
Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
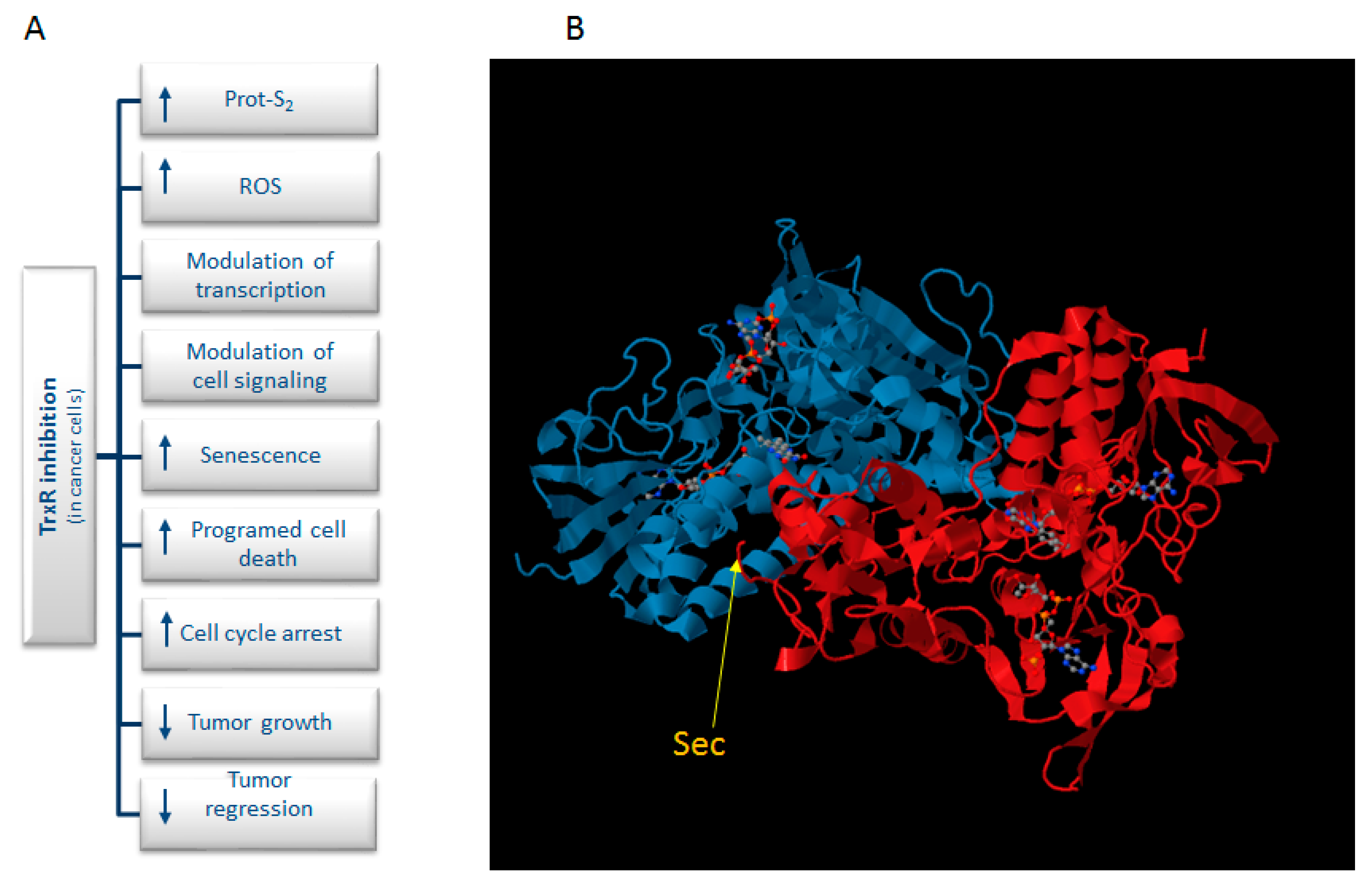
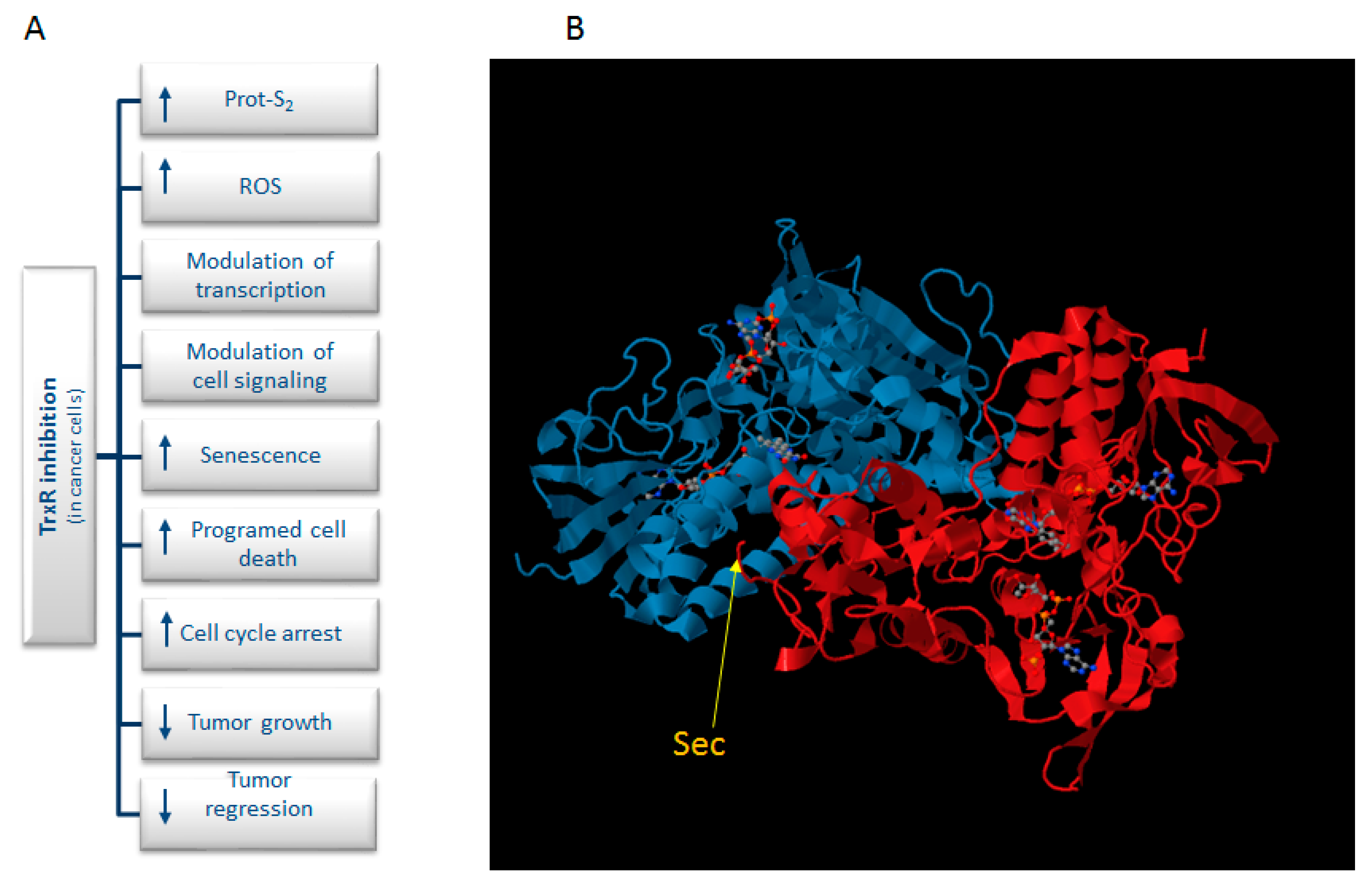
:1. Introduction
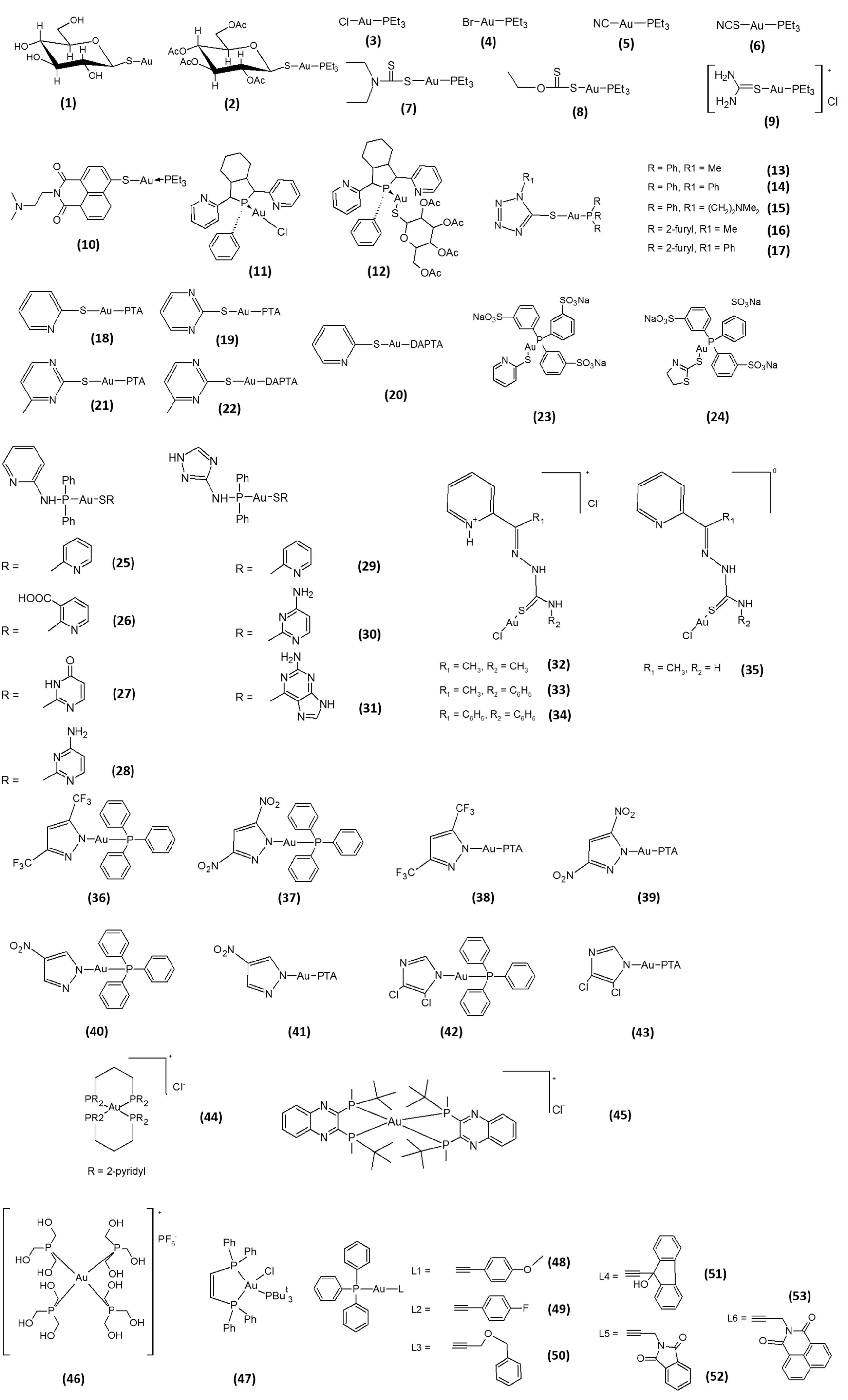
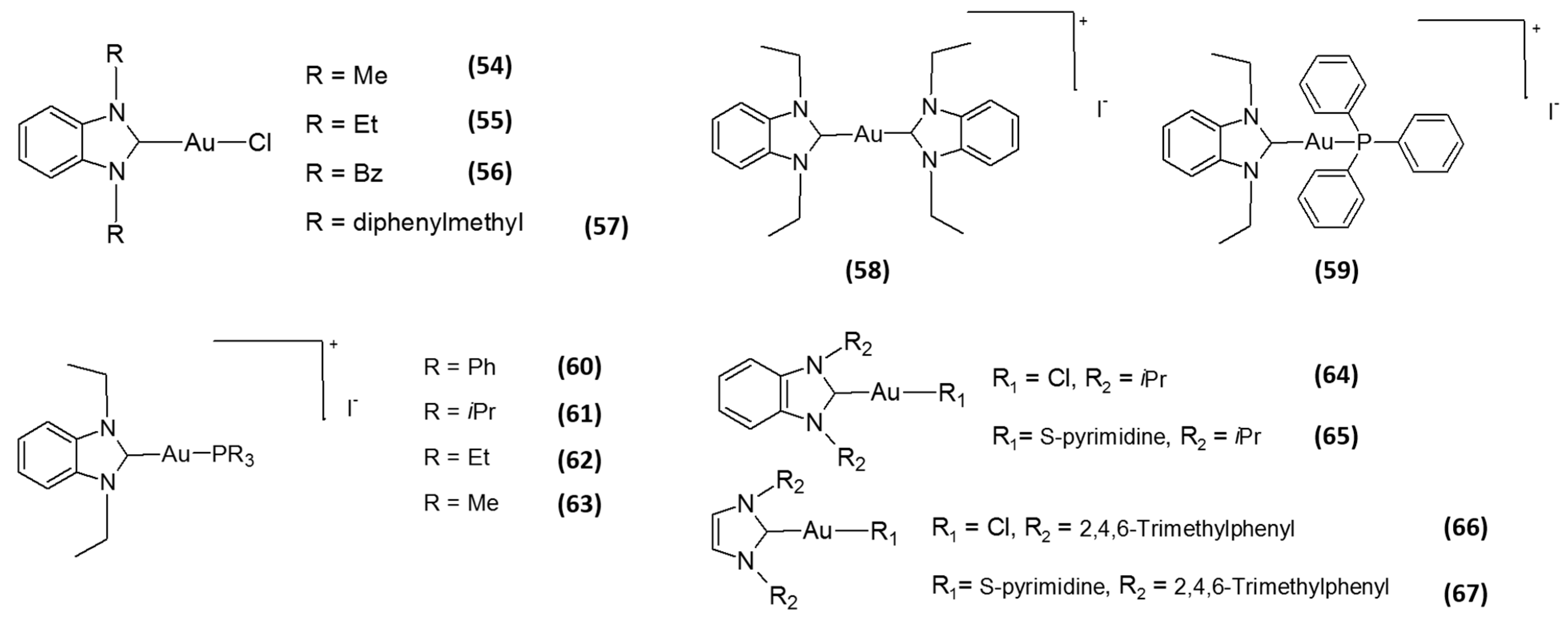
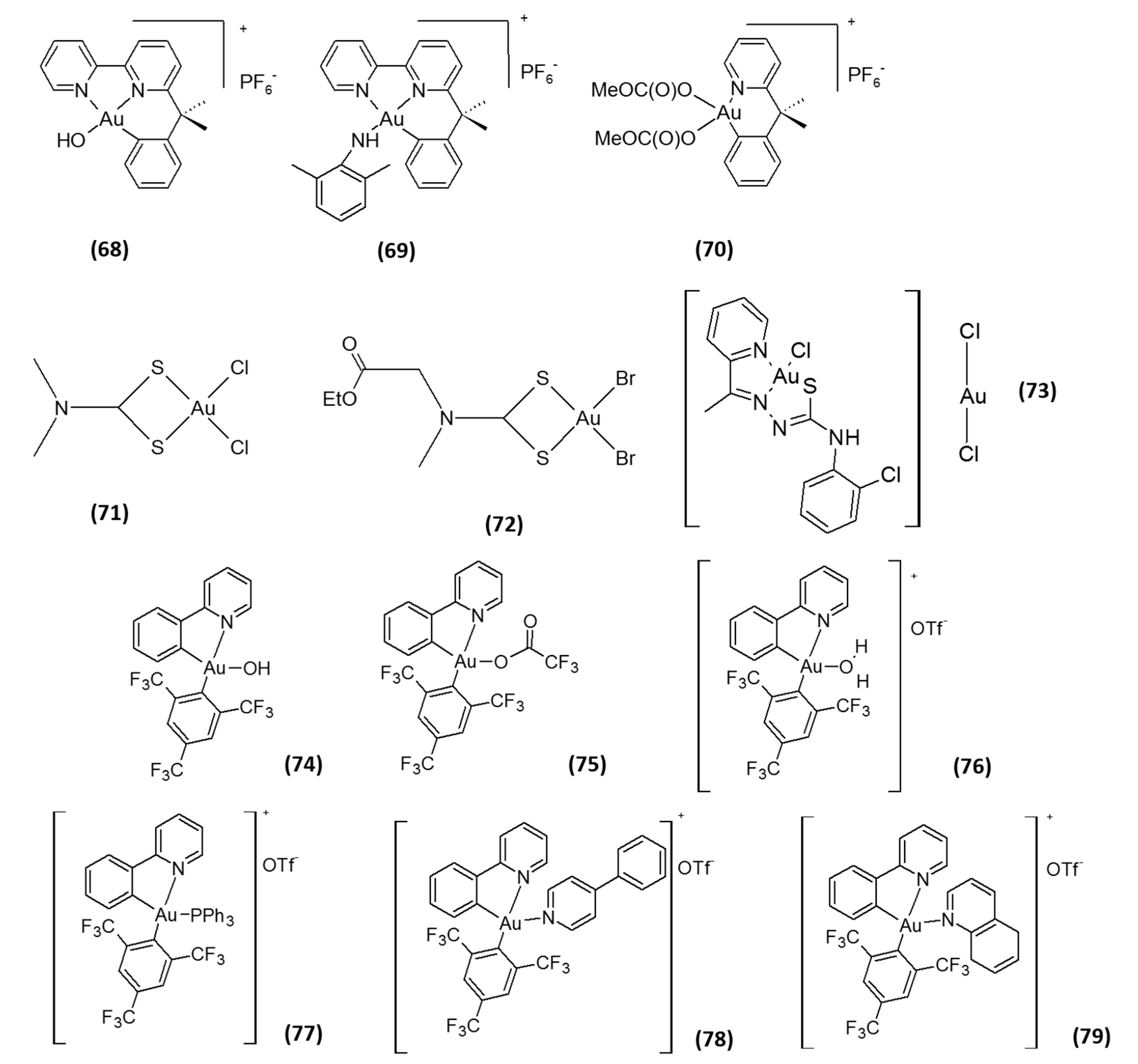
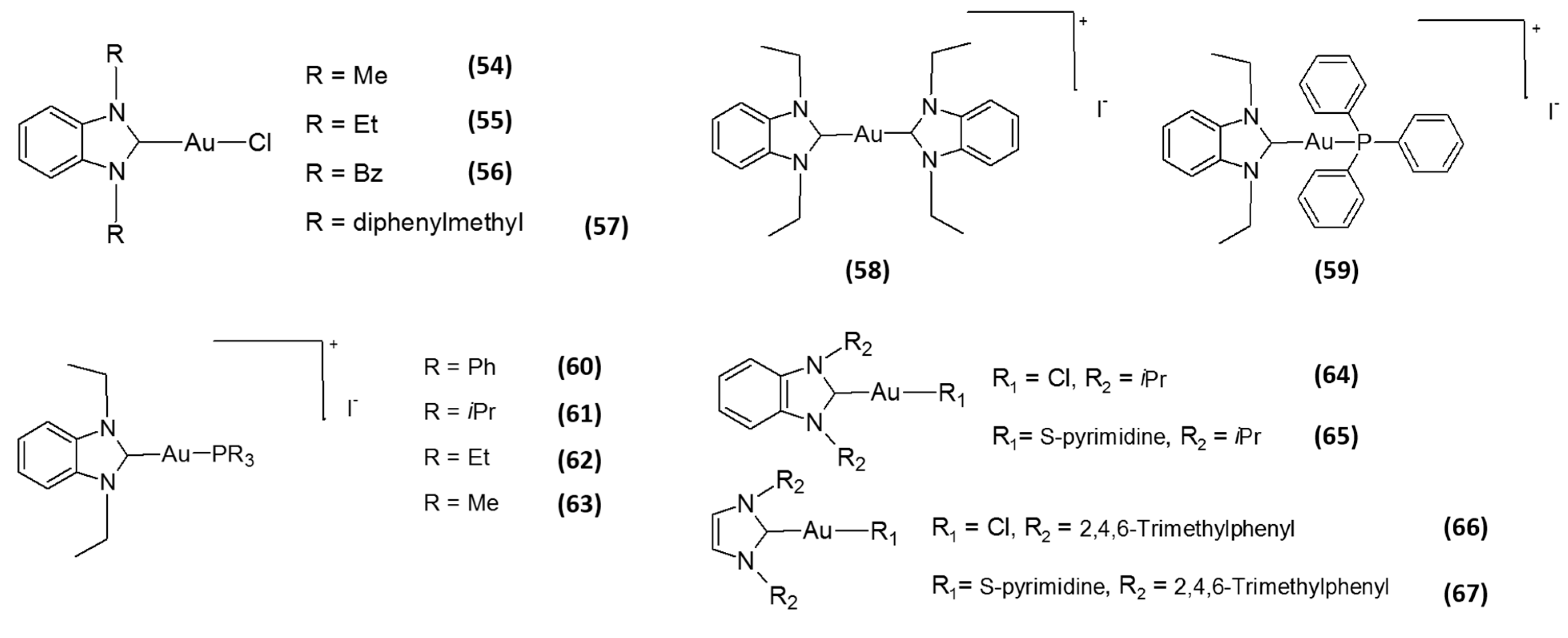
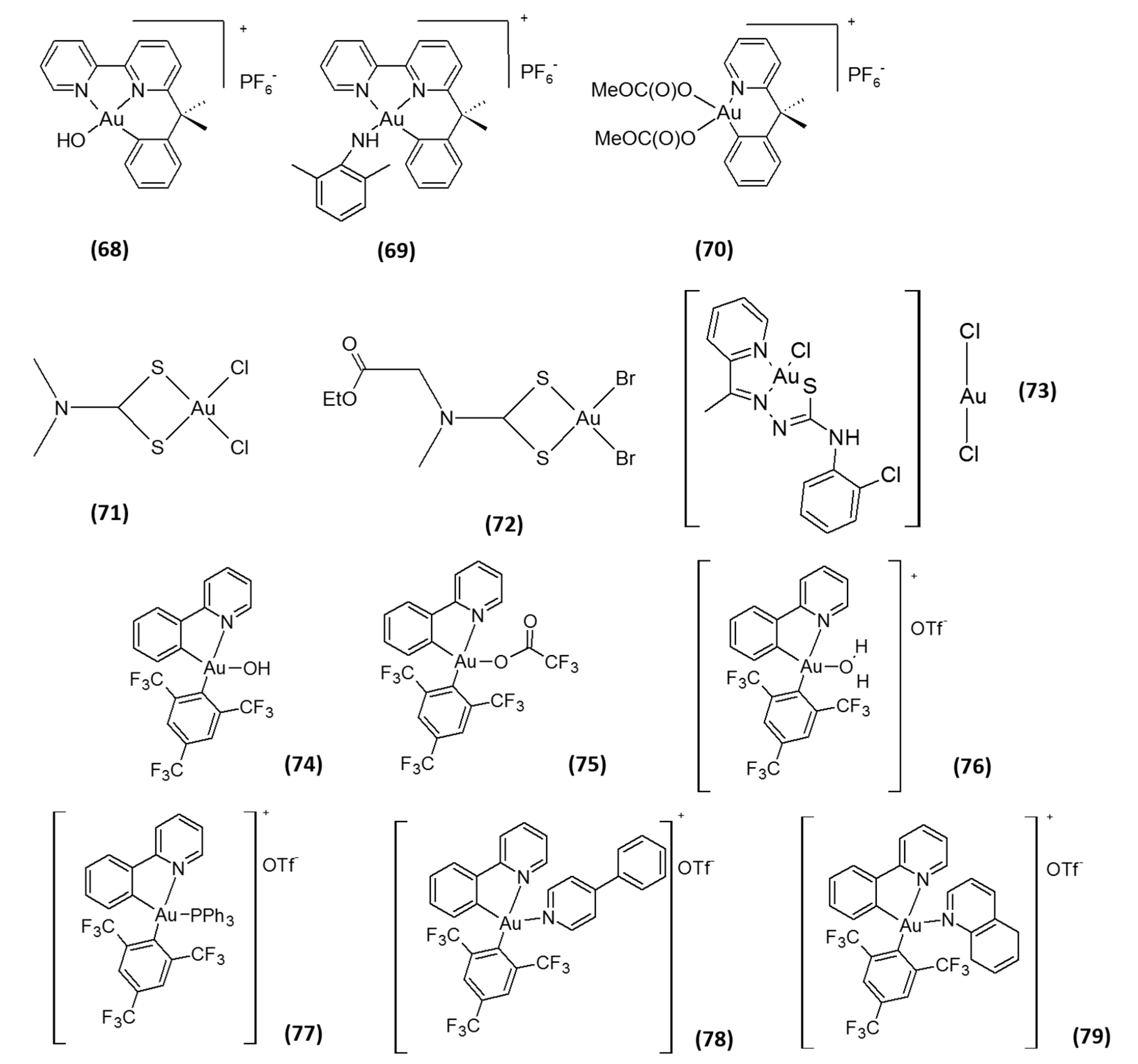
2. Gold-Based Inhibitors

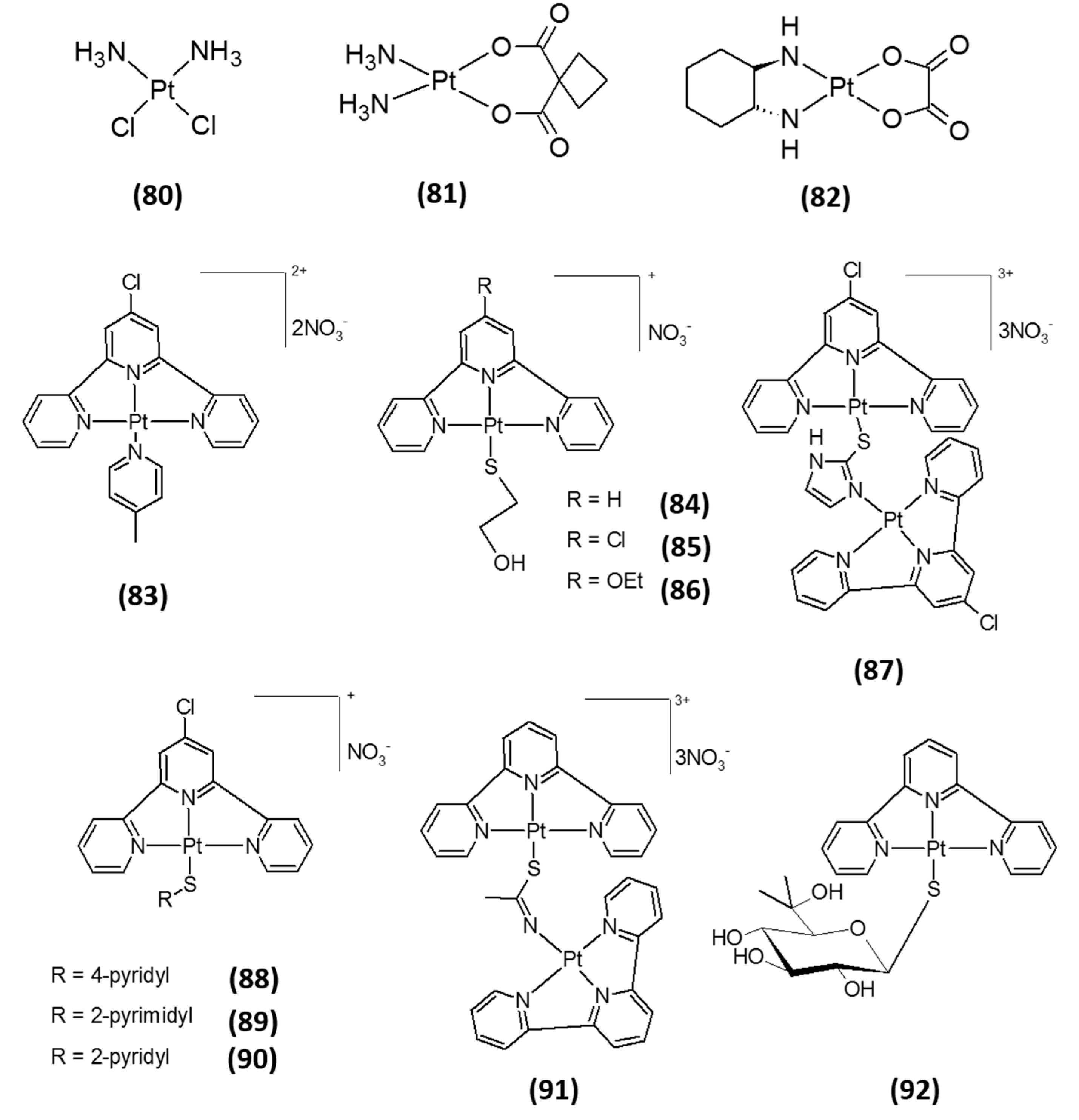
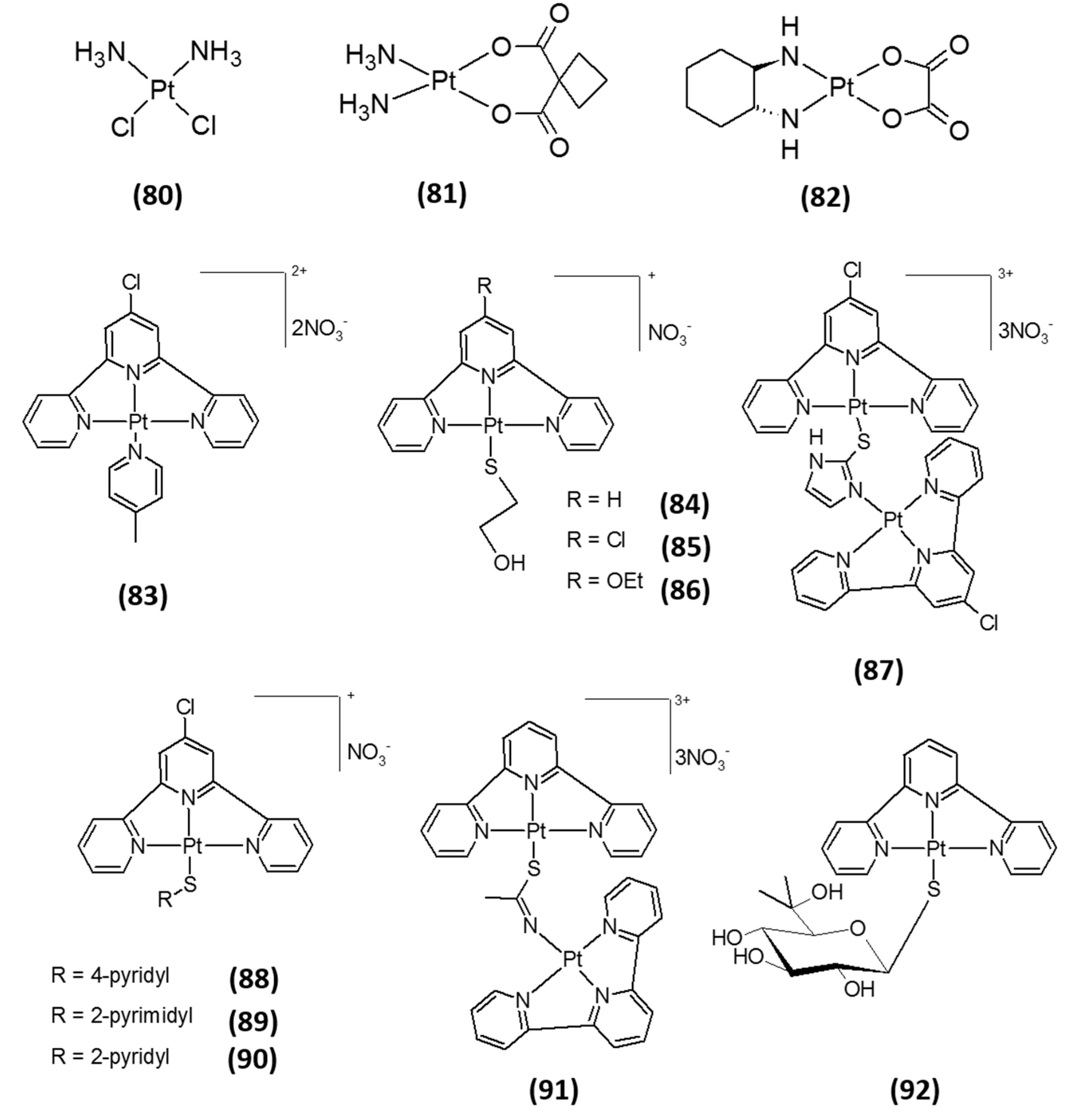
3. Platinum-Based Inhibitors
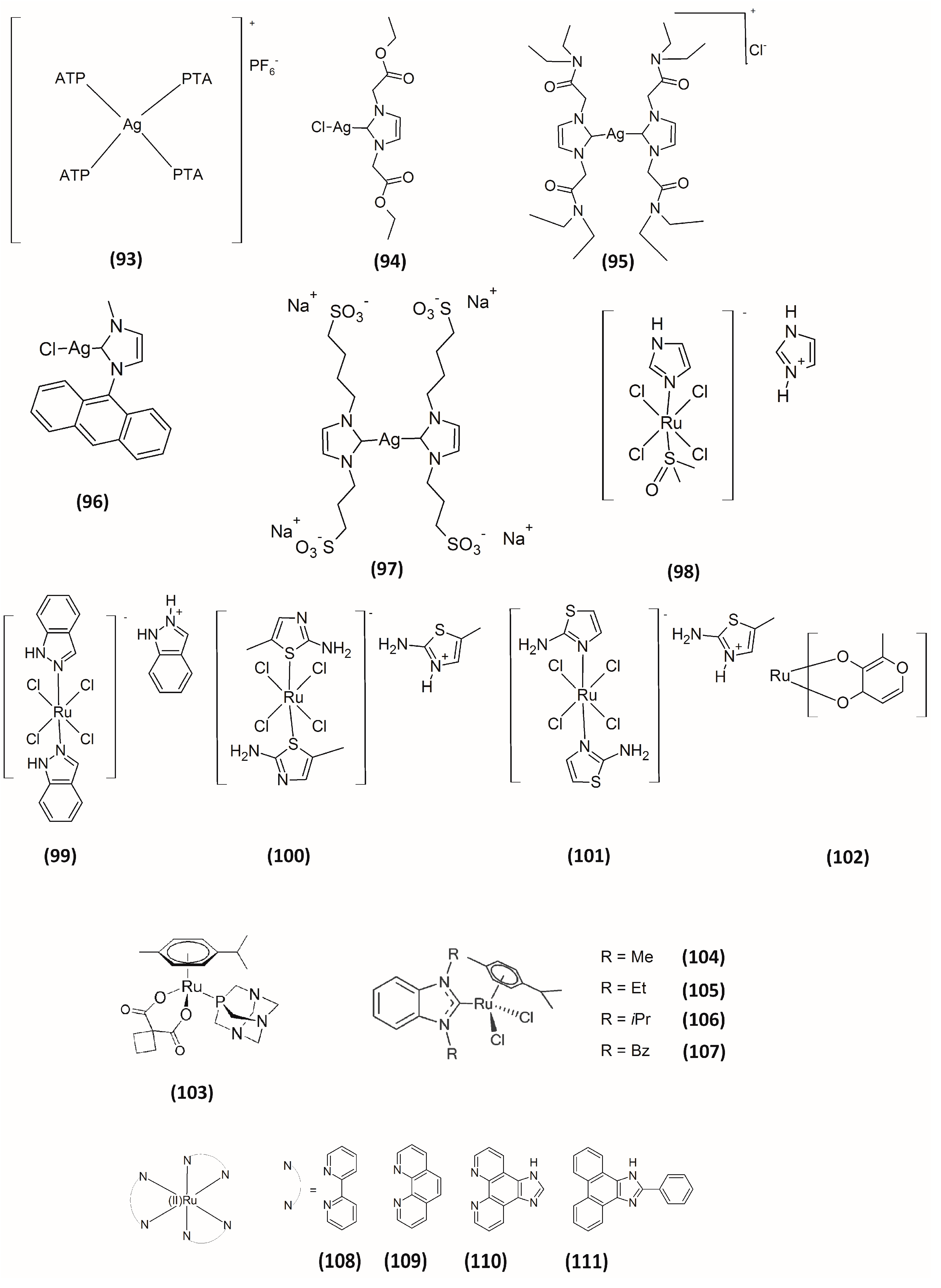
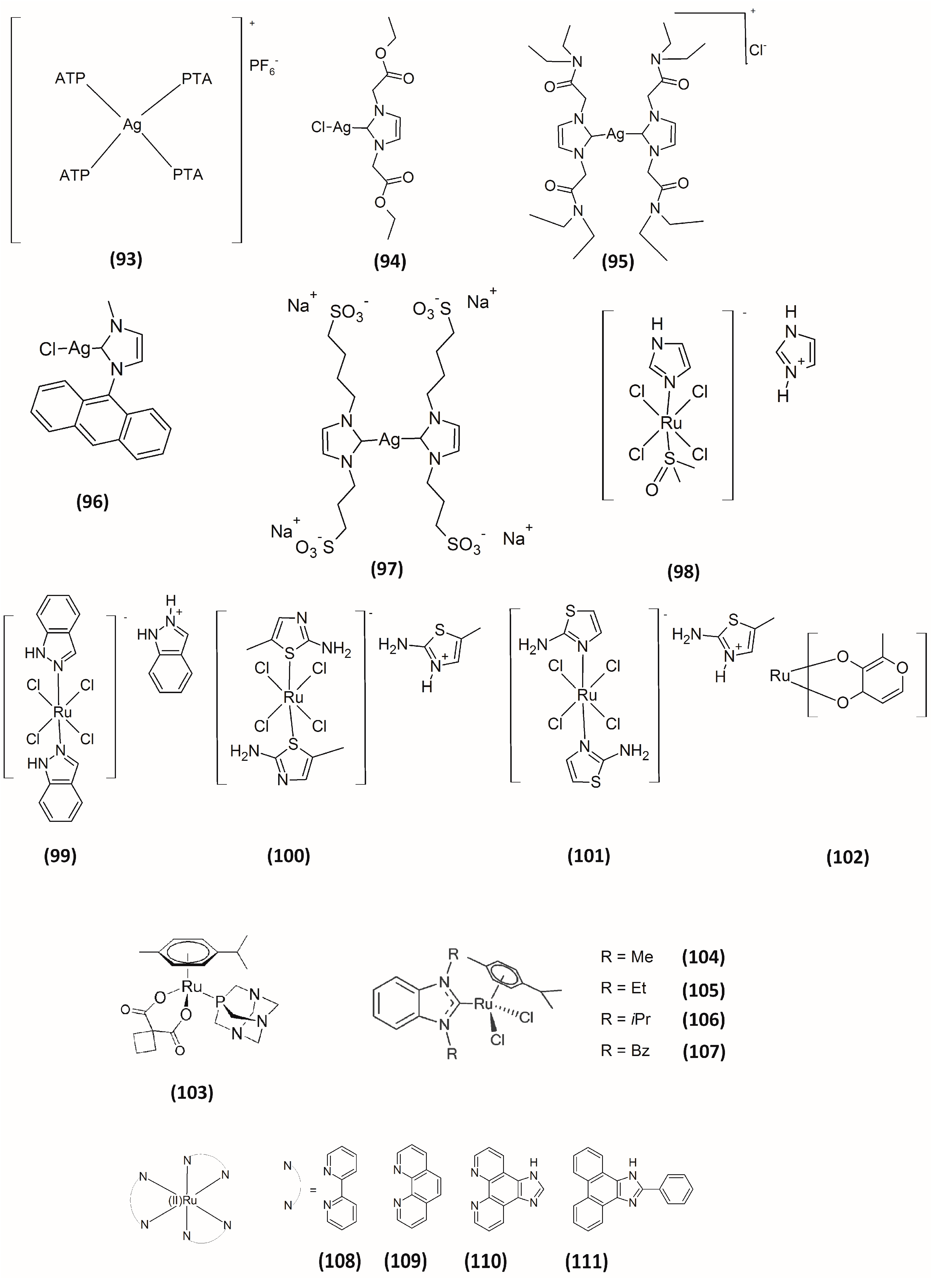
4. Silver-Based Inhibitors
5. Ruthenium-Based Inhibitors
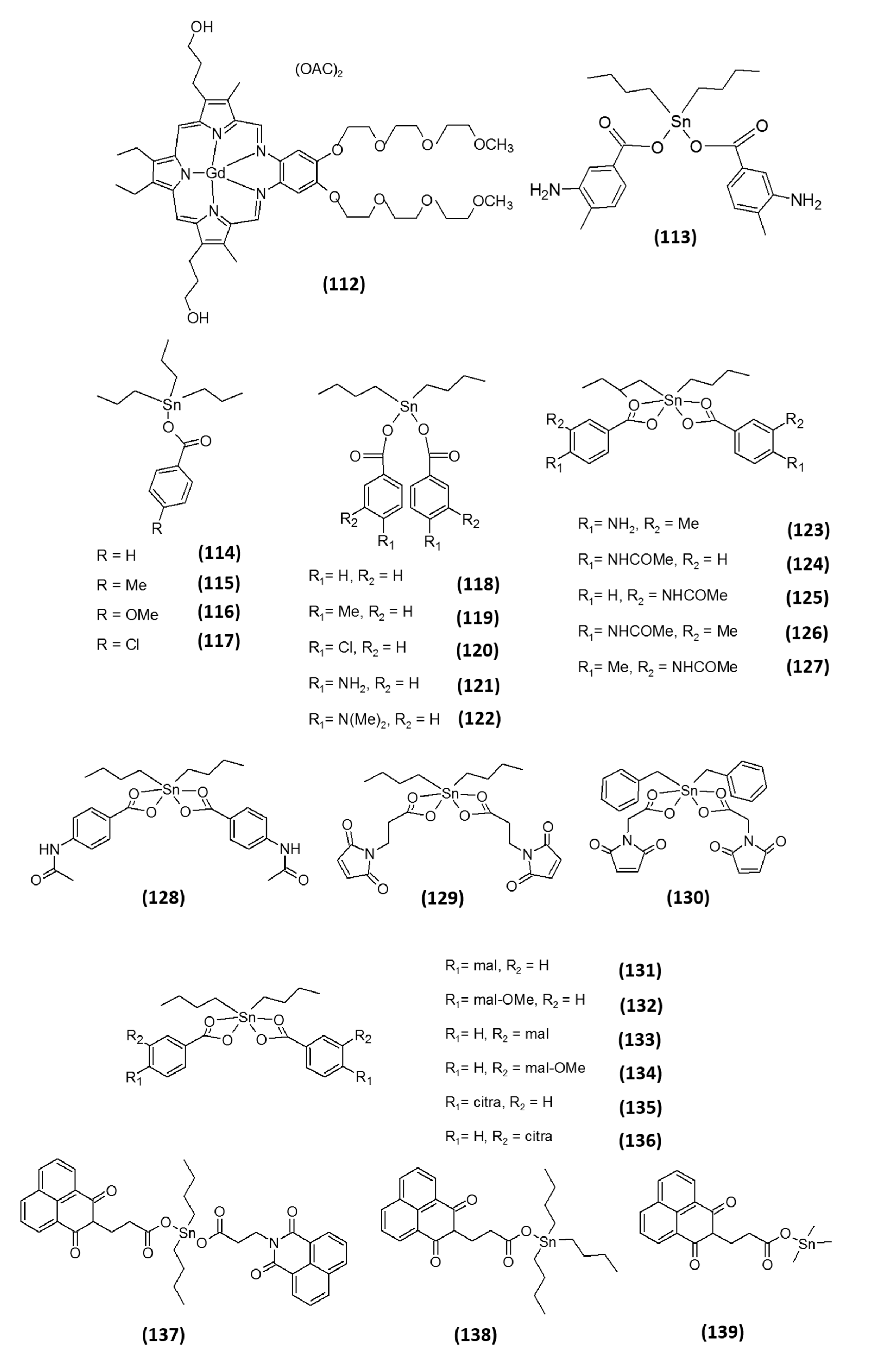
6. Other Metal-Containing Inhibitors
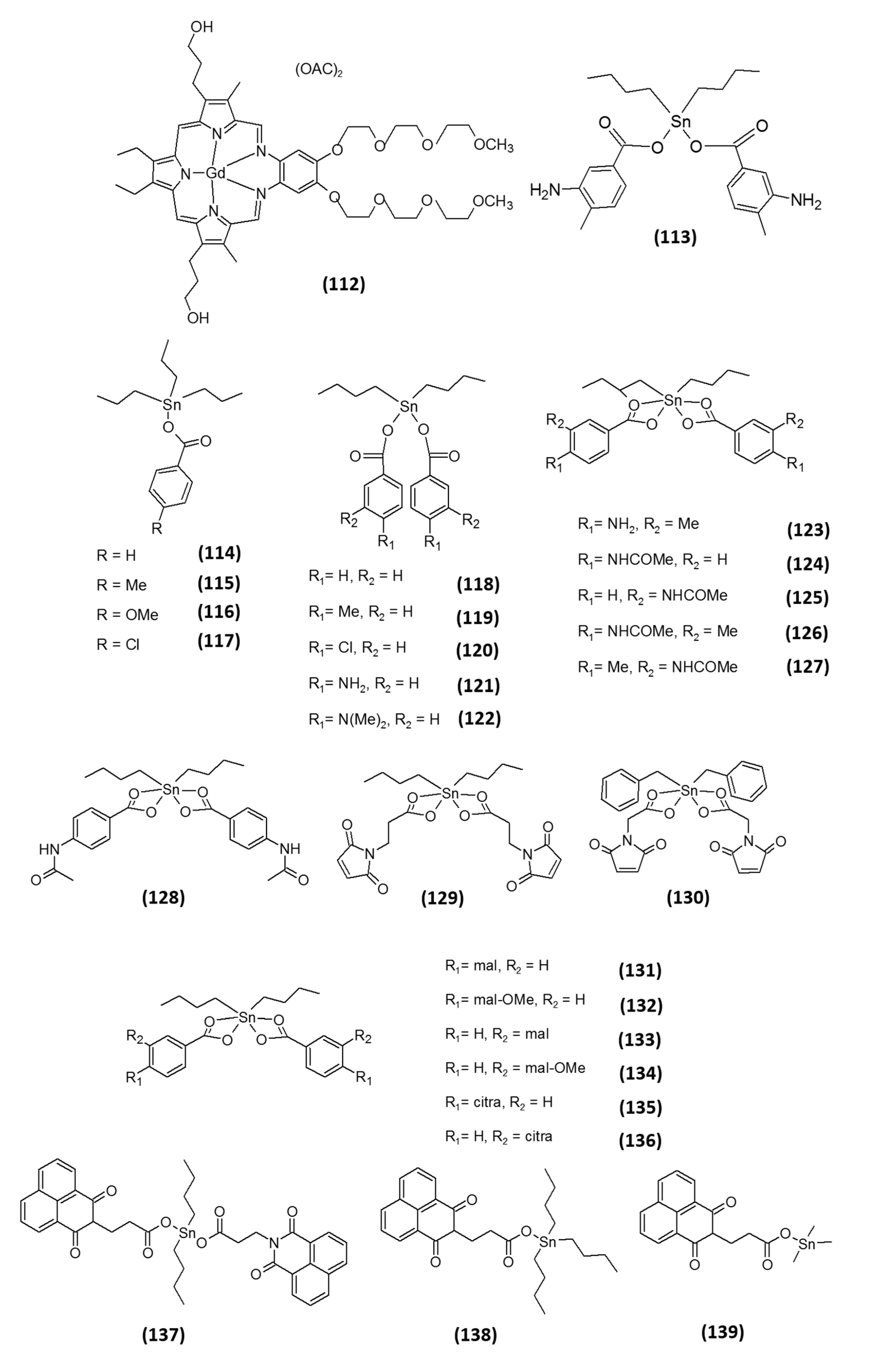
7. Semimetal-Based Inhibitors
