 12769-12786. https://doi.org/10.3390/molecules200712769
Jiang L, Zhang X, Chen X, He Y, Qiao L, Zhang Y, Li G, Xiang Y. Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs. Molecules. 2015; 20(7):12769-12786. https://doi.org/10.3390/molecules200712769
Chicago/Turabian StyleJiang, Ludi, Xianbao Zhang, Xi Chen, Yusu He, Liansheng Qiao, Yanling Zhang, Gongyu Li, and Yuhong Xiang. 2015. "Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs" Molecules 20, no. 7: 12769-12786. https://doi.org/10.3390/molecules200712769
APA StyleJiang, L., Zhang, X., Chen, X., He, Y., Qiao, L., Zhang, Y., Li, G., & Xiang, Y. (2015). Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs. Molecules, 20(7), 12769-12786. https://doi.org/10.3390/molecules200712769