
Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo


Abstract
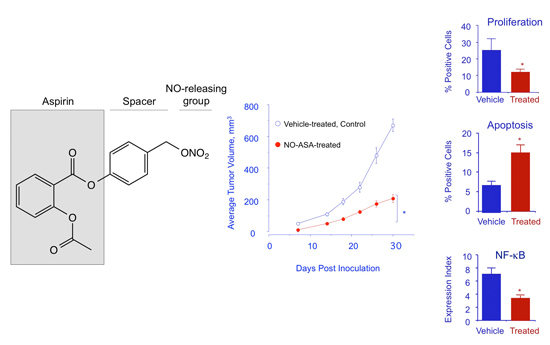
:
1. Introduction
2. Results and Discussion
2.1. NO-ASA Inhibits Breast Cancer Cell Growth

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50, μM, 24 h | IC50, μM, 48 h | ||
|---|---|---|---|---|
| MDA-MB-231 | SK BR-3 | MDA-MB-231 | SK BR-3 | |
| ASA | >3000 † | >3000 † | 2200 ± 185 | 2550 ± 210 |
| p-NO-ASA | 13 ± 2 * | 17 ± 2 * | 3.3 ± 0.4 | 5.3 ± 0.8 |
| m-NO-ASA | 173 ± 15 * | 185 ± 12 * | 95 ± 10 | 110 ± 15 |
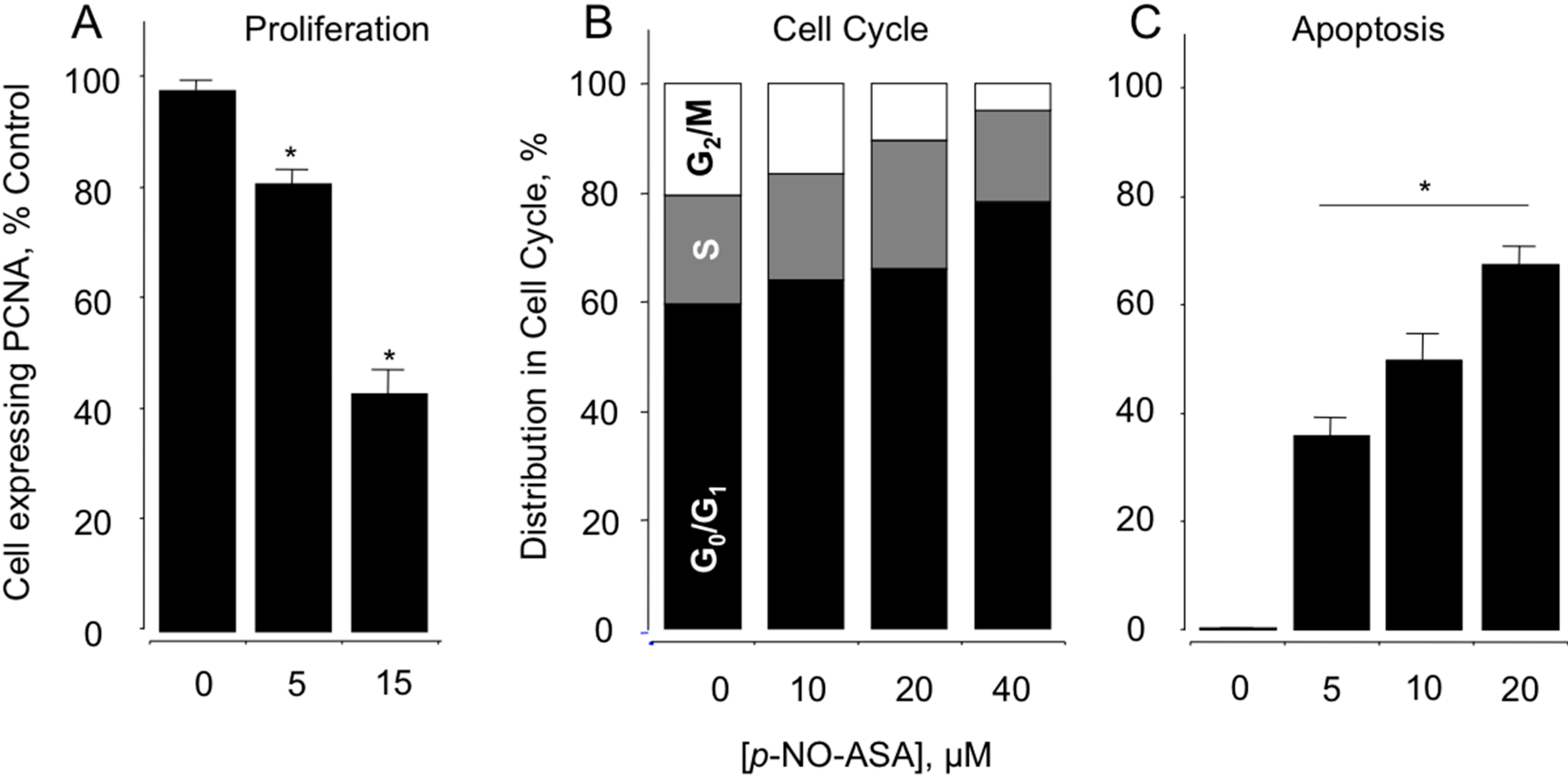
2.2. NO-ASA Inhibits Cellular Proliferation, Alters Cell Cycle Phases and Induces Cell Death
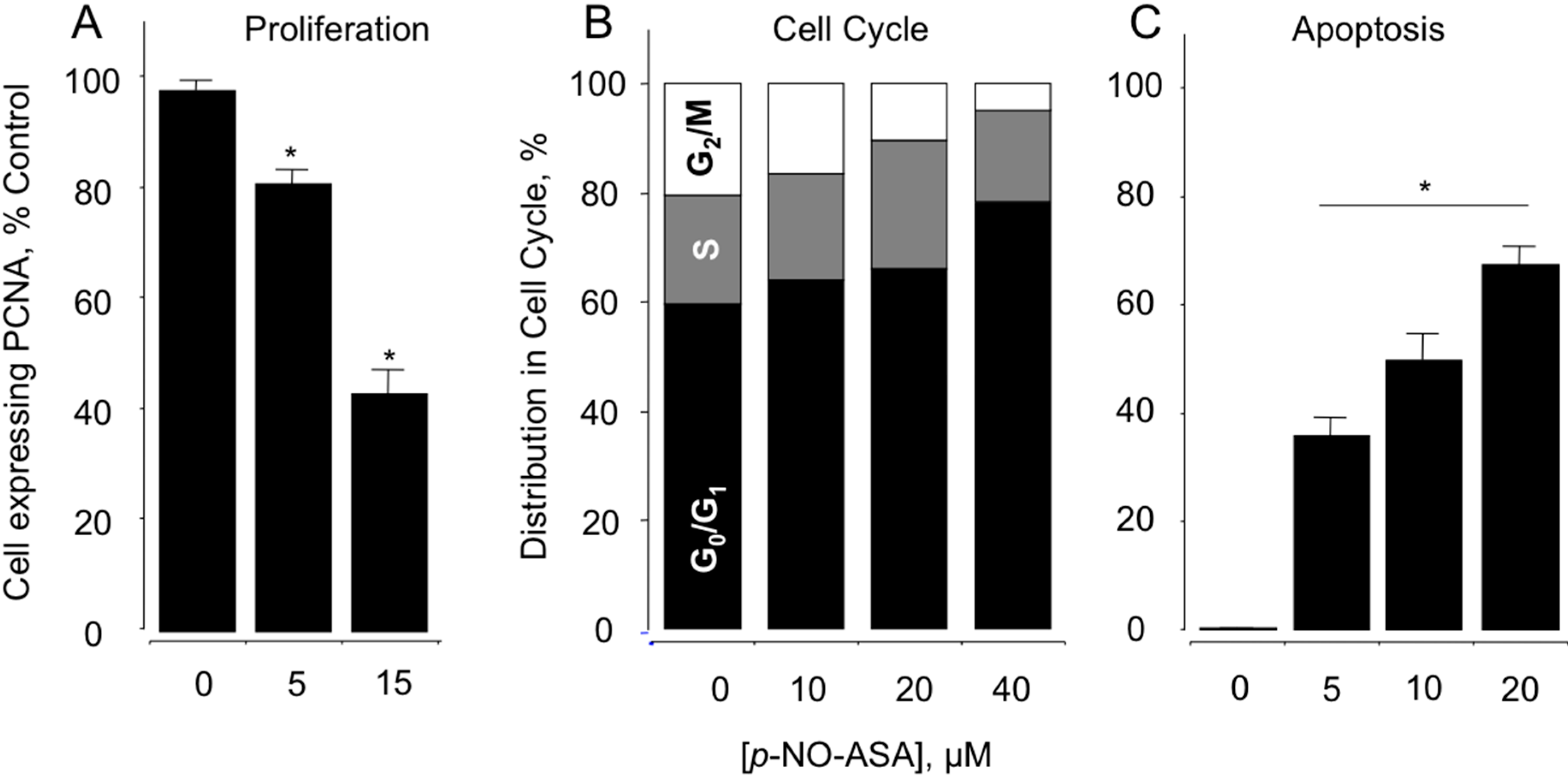
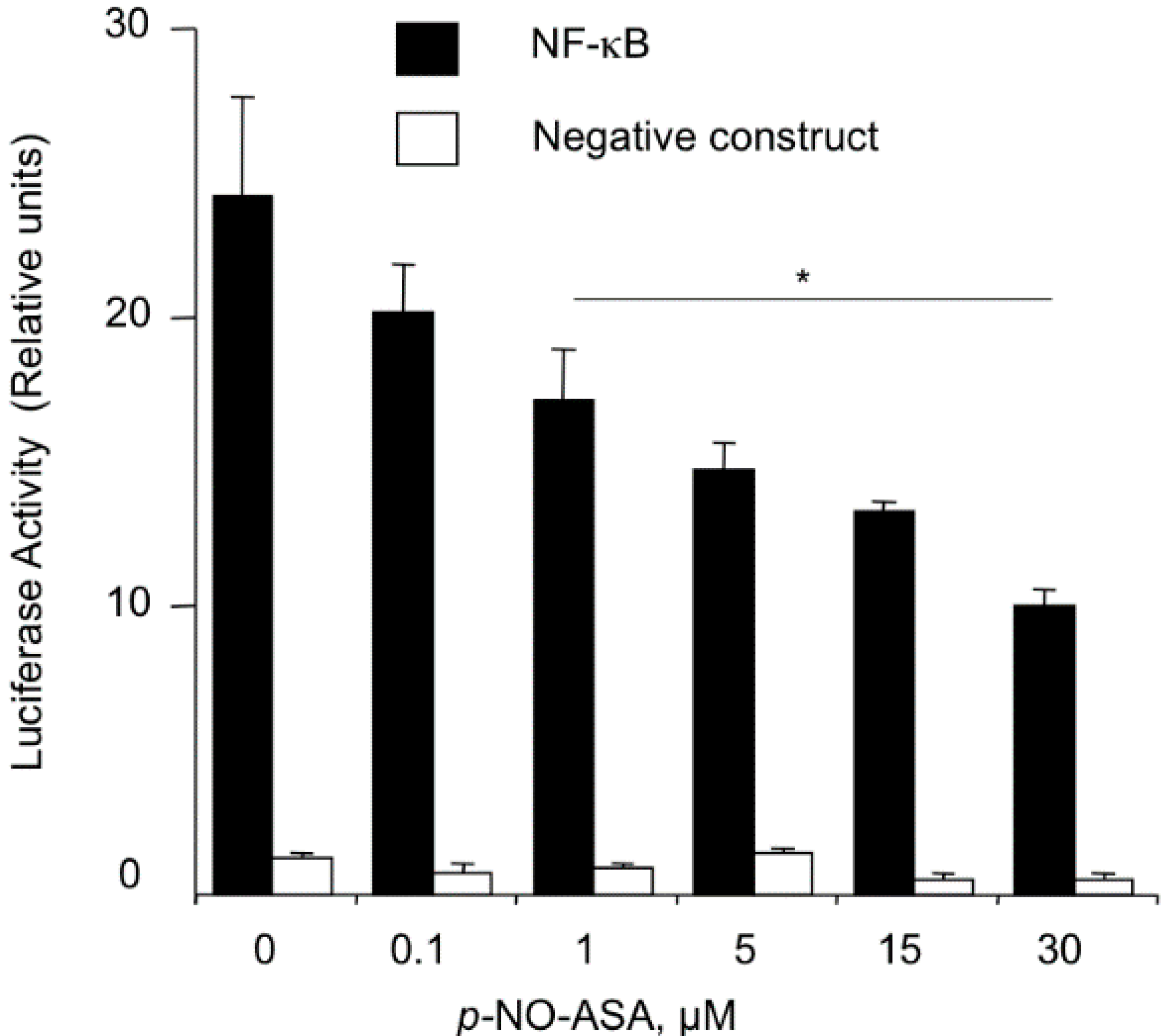
2.3. NO-ASA Inhibits NF-κB Signaling Pathway
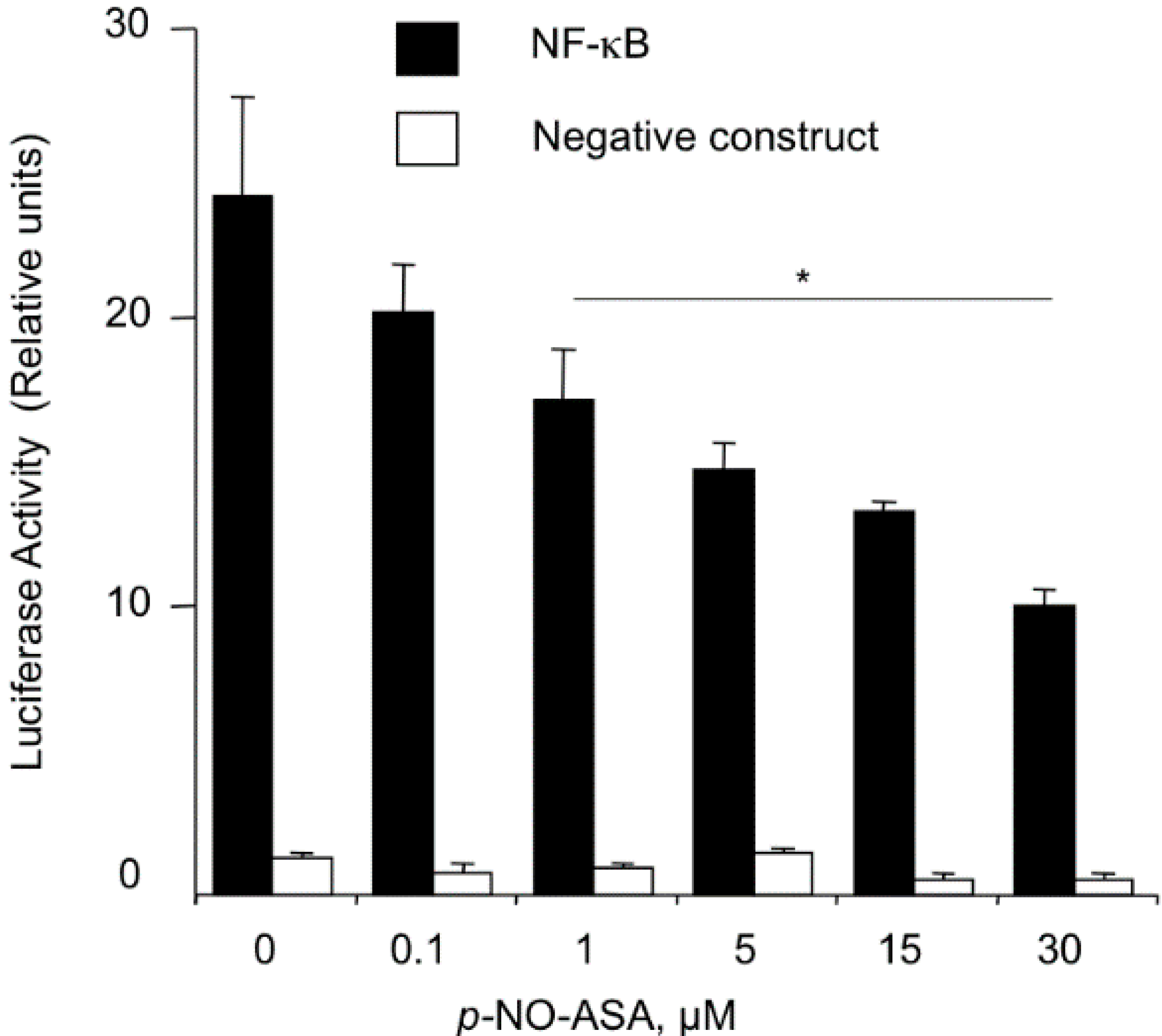
2.4. NO-ASA Inhibits NF-κB DNA-Binding Activity
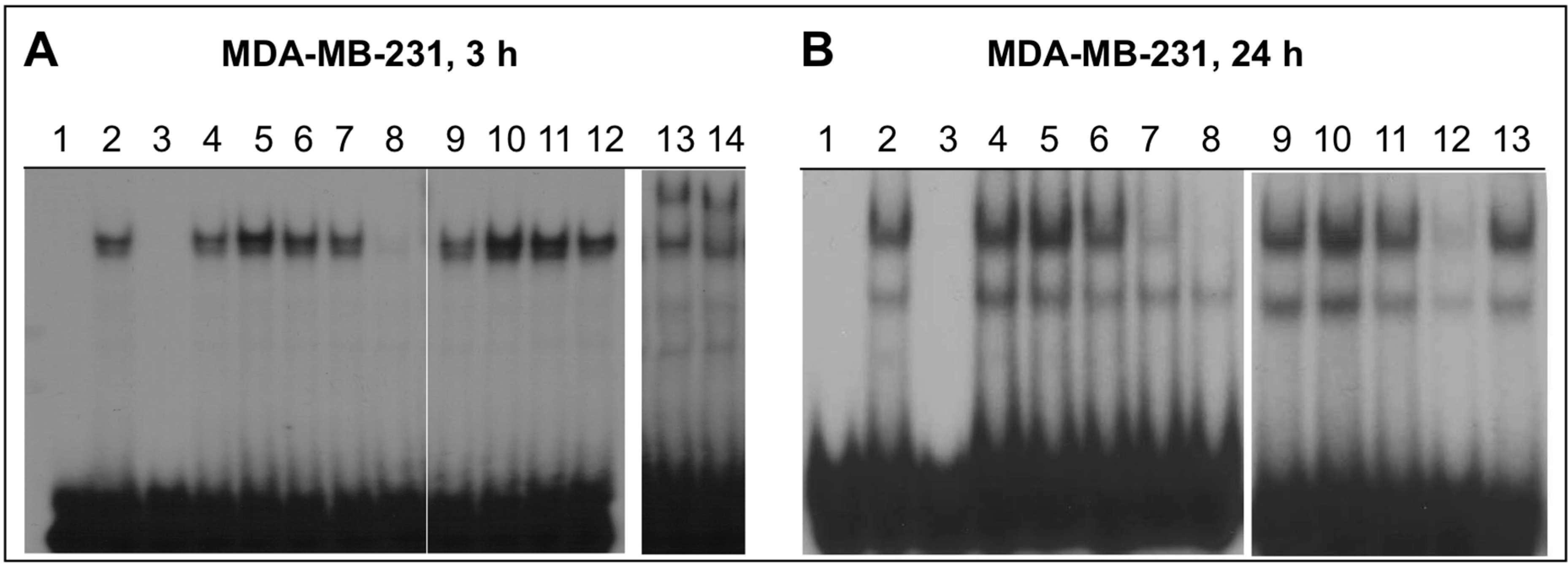
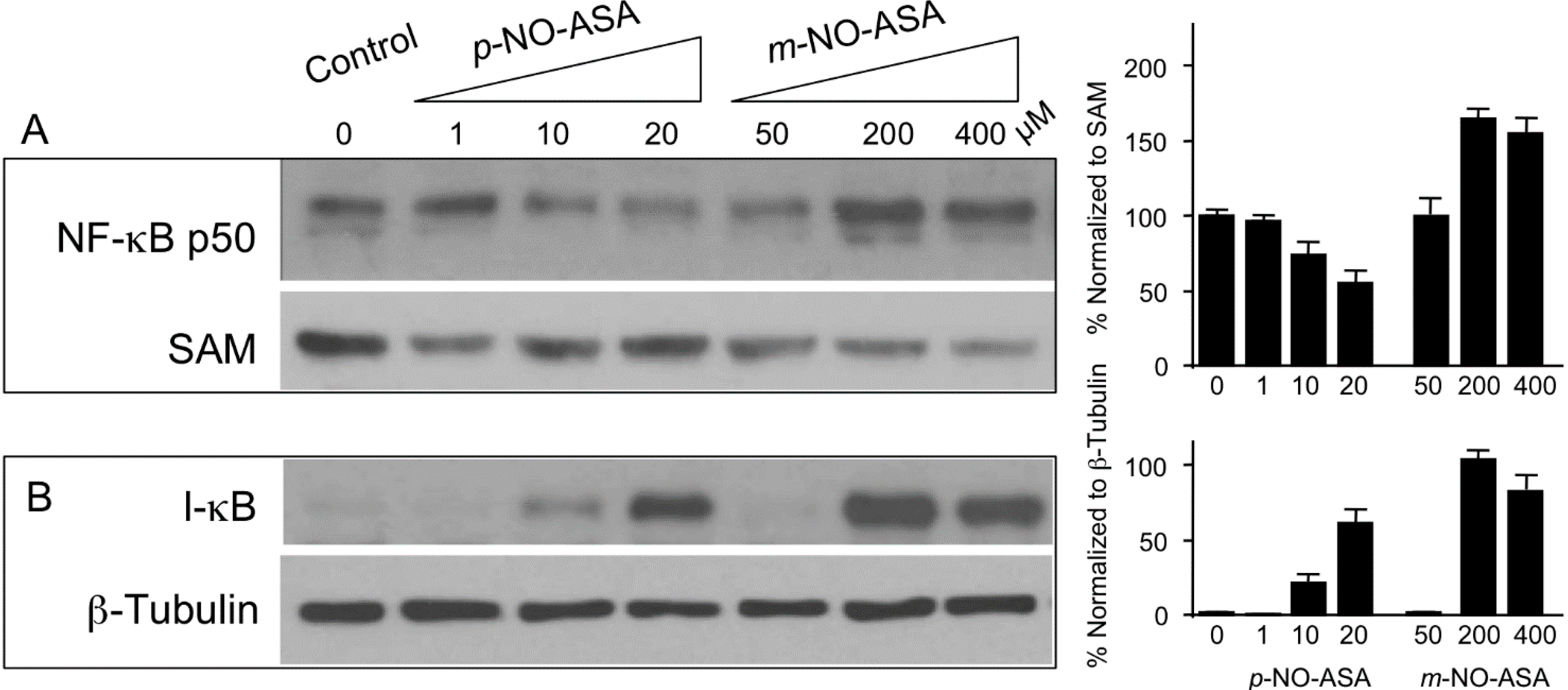
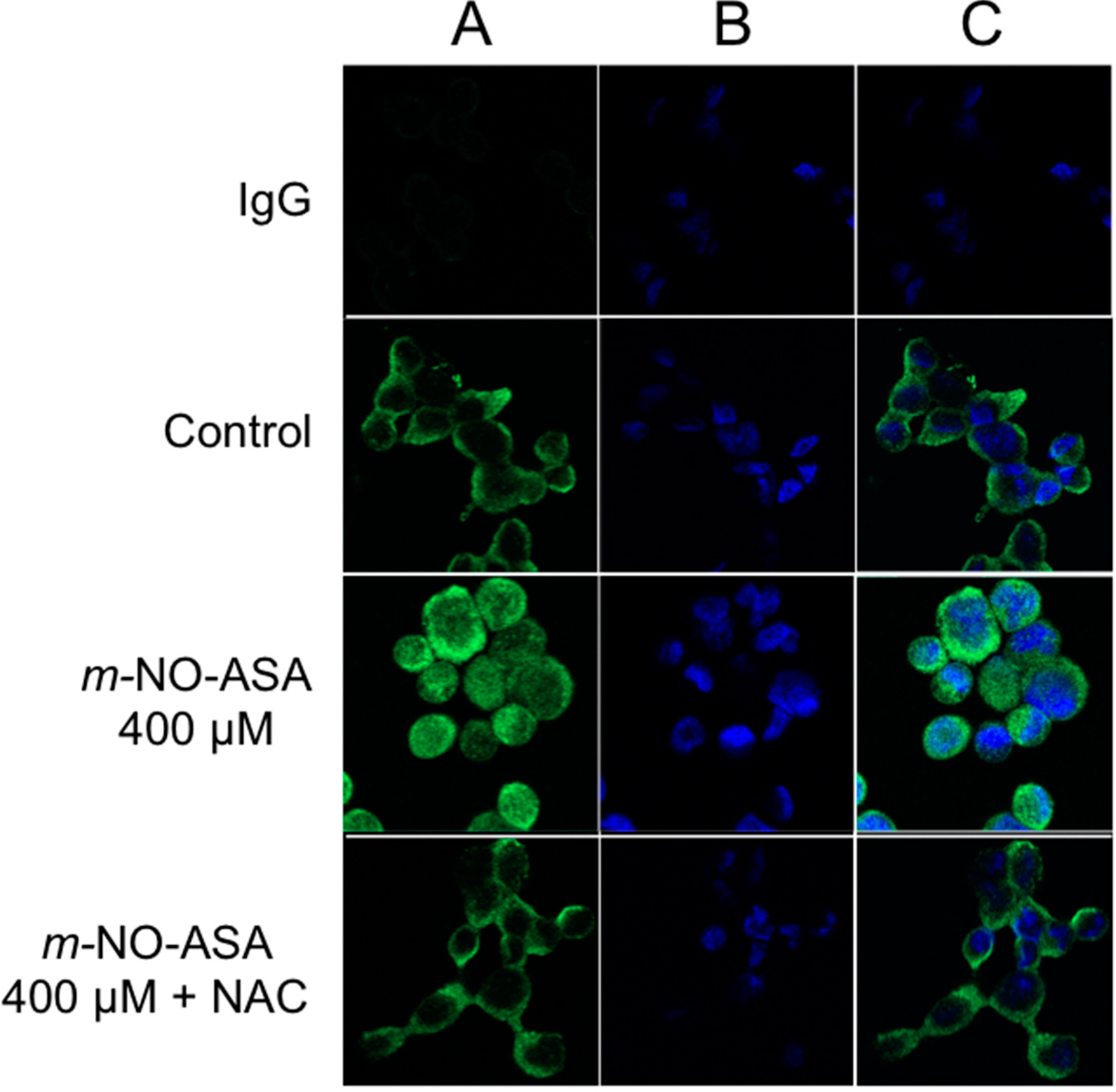
2.5. Effect of NO-ASA on Protein Levels of NF-κB Subunit p50
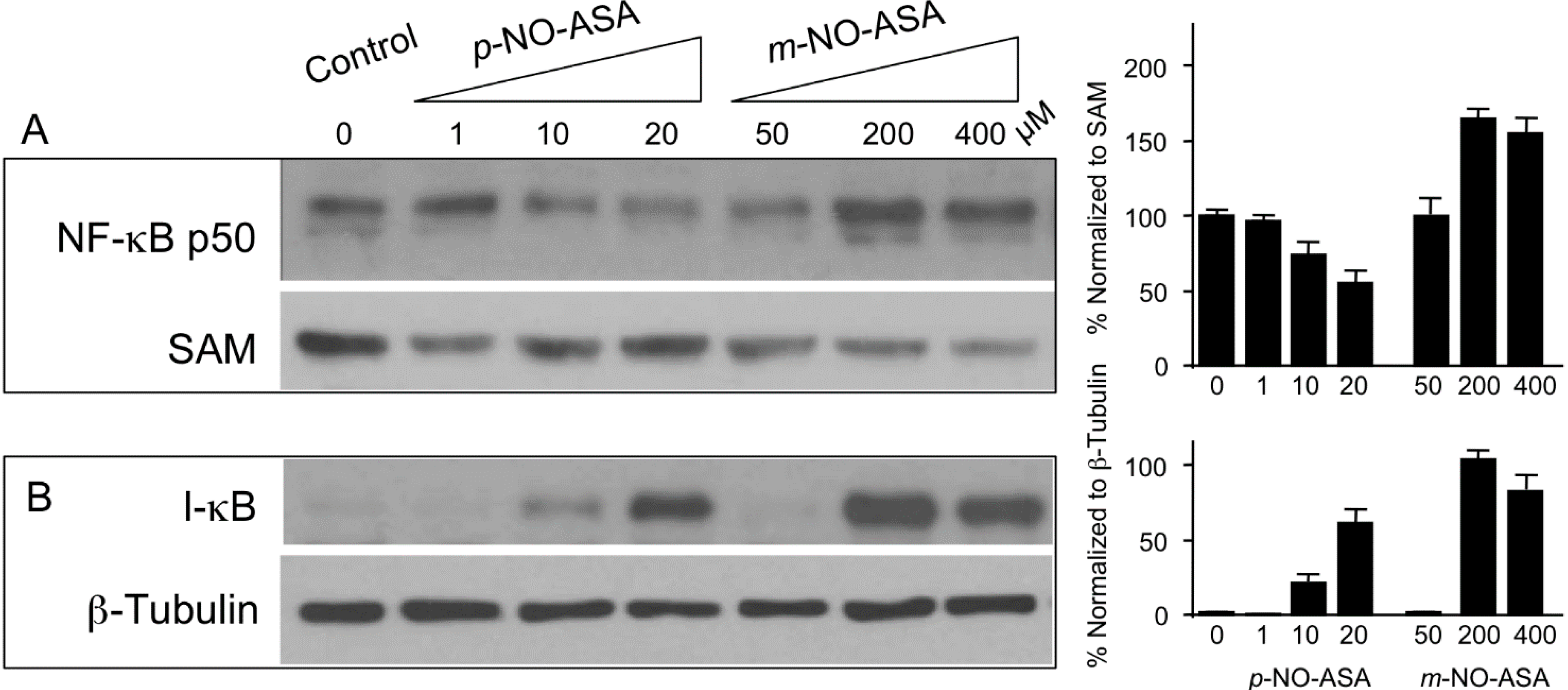
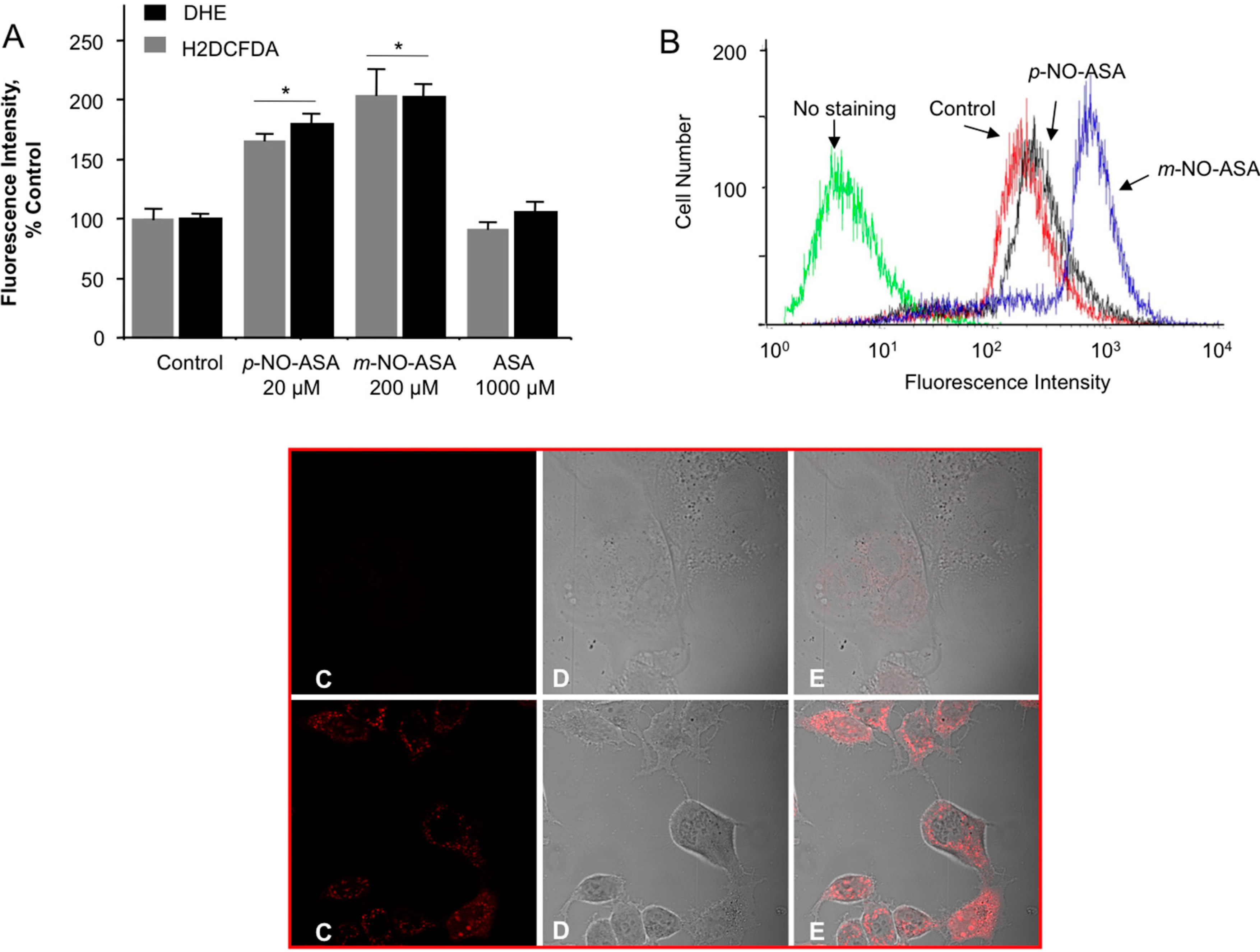
2.6. NO-ASA Induces ROS
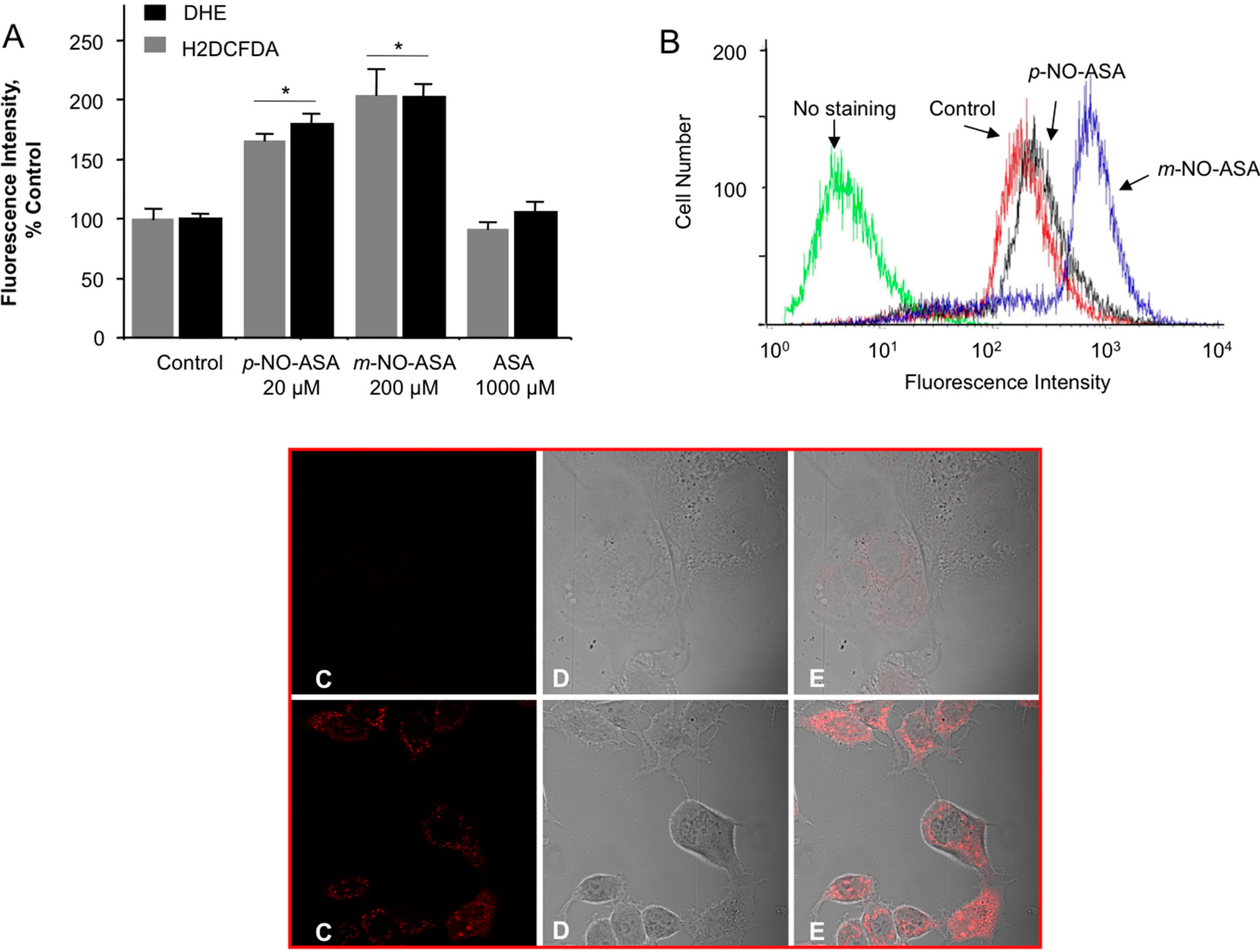