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Gromer, S.; Urig, S.; Becker, K. The thioredoxin system—From science to clinic. Med. Res. Rev. 2004, 24, 40–89. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Yoon, H.W.; Lee, S.R.; Rhee, S.G. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J. Biol. Chem. 2004, 279, 3142–3150. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Awwad, R.T.; Smart, D.D.; Spitz, D.R.; Gius, D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. 2006, 236, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Prast-Nielsen, S.; Flaberg, E.; Szekely, L.; Arner, E.S. High levels of thioredoxin reductase 1 modulate drug-specific cytotoxic efficacy. Free Radic. Biol. Med. 2009, 47, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Grogan, T.M.; Fenoglio-Prieser, C.; Zeheb, R.; Bellamy, W.; Frutiger, Y.; Vela, E.; Stemmerman, G.; Macdonald, J.; Richter, L.; Gallegos, A.; et al. Thioredoxin, a putative oncogene product, is overexpressed in gastric carcinoma and associated with increased proliferation and increased cell survival. Hum. Pathol. 2000, 31, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Raffel, J.; Bhattacharyya, A.K.; Gallegos, A.; Cui, H.; Einspahr, J.G.; Alberts, D.S.; Powis, G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J. Lab. Clin. Med. 2003, 142, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Penney, R.B.; Roy, D. Thioredoxin-mediated redox regulation of resistance to endocrine therapy in breast cancer. Biochim. Biophys. Acta 2013, 1836, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Yu, S.; Zhao, G. Recent advances in the development of thioredoxin reductase inhibitors as anticancer agents. Curr. Drug Targets 2012, 13, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Saccoccia, F.; Angelucci, F.; Boumis, G.; Carotti, D.; Desiato, G.; Miele, A.E.; Bellelli, A. Thioredoxin