
A New and Efficient Synthesis of 6-O-Methylscutellarein, the Major Metabolite of the Natural Medicine Scutellarin

Abstract
:1. Introduction
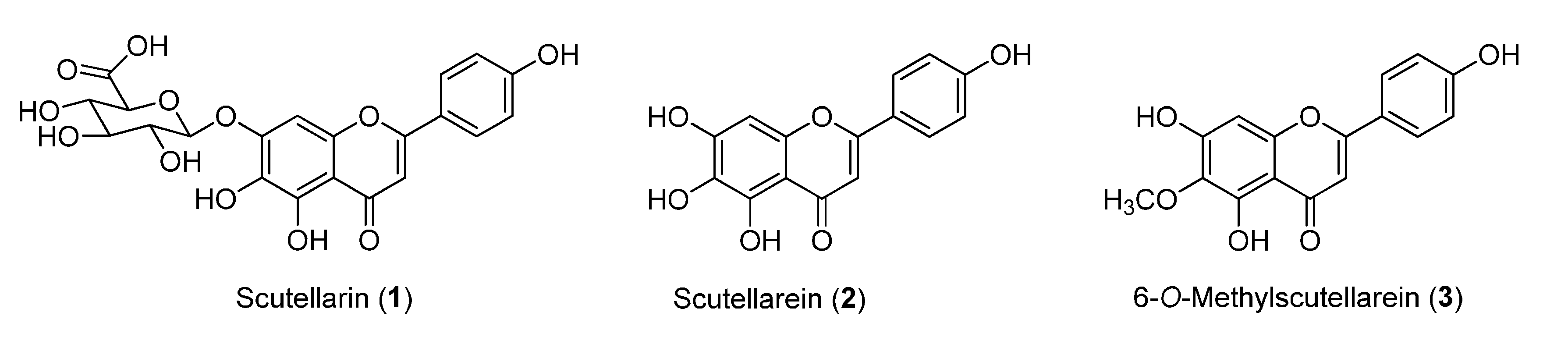

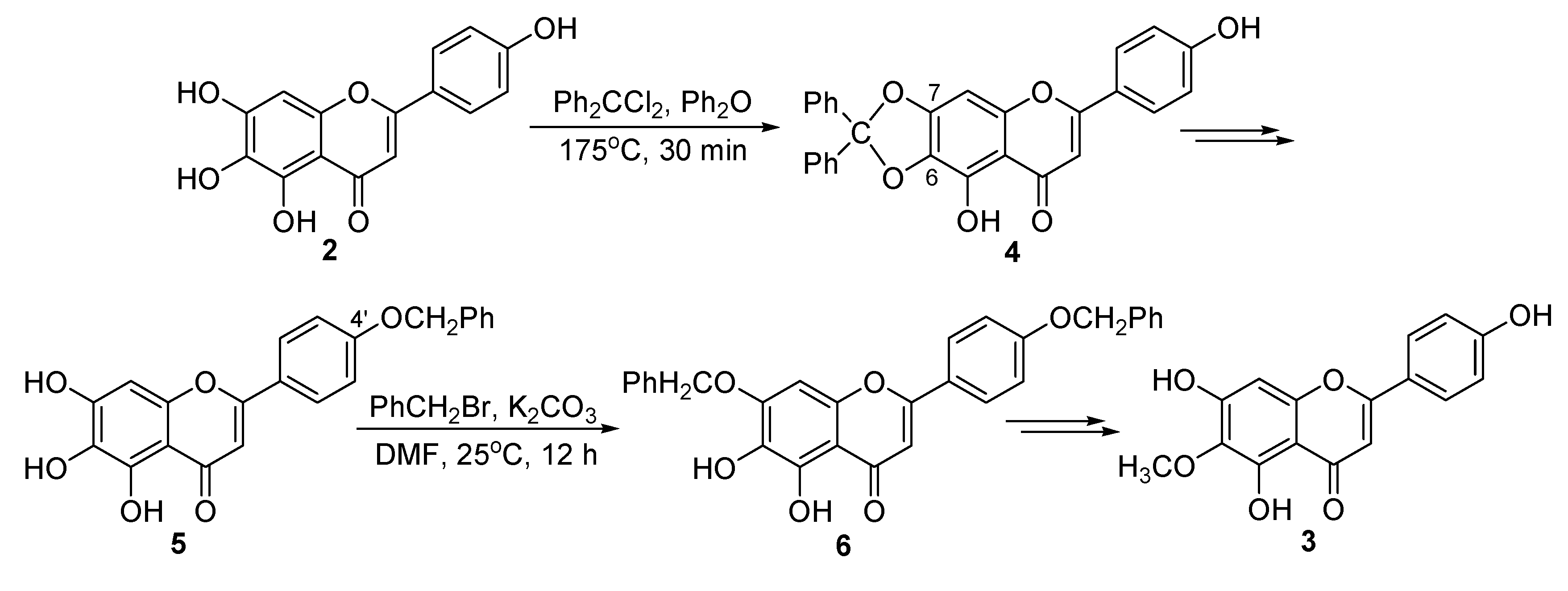
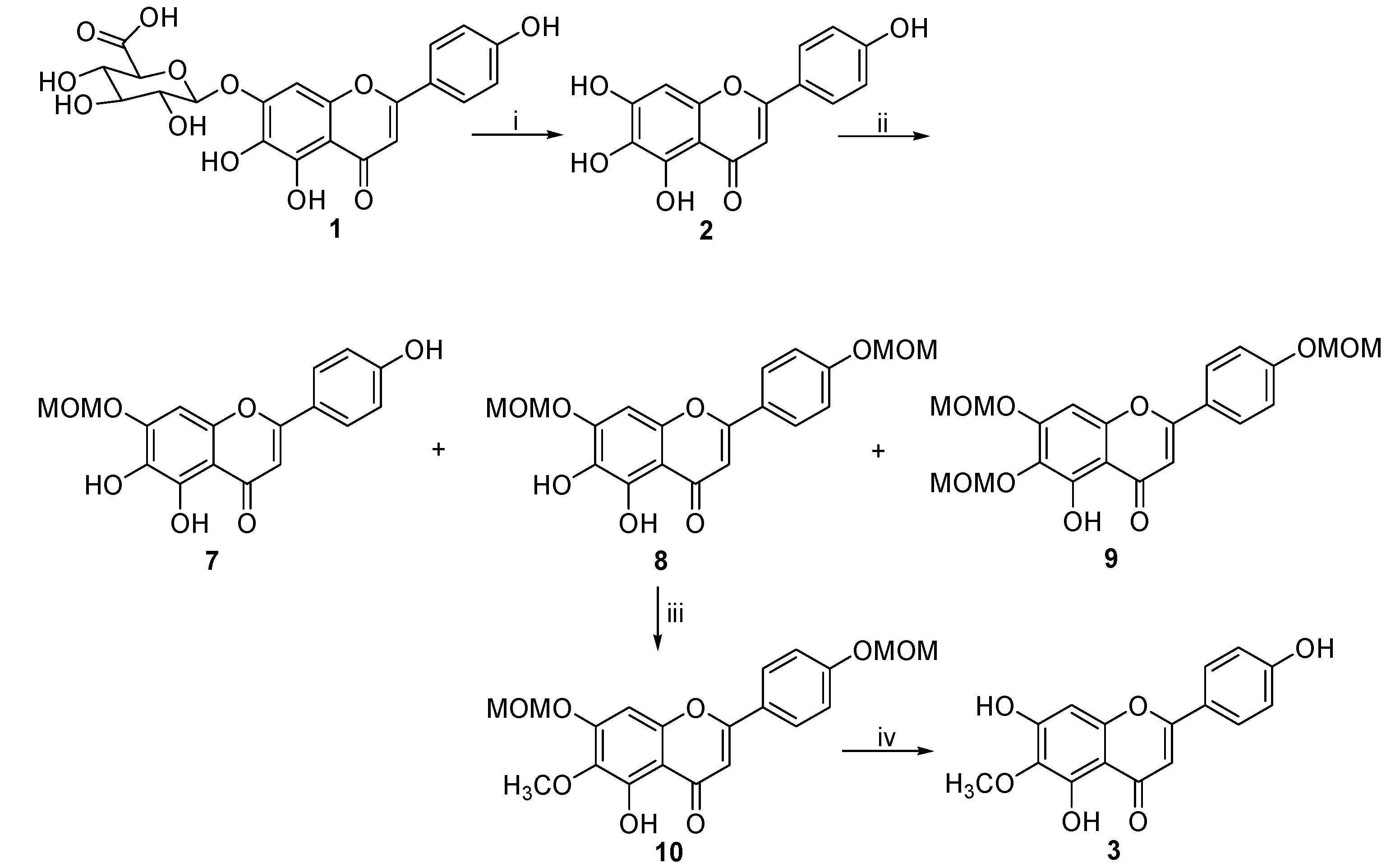
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Run | Reaction Conditions | Yield (%) |
|---|---|---|
| 1 | MOMCl (4.0 equiv.), K2CO3 (4.2 equiv.), acetone, reflux, N2, 8 h | 7 (14.8), 8 (28.9), 9 (34.2) |
| 2 | MOMCl (4.2 equiv.), K2CO3 (4.4 equiv.), acetone, reflux, N2, 8 h | 7 (12.1), 8 (24.3), 9 (38.7) |
| 3 | MOMCl (4.4 equiv.), K2CO3 (4.6 equiv.), acetone, reflux, N2, 8 h | 7 (10.2), 8 (21.6), 9 (41.8) |
| 4 | MOMCl (3.8 equiv.), K2CO3 (4.0 equiv.), acetone, reflux, N2, 8 h | 7 (16.4), 8 (32.1), 9 (31.6) |
| 5 | MOMCl (3.6 equiv.), K2CO3 (3.8 equiv.), acetone, reflux, N2, 8 h | 7 (18.2), 8 (35.8), 9 (26.5) |