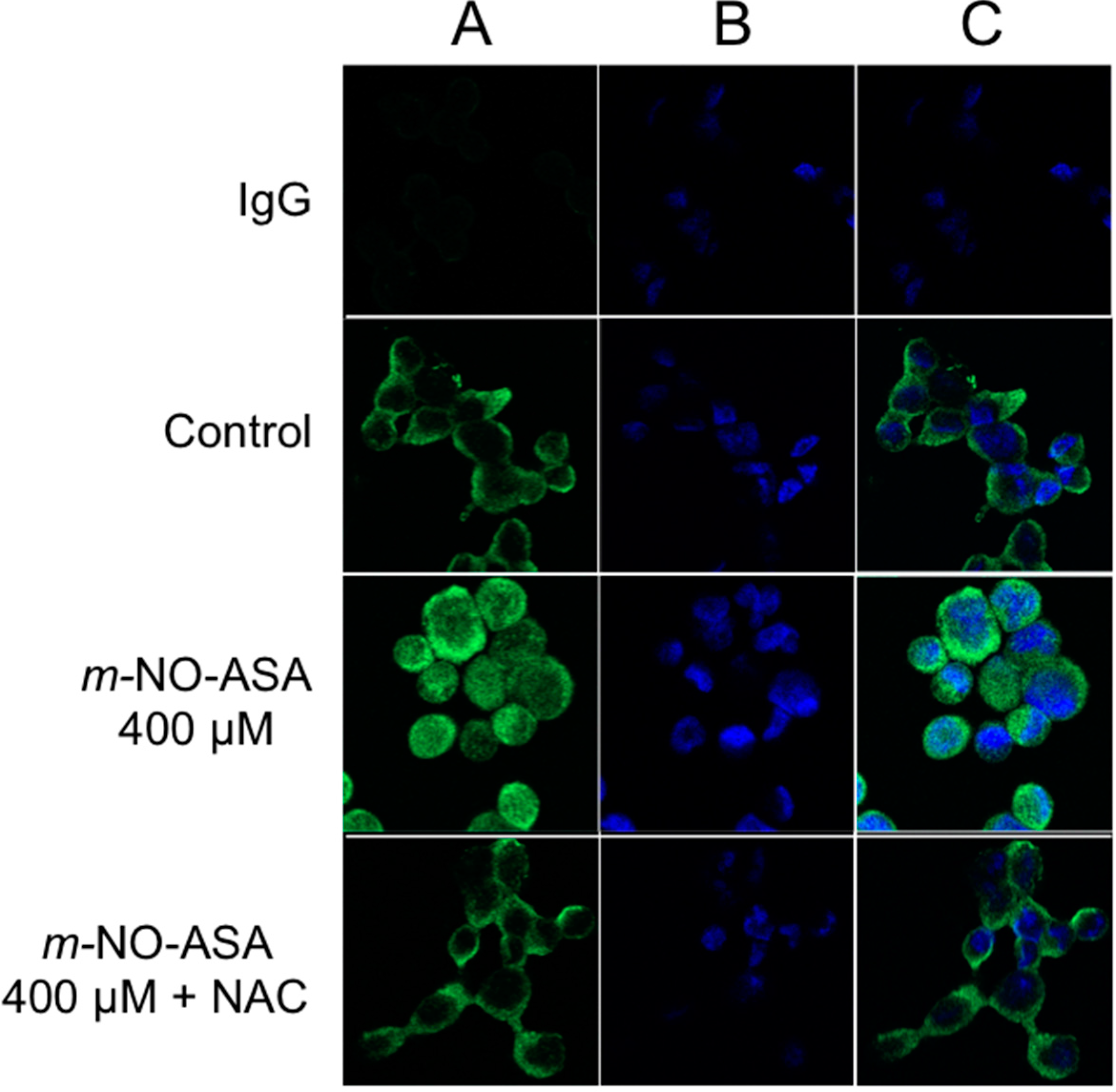
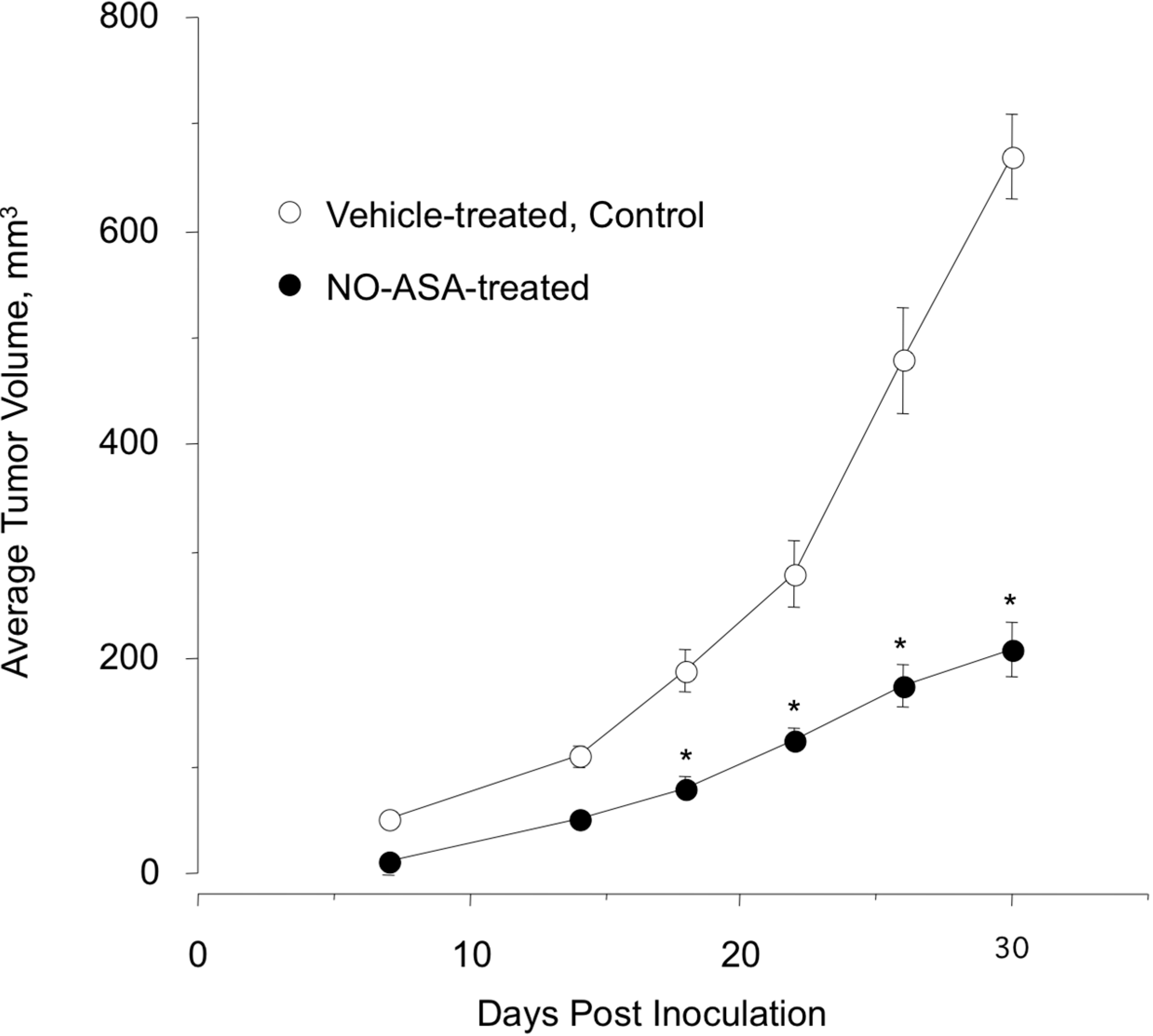
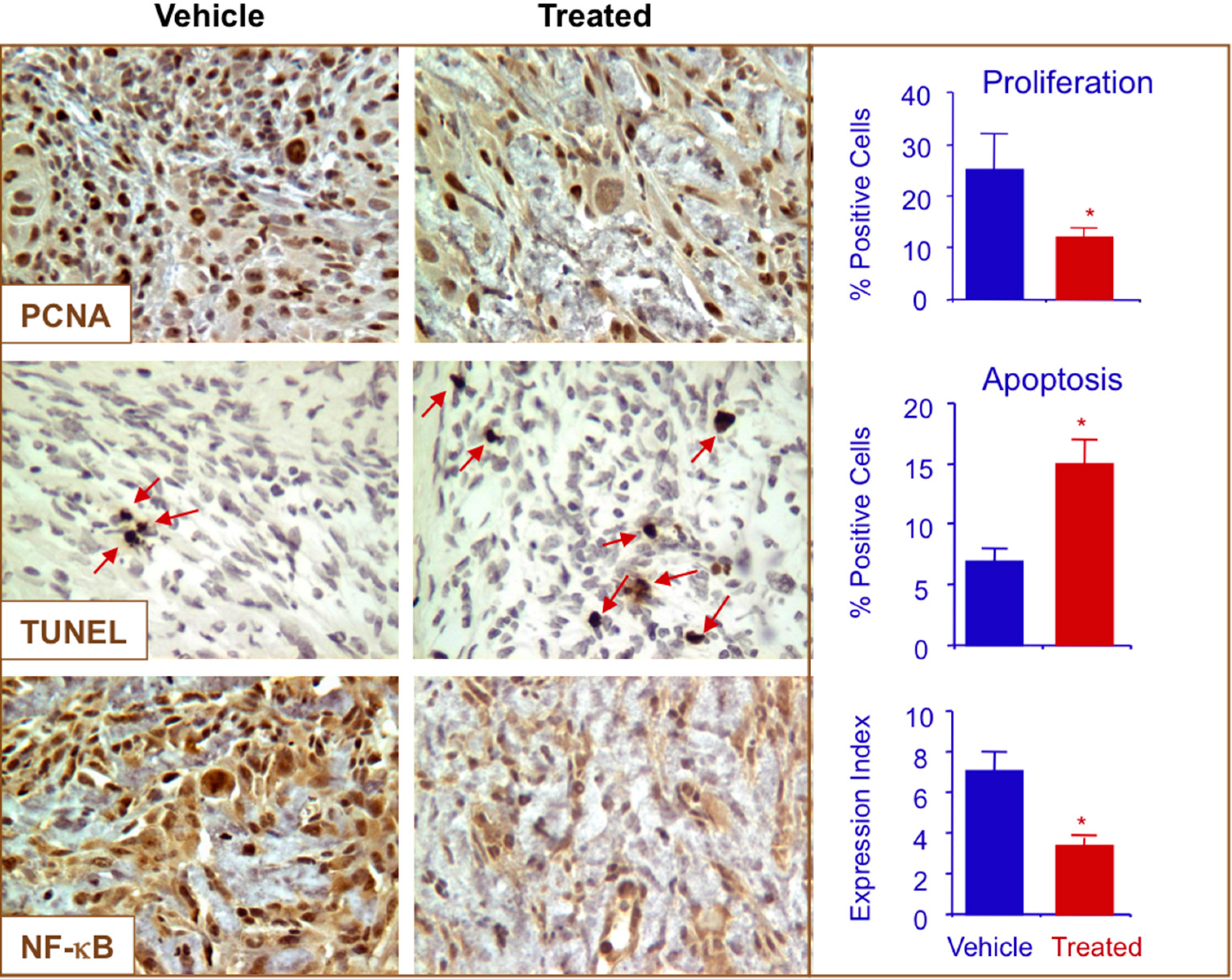
2.7. NO-ASA Modulates Cell Proliferation and Inhibits NF-κB Activation in a ER(−) Xenograft Model
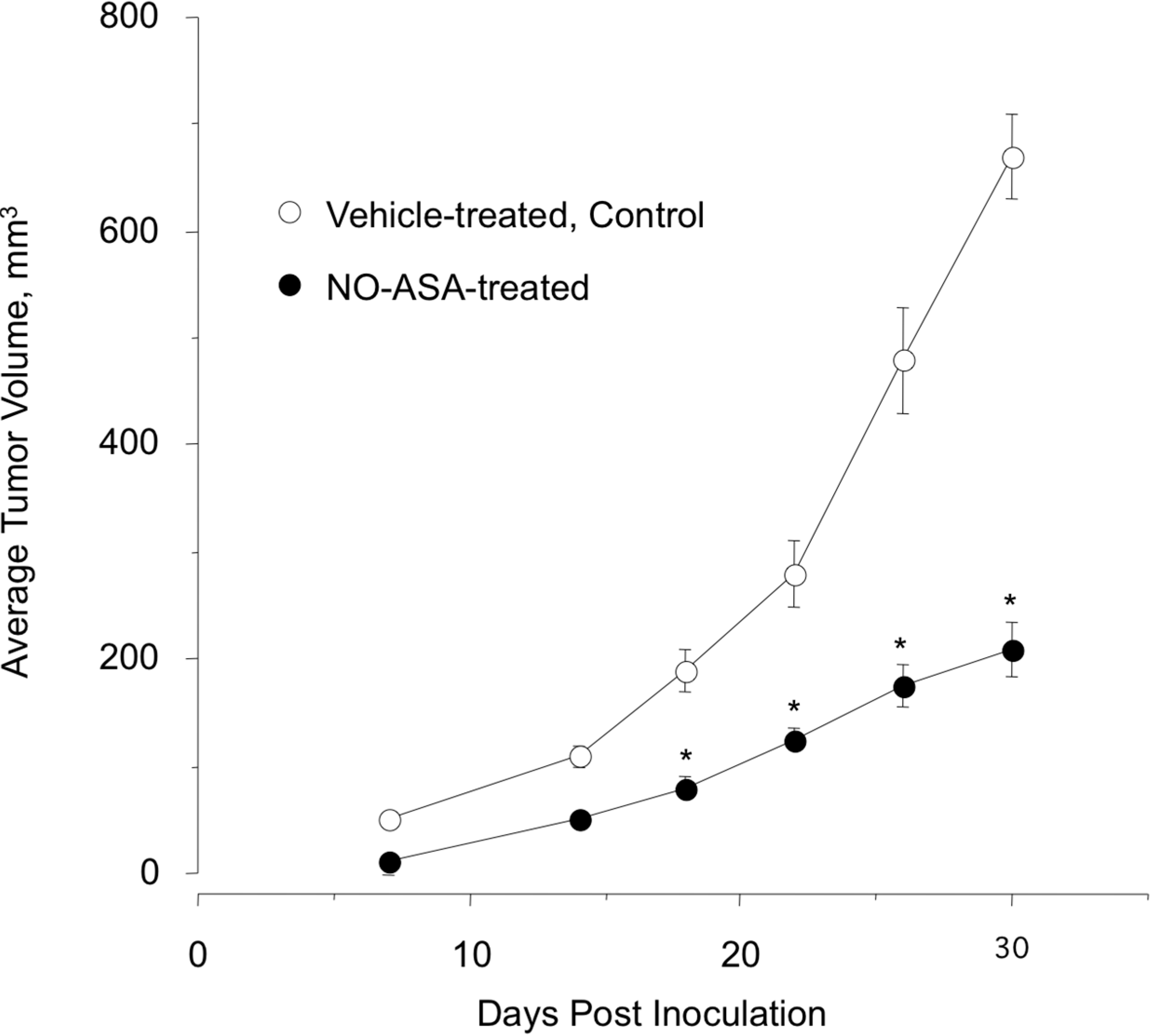
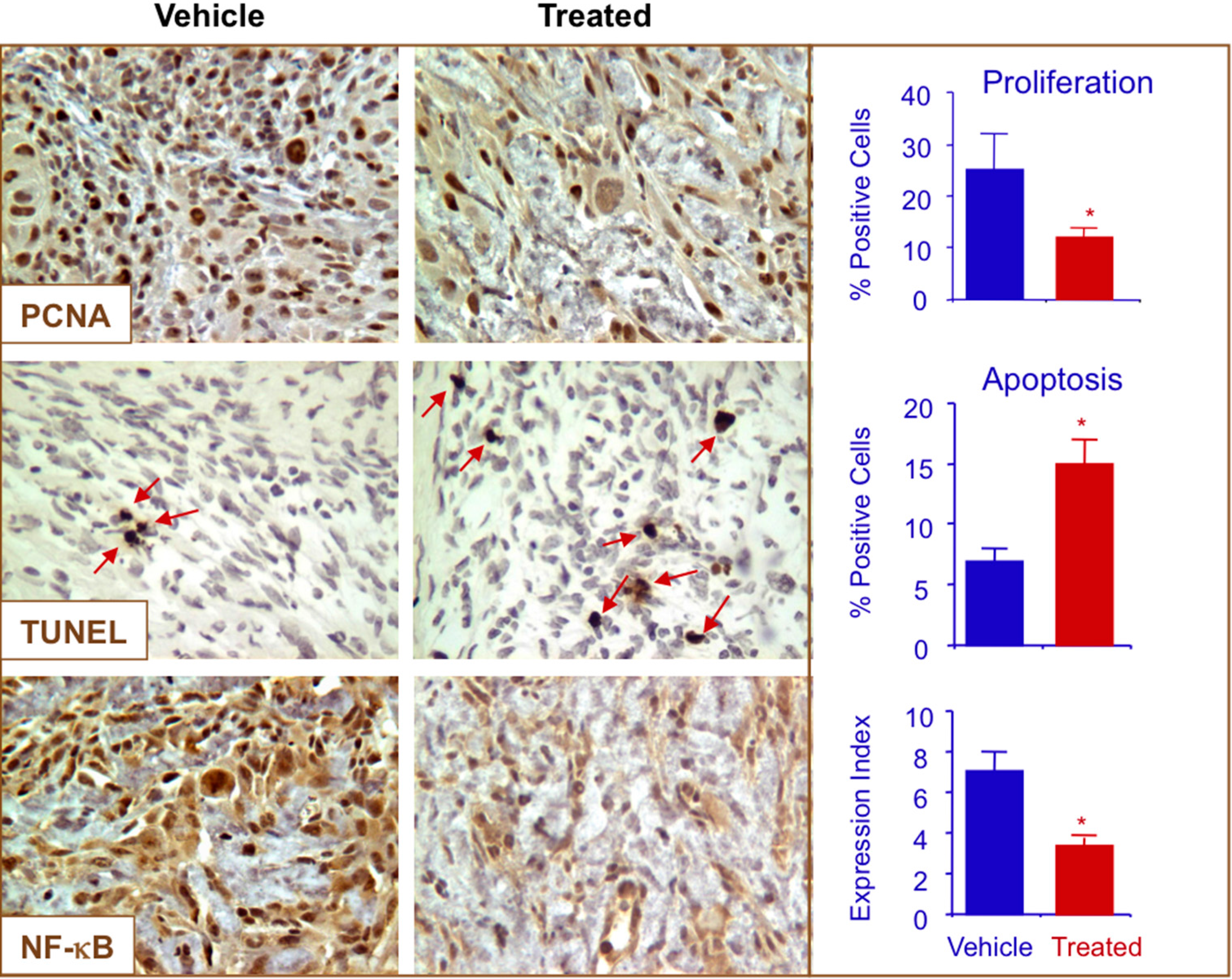
3. Experimental Section
3.1. Reagents
3.2. Breast Cancer Cells
3.3. Cell Growth Inhibition Assay
3.4. Cell Proliferation
3.5. Flow Cytometry for Phase Distribution in the Cell Cycle and Detection of Apoptosis
3.6. Apoptosis Assay
3.7. Cell Transfection and NF-κB Reporter Assays
3.8. Electrophoretic Mobility Shift Assays (EMSA)
3.9. Western Blotting
3.10. Determination of Reactive Oxygen Species (ROS)
3.11. Xenografts
3.12. Immunohistochemistry
3.13. TUNEL Staining
3.14. Scoring the Expression of Biomarkers
3.15. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA A Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Schnitt, S.J.; Guidi, A.J. Diseases of the Breast; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Reeder, J.G.; Vogel, V.G. Breast cancer risk management. Clin. Breast Cancer 2007, 7, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.H.; Lippman, S.M. Chemoprevention of breast cancer. Breast Cancer Res. Treat. 