reductase and its inhibitors. Curr. Protein Pept. Sci. 2014, 15, 621–646. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; McCollum, G.W.; Boeglin, M.E.; Burk, R.F. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem. Biophys. Res. Commun. 1997, 234, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Gromer, S.; Arscott, L.D.; Williams, C.H., Jr.; Schirmer, R.H.; Becker, K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998, 273, 20096–20101. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Boscolo, R.; Bindoli, A. Induction of mitochondrial permeability transition by auranofin, a gold(I)-phosphine derivative. Br. J. Pharmacol. 2002, 136, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Folda, A.; Bindoli, A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem. Pharmacol. 2004, 67, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Folda, A.; Dani, B.; Menabo, R.; Scutari, G.; Bindoli, A. Gold(I) complexes determine apoptosis with limited oxidative stress in Jurkat T cells. Eur. J. Pharmacol. 2008, 582, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Chaudiere, J.; Tappel, A.L. Interaction of gold(I) with the active site of selenium-glutathione peroxidase. J. Inorg. Biochem. 1984, 20, 313–325. [Google Scholar] [CrossRef]
- Urig, S.; Fritz-Wolf, K.; Reau, R.; Herold-Mende, C.; Toth, K.; Davioud-Charvet, E.; Becker, K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew. Chem. Int. Ed. 2006, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Saccoccia, F.; Angelucci, F.; Boumis, G.; Brunori, M.; Miele, A.E.; Williams, D.L.; Bellelli, A. On the mechanism and rate of gold incorporation into thiol-dependent flavoreductases. J. Inorg. Biochem. 2012, 108, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, A.; Gabbiani, C.; Ginanneschi, M.; Messori, L. Reactions of medicinally relevant gold compounds with the C-terminal motif of thioredoxin reductase elucidated by MS analysis. Chem. Commun. 2010, 46, 7001–7003. [Google Scholar] [CrossRef] [PubMed]
- Millet, R.; Urig, S.; Jacob, J.; Amtmann, E.; Moulinoux, J.P.; Gromer, S.; Becker, K.; Davioud-Charvet, E. Synthesis of 5-nitro-2-furancarbohydrazides and their cis-diamminedichloroplatinum complexes as bitopic and irreversible human thioredoxin reductase inhibitors. J. Med. Chem. 2005, 48, 7024–7039. [Google Scholar] [CrossRef] [PubMed]
- Auranofin in Treating Patients With Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. Available online: https://clinicaltrials.gov/show/NCT01747798 (accessed on 8 July 2015).
- Meyers, S. “Concierge” medicine. Who really pays for gold standard access to doctors? Trustee 2003, 56, 12–14. [Google Scholar] [PubMed]
- Christodoulou, J.; Sadler, P.J.; Tucker, A. A new structural transition of serum albumin dependent on the state of Cys34. Detection by 1H-NMR spectroscopy. Eur. J. Biochem. 1994, 225, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.R.; Xiao, J.; Schliesman, B.; Parsons, D.J.; Shaw, C.F., III. Kinetics and Mechanism of the Reaction between Serum Albumin and Auranofin (and Its Isopropyl Analogue) in Vitro. Inorg. Chem. 1996, 35, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Fernandes, A.P.; Rigobello, M.P.; Dani, B.; Sorrentino, F.; Tisato, F.; Björnstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; et al. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem. Pharmacol. 2010, 79, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Qian, X.; Xu, Y.; Vlecken, D.H.; Marques, I.J.; Kubutat, D.; Will, J.; Sheldrick, W.S.; Jesse, P.; Prokop, A.; et al. A gold(I) phosphine complex containing a naphthalimide ligand functions as a TrxR inhibiting antiproliferative agent and angiogenesis inhibitor. J. Med. Chem. 2009, 52, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Battaglia, E.; Deborde, V.; Muller, T.; Reau, R.; Davioud-Charvet, E.; Bagrel, D. A sugar-modified phosphole gold complex with antiproliferative properties acting as a thioredoxin reductase inhibitor in MCF-7 cells. ChemMedChem 2008, 3, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Farhadi, M.; Ahmadi, R.; Toth, K.; Lohr, J.; Helmke, B.M.; Kehr, S.; Unterberg, A.; Ott, I.; Gust, R.; et al. Antiglioma activity of GoPI-sugar, a novel gold(I)-phosphole inhibitor: Chemical synthesis, mechanistic studies, and effectiveness in vivo. Biochim. Biophys. Acta 2014, 1844, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Serebryanskaya, T.V.; Lyakhov, A.S.; Ivashkevich, L.S.; Schur, J.; Frias, C.; Prokop, A.; Ott, I. Gold(I) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents. Dalton Trans. 2015, 44, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Vergara, E.; Casini, A.; Sorrentino, F.; Zava, O.; Cerrada, E.; Rigobello, M.P.; Bindoli, A.; Laguna, M.; Dyson, P.J. Anticancer therapeutics that target selenoenzymes: Synthesis, characterization, in vitro cytotoxicity, and thioredoxin reductase inhibition of a series of gold(I) complexes containing hydrophilic phosphine ligands. ChemMedChem 2010, 5, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Ortego, L.; Cardoso, F.; Martins, S.; Fillat, M.F.; Laguna, A.; Meireles, M.; Villacampa, M.D.; Gimeno, M.C. Strong inhibition of thioredoxin reductase by highly cytotoxic gold(I) complexes. DNA binding studies. J. Inorg. Biochem. 2014, 130, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Lessa, J.A.; Guerra, J.C.; de Miranda, L.F.; Romeiro, C.F.; Da Silva, J.G.; Mendes, I.C.; Speziali, N.L.; Souza-Fagundes, E.M.; Beraldo, H. Gold(I) complexes with thiosemicarbazones: Cytotoxicity against human tumor cell lines and inhibition of thioredoxin reductase activity. J. Inorg. Biochem. 2011, 105, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Galassi, R.; Burini, A.; Ricci, S.; Pellei, M.; Rigobello, M.P.; Citta, A.; Dolmella, A.; Gandin, V.; Marzano, C. Synthesis and characterization of azolate gold(I) phosphane complexes as thioredoxin reductase inhibiting antitumor agents. Dalton Trans. 2012, 41, 5307–5318. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Nichols, S.J.; Leedman, P.J.; Berners-Price, S.J.; Filipovska, A. A gold(I) phosphine complex selectively induces apoptosis in breast cancer cells: Implications for anticancer therapeutics targeted to mitochondria. Biochem. Pharmacol. 2007, 74, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, C.; Kunz, P.C.; Kassack, M.U.; Hamacher, A.; Bohler, P.; Watjen, W.; Ott, I.; Rubbiani, R.; Spingler, B. Gold(I) complexes of water-soluble diphos-type ligands: Synthesis, anticancer activity, apoptosis and thioredoxin reductase inhibition. Dalton Trans. 2011, 40, 9212–9220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Cao, R.; Zhang, W.; Yin, M.; Xiao, X.; Liu, Q.; Huang, N. A soluble bis-chelated gold(I) diphosphine compound with strong anticancer activity and low toxicity. J. Med. Chem. 2013, 56, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Papini, G.; Morresi, B.; Galassi, R.; Ricci, S.; Tisato, F.; Porchia, M.; Rigobello, M.P.; Gandin, V.; et al. In vitro antitumour activity of water soluble Cu(I), Ag(I) and Au(I) complexes supported by hydrophilic alkyl phosphine ligands. J. Inorg. Biochem. 2011, 105, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Lupidi, G.; Avenali, L.; Bramucci, M.; Quassinti, L.; Pettinari, R.; Khalife, H.K.; Gali-Muhtasib, H.; Marchetti, F.; Pettinari, C. Synthesis, properties, and antitumor effects of a new mixed phosphine gold(I) compound in human colon cancer cells. J. Inorg. Biochem. 2013, 124, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Bagowski, C.P.; Kokoschka, M.; Stefanopoulou, M.; Alborzinia, H.; Can, S.; Vlecken, D.H.; Sheldrick, W.S.; Wolfl, S.; Ott, I. On the biological properties of alkynyl phosphine gold(I) complexes. Angew. Chem. Int. Ed. 2012, 51, 8895–8899. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.L.; Ruhayel, R.A.; Barnard, P.J.; Baker, M.V.; Berners-Price, S.J.; Filipovska, A. Mitochondria-targeted chemotherapeutics: The rational design of gold(I) N-heterocyclic carbene complexes that are selectively toxic to cancer cells and target protein selenols in preference to thiols. J. Am. Chem. Soc. 2008, 130, 12570–12571. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G.; et al. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Monchgesang, S.; Sheldrick, W.S.; Wolfl, S.; Ott, I. Comparative in vitro evaluation of N-heterocyclic carbene gold(I) complexes of the benzimidazolylidene type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Salassa, L.; de Almeida, A.; Casini, A.; Ott, I. Cytotoxic gold(I) N-heterocyclic carbene complexes with phosphane ligands as potent enzyme inhibitors. ChemMedChem 2014, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Oehninger, L.; Geldmacher, Y.; Alborzinia, H.; Wolfl, S.; Sheldrick, W.S.; Ott, I. Gold(I) N-heterocyclic carbene complexes with naphthalimide ligands as combined thioredoxin reductase inhibitors and DNA intercalators. ChemMedChem 2014, 9, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Pfluger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Shearwood, A.M.; Thyer, R.; McNamara, E.; Davies, S.M.; Callus, B.A.; Miranda-Vizuete, A.; Berners-Price, S.J.; Cheng, Q.; Arner, E.S.; et al. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radic. Biol. Med. 2011, 50, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Coronnello, M.; Mini, E.; Caciagli, B.; Cinellu, M.A.; Bindoli, A.; Gabbiani, C.; Messori, L. Mechanisms of cytotoxicity of selected organogold(III) compounds. J. Med. Chem. 2005, 48, 6761–6765. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Messori, L.; Marcon, G.; Agostina Cinellu, M.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold complexes inhibit mitochondrial thioredoxin reductase: Consequences on mitochondrial functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; McNaughton, M.; Gajewska, M.; Kumar, S.; Birmingham, A.; Powis, G. Thioredoxin reductase and cancer cell growth inhibition by organogold(III) compounds. Anticancer Drugs 2006, 17, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, L.; Fregona, D.; Mongiat, M.; Ronconi, L.; Fassina, A.; Colombatti, A.; Aldinucci, D. Antitumor activity of gold(III)-dithiocarbamato derivatives on prostate cancer cells and xenografts. Int. J. Cancer 2011, 128, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Parrilha, G.L.; Ferraz, K.S.; Lessa, J.A.; de Oliveira, K.N.; Rodrigues, B.L.; Ramos, J.P.; Souza-Fagundes, E.M.; Ott, I.; Beraldo, H. Metal complexes with 2-acetylpyridine-N(4)-orthochlorophenylthiosemicarbazone: Cytotoxicity and effect on the enzymatic activity of thioredoxin reductase and glutathione reductase. Eur. J. Med. Chem. 2014, 84, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Zehnder, T.N.; Mari, C.; Blacque, O.; Venkatesan, K.; Gasser, G. Anticancer profile of a series of gold(III) (2-phenyl)pyridine complexes. ChemMedChem 2014, 9, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P.J.; Keppler, B.K.; Messori, L. Gold(III) compounds as anticancer agents: Relevance of gold-protein interactions for their mechanism of action. J. Inorg. Biochem. 2008, 102, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Cinellu, M.A.; Minghetti, G.; Gabbiani, C.; Coronnello, M.; Mini, E.; Messori, L. Structural and solution chemistry, antiproliferative effects, and DNA and protein binding properties of a series of dinuclear gold(III) compounds with bipyridyl ligands. J. Med. Chem. 2006, 49, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Ronconi, L.; Fregona, D. The Midas touch in cancer chemotherapy: From platinum- to gold-dithiocarbamato complexes. Dalton Trans. 2009, 10670–10680. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Martinez, S.M.; Prieto-Garcia, L.; Prieto, M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Subcellular targets of cisplatin cytotoxicity: an integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Prast-Nielsen, S.; Cebula, M.; Pader, I.; Arner, E.S. Noble metal targeting of thioredoxin reductase—Covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radic. Biol. Med. 2010, 49, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Sasada, T.; Nakamura, H.; Ueda, S.; Sato, N.; Kitaoka, Y.; Gon, Y.; Takabayashi, A.; Spyrou, G.; Holmgren, A.; Yodoi, J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II). Free Radic. Biol. Med. 1999, 27, 504–514. [Google Scholar] [CrossRef]
- Arnér, E.S.; Nakamura, H.; Sasada, T.; Yodoi, J.; Holmgren, A.; Spyrou, G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum(II) and its major metabolite, the glutathione-platinum complex. Free Radic. Biol. Med. 2001, 31, 1170–1178. [Google Scholar] [CrossRef]
- Witte, A.B.; Anestål, K.; Jerremalm, E.; Ehrsson, H.; Arnér, E.S. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 2005, 39, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Jennette, K.W.; Lippard, S.J.; Vassiliades, G.A.; Bauer, W.R. Metallointercalation reagents. 2-hydroxyethanethiolato(2,2′,2′-terpyridine)-platinum(II) monocation binds strongly to DNA by intercalation. Proc. Natl. Acad. Sci. USA 1974, 71, 3839–3843. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Herold-Mende, C.; Park, J.J.; Lowe, G.; Schirmer, R.H. Human thioredoxin reductase is efficiently inhibited by (2,2′:6′,2′′-terpyridine)platinum(II) complexes. Possible implications for a novel antitumor strategy. J. Med. Chem. 2001, 44, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Ko, T.P.; Su, W.C.; Su, T.L.; Wang, A.H. Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1. J. Inorg. Biochem. 2009, 103, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, R.; Urig, S.; Hartmann, M.; Helmke, B.M.; Koncarevic, S.; Allenberger, B.; Kienhoefer, C.; Neher, M.; Steiner, H.H.; Unterberg, A.; et al. Antiglioma activity of 2,2′:6′,2′′-terpyridineplatinum(II) complexes in a rat model—Effects on cellular redox metabolism. Free Radic. Biol. Med. 2006, 40, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Banti, C.N.; Hadjikakou, S.K. Anti-proliferative and anti-tumor activity of silver(I) compounds. Metallomics 2013, 5, 569–596. [Google Scholar] [CrossRef] [PubMed]
- Aaseth, J.; Olsen, A.; Halse, J.; Hovig, T. Argyria-tissue deposition of silver as selenide. Scand. J. Clin. Lab. Investig. 1981, 41, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Pellei, M.; Gandin, V.; Marinelli, M.; Marzano, C.; Yousufuddin, M.; Dias, H.V.; Santini, C. Synthesis and biological activity of ester- and amide-functionalized imidazolium salts and related water-soluble coinage metal N-heterocyclic carbene complexes. Inorg. Chem. 2012, 51, 9873–9882. [Google Scholar] [CrossRef] [PubMed]
- Citta, A.; Schuh, E.; Mohr, F.; Folda, A.; Massimino, M.L.; Bindoli, A.; Casini, A.; Rigobello, M.P. Fluorescent silver(I) and gold(I)-N-heterocyclic carbene complexes with cytotoxic properties: Mechanistic insights. Metallomics 2013, 5, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Pellei, M.; Marinelli, M.; Marzano, C.; Dolmella, A.; Giorgetti, M.; Santini, C. Synthesis and in vitro antitumor activity of water soluble sulfonate- and ester-functionalized silver(I) N-heterocyclic carbene complexes. J. Inorg. Biochem. 2013, 129, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.; Beijnen, J.H.; Sava, G. Approaching tumour therapy beyond platinum drugs: Status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Komeda, S.; Casini, A. Next-generation anticancer metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Camalli, M.; Bindoli, A.; Sorrentino, F.; Casini, A.; Gabbiani, C.; Corsini, M.; Zanello, P.; Rigobello, M.P.; Messori, L. Activity of rat cytosolic thioredoxin reductase is strongly decreased by trans-[bis(2-amino-5- methylthiazole)tetrachlororuthenate(III)]: First report of relevant thioredoxin reductase inhibition for a ruthenium compound. J. Med. Chem. 2007, 50, 5871–5874. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging protein targets for anticancer metallodrugs: Inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef] [PubMed]
- Oehninger, L.; Stefanopoulou, M.; Alborzinia, H.; Schur, J.; Ludewig, S.; Namikawa, K.; Munoz-Castro, A.; Koster, R.W.; Baumann, K.; Wolfl, S.; et al. Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans. 2013, 42, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yu, L.; Yang, F.; Zhao, Z.; Yu, B.; Lai, H.; Wong, K.H.; Ngai, S.M.; Zheng, W.; Chen, T. Ruthenium polypyridyl complexes as inducer of ROS-mediated apoptosis in cancer cells by targeting thioredoxin reductase. Metallomics 2014, 6, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Magda, D.; Miller, R.A. Motexafin gadolinium: A novel redox active drug for cancer therapy. Semin. Cancer Biol. 2006, 16, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M. Motexafin gadolinium: A redox-active tumor selective agent for the treatment of cancer. Curr. Opin. Oncol. 2004, 16, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Hashemy, S.I.; Ungerstedt, J.S.; Zahedi Avval, F.; Holmgren, A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006, 281, 10691–10697. [Google Scholar] [CrossRef] [PubMed]
- Navakoski de Oliveira, K.; Andermark, V.; von Grafenstein, S.; Onambele, L.A.; Dahl, G.; Rubbiani, R.; Wolber, G.; Gabbiani, C.; Messori, L.; Prokop, A.; et al. Butyltin(IV) benzoates: Inhibition of thioredoxin reductase, tumor cell growth inhibition, and interactions with proteins. ChemMedChem 2013, 8, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Navakoski de Oliveira, K.; Andermark, V.; Onambele, L.A.; Dahl, G.; Prokop, A.; Ott, I. Organotin complexes containing carboxylate ligands with maleimide and naphthalimide derived partial structures: TrxR inhibition, cytotoxicity and activity in resistant cancer cells. Eur. J. Med. Chem. 2014, 87, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Flemming, A. On the specific antibacterial properties of penicillin and potassium tellurite. J. Pathol. Bacteriol. 1932, 35, 831–842. [Google Scholar] [CrossRef]
- Engman, L.; Kandra, T.; Gallegos, A.; Williams, R.; Powis, G. Water-soluble organotellurium compounds inhibit thioredoxin reductase and the growth of human cancer cells. Anticancer Drug Des. 2000, 15, 323–330. [Google Scholar] [PubMed]
- Engman, L.; Al-Maharik, N.; McNaughton, M.; Birmingham, A.; Powis, G. Thioredoxin reductase and cancer cell growth inhibition by organotellurium antioxidants. Anticancer Drugs 2003, 14, 153–161. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, M.; Engman, L.; Birmingham, A.; Powis, G.; Cotgreave, I.A. Cyclodextrin-derived diorganyl tellurides as glutathione peroxidase mimics and inhibitors of thioredoxin reductase and cancer cell growth. J. Med. Chem. 2004, 47, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; Al-Maharik, N.; McNaughton, M.; Birmingham, A.; Powis, G. Thioredoxin reductase and cancer cell growth inhibition by organotellurium compounds that could be selectively incorporated into tumor cells. Bioorg. Med. Chem. 2003, 11, 5091–5100. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Zhu, J.; Shi, X.G.; Ni, J.H.; Zhong, H.J.; Si, G.Y.; Jin, X.L.; Tang, W.; Li, X.S.; Xong, S.M.; et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RARα/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar] [PubMed]
- Gazitt, Y.; Akay, C. Arsenic trioxide: An anti cancer missile with multiple warheads. Hematology 2005, 10, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fang, J.; Dong, Y.; Chen, S.J.; Chen, Z. Arsenic in cancer therapy. Anticancer Drugs 2005, 16, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Douer, D.; Tallman, M.S. Arsenic trioxide: New clinical experience with an old medication in hematologic malignancies. J. Clin. Oncol. 2005, 23, 2396–2410. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.H., Jr.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002, 62, 3893–3903. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandin, V.; Fernandes, A.P. Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents. Molecules 2015, 20, 12732-12756. https://doi.org/10.3390/molecules200712732
Gandin V, Fernandes AP. Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents. Molecules. 2015; 20(7):12732-12756. https://doi.org/10.3390/molecules200712732
Chicago/Turabian StyleGandin, Valentina, and Aristi P. Fernandes. 2015. "Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents" Molecules 20, no. 7: 12732-12756. https://doi.org/10.3390/molecules200712732
APA StyleGandin, V., & Fernandes, A. P. (2015). Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents. Molecules, 20(7), 12732-12756. https://doi.org/10.3390/molecules200712732