| 6 | MOMCl (3.4 equiv.), K2CO3 (3.6 equiv.), acetone, reflux, N2, 8 h | 7 (19.2), 8 (43.2), 9 (18.7) |
| 7 | MOMCl (3.2 equiv.), K2CO3 (3.4 equiv.), acetone, reflux, N2, 8 h | 7 (20.6), 8 (38.4), 9 (16.4) |
| 8 | MOMCl (3.0 equiv.), K2CO3 (3.2 equiv.), acetone, reflux, N2, 8 h | 7 (21.3), 8 (36.2), 9 (14.2) |

3. Experimental Section
General Information
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Lapikova, E.S.; Drozd, N.N.; Tolstenkov, A.S.; Makarov, V.A.; Zvyagintseva, T.N.; Shevchenko, N.M.; Bakunina, I.U.; Besednova, N.N.; Kuznetsova, T.A. Inhibition of thrombin and factor Xa by Fucus evanescens fucoidan and its modified analogs. Bull. Exp. Biol. Med. 2008, 146, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Simard, D.; Bayrakdarian, M.; Therrien, E.; Nilsson, I.; Fjellström, O. Design, synthesis, and thrombin-inhibitory activity of pyridin-2-ones as P2/P3 core motifs. Bioorg. Med. Chem. Lett. 2008, 18, 1972–1976. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Riley, D.P.; Caputi, A.P.; Salvemini, D. Antioxidant therapy: A new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol. Rev. 2001, 53, 135–159. [Google Scholar] [PubMed]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric oxide and atherosclerosis: An update. Nitric Oxide 2006, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.W.; Feng, T.M.; Shan, L.C.; Cai, B.Z.; Chu, W.F.; Niu, H.L.; Lu, Y.J.; Yang, B.F. Scutellarin-induced endothelium-independent relaxation in rat aorta. Phytother. Res. 2008, 22, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; He, W.; Lu, W.H.; Zeng, F.D. Effects of scutellarin on liver function after brain ischemia/reperfusion in rats. Acta. Pharmacol. Sin. 2003, 24, 1118–1124. [Google Scholar] [PubMed]
- Hong, H.; Liu, G.Q. Scutellarin