2000, 62, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.; Massarweh, S.; Shou, J.; Osborne, C.K. Breast cancer endocrine resistance: How growth factor signaling and estrogen receptor coregulators modulate response. Clin. Cancer Res. 2003, 9, 447S–454S. [Google Scholar] [PubMed]
- Kaza, C.S.; Kashfi, K.; Rigas, B. Colon cancer prevention with NO-releasing NSAIDs. Prostaglandins Other Lipid Mediat. 2002, 67, 107–120. [Google Scholar] [CrossRef]
- Rigas, B.; Kashfi, K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol. Med. 2004, 10, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.L.; Kashfi, K.; Ouyang, N.; del Soldato, P.; Kopelovich, L.; Rigas, B. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem. Biophys. Res. Commun. 2004, 313, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, N.; Williams, J.L.; Tsioulias, G.J.; Gao, J.; Iatropoulos, M.J.; Kopelovich, L.; Kashfi, K.; Rigas, B. Nitric oxide-donating aspirin prevents pancreatic cancer in a hamster tumor model. Cancer Res. 2006, 66, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Reuter, B.; Cicala, C.; McKnight, W.; Grisham, M.B.; Cirino, G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology 1994, 107, 173–179. [Google Scholar] [PubMed]
- Fiorucci, S.; Santucci, L.; Gresele, P.; Faccino, R.M.; del Soldato, P.; Morelli, A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: A proof of concept endoscopic study. Gastroenterology 2003, 124, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [PubMed]
- Biswas, D.K.; Dai, S.C.; Cruz, A.; Weiser, B.; Graner, E.; Pardee, A.B. The nuclear factor kappa B (NF-kappa B): A potential therapeutic target for estrogen receptor negative breast cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 10386–10391. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.L.; Nath, N.; Chen, J.; Hundley, T.R.; Gao, J.; Kopelovich, L.; Kashfi, K.; Rigas, B. Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: Implications for chemoprevention. Cancer Res. 2003, 63, 7613–7618. [Google Scholar] [PubMed]
- Williams, J.L.; Ji, P.; Ouyang, N.; Liu, X.; Rigas, B. NO-donating aspirin inhibits the activation of NF-kappaB in human cancer cell lines and Min mice. Carcinogenesis 2008, 29, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.I.; Cisterne, A.; Baraz, R.; Bradstock, K.F.; Bendall, L.J. para-NO-Aspirin inhibits NF-kappaB and induces apoptosis in B-cell progenitor acute lymphoblastic leukemia. Exp. Hematol. 2012, 40, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Williams, J.L. Nitric oxide-donating aspirin induces G2/M phase cell cycle arrest in human cancer cells by regulating phase transition proteins. Int. J. Oncol. 2012, 41, 325–330. [Google Scholar] [PubMed]
- Zhou, H.; Huang, L.; Sun, Y.; Rigas, B. Nitric oxide-donating aspirin inhibits the growth of pancreatic cancer cells through redox-dependent signaling. Cancer Lett. 2009, 273, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, J.; Rigas, B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis 2009, 30, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Vassell, R.; Chattopadhyay, M.; Kogan, M.; Kashfi, K. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: Effects on COX-2 expression and Wnt/beta-catenin/TCF-4 signaling. Biochem. Pharmacol. 2009, 78, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Kodela, R.; Chattopadhyay, M.; Kashfi, K. Synthesis and biological activity of NOSH-naproxen (AVT-219) and NOSH-sulindac (AVT-18A) as potent anti-inflammatory agents with chemotherapeutic potential. Med. Chem. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Ryan, Y.; Qiao, L.L.; Williams, J.L.; Chen, J.; del Soldato, P.; Traganos, F.; Rigas, B. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: Evidence of a tissue type-independent effect. J. Pharmacol. Exp. Ther. 2002, 303, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Ito, T.; Azuma, S.; Ito, E.; Honma, R.; Yanagisawa, Y.; Nishikawa, A.; Kawamura, M.; Imai, J.; Watanabe, S.; et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009, 100, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.A.; Parce, J.W.; Dechatelet, L.R.; Szejda, P.; Seeds, M.C.; Thomas, M. Flow cytometric studies of oxidative product formation by neutrophils: A graded response to membrane stimulation. J. Immunol. 1983, 130, 1910–1917. [Google Scholar] [PubMed]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.B.; vanden Hoek, T.L.; Shao, Z.H.; Li, C.Q.; Schumacker, P.T. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am. J. Physiol. 1999, 277, H2240–H2246. [Google Scholar] [PubMed]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anti-Cancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef]
- Jhorar, R.; Sharma, R.; Kaur, A.; Mukherjee, T.K. Role of reactive oxygen species in estrogen dependent breast cancer complication. Anti-Cancer Agents Med. Chem. 2015. [Google Scholar] [CrossRef]
- Rao, C.V.; Reddy, B.S.; Steele, V.E.; Wang, C.X.; Liu, X.; Ouyang, N.; Patlolla, J.M.; Simi, B.; Kopelovich, L.; Rigas, B. Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: Effects on molecular targets. Mol. Cancer Ther. 2006, 5, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Joseph, S.; Gao, L.; Patlolla, J.M.; Choi, C.I.; Kopelovich, L.; Steele, V.E.; Rigas, B. Pharmacokinetic and pharmacodynamic study of NO-donating aspirin in F344 rats. Int. J. Oncol. 2008, 33, 799–805. [Google Scholar] [PubMed]
- Kiessling, M.K.; Klemke, C.D.; Kaminski, M.M.; Galani, I.E.; Krammer, P.H.; Gulow, K. Inhibition of constitutively activated nuclear factor-kappaB induces reactive oxygen species- and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res. 2009, 69, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [PubMed]
- Moodie, F.M.; Marwick, J.A.; Anderson, C.S.; Szulakowski, P.; Biswas, S.K.; Bauter, M.R.; Kilty, I.; Rahman, I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004, 18, 1897–1899. [Google Scholar] [PubMed]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Borgo, S.; Williams, J.L.; Chen, J.; Gao, J.; Glekas, A.; Benedini, F.; del Soldato, P.; Rigas, B. Positional isomerism markedly affects the growth inhibition of colon cancer cells by nitric oxide-donating aspirin in vitro and in vivo. J. Pharmacol. Exp. Ther. 2005, 312, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors upon request.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nath, N.; Chattopadhyay, M.; Rodes, D.B.; Nazarenko, A.; Kodela, R.; Kashfi, K. Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo. Molecules 2015, 20, 12481-12499. https://doi.org/10.3390/molecules200712481
Nath N, Chattopadhyay M, Rodes DB, Nazarenko A, Kodela R, Kashfi K. Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo. Molecules. 2015; 20(7):12481-12499. https://doi.org/10.3390/molecules200712481
Chicago/Turabian StyleNath, Niharika, Mitali Chattopadhyay, Deborah B. Rodes, Anna Nazarenko, Ravinder Kodela, and Khosrow Kashfi. 2015. "Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo" Molecules 20, no. 7: 12481-12499. https://doi.org/10.3390/molecules200712481
APA StyleNath, N., Chattopadhyay, M., Rodes, D. B., Nazarenko, A., Kodela, R., & Kashfi, K. (2015). Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo. Molecules, 20(7), 12481-12499. https://doi.org/10.3390/molecules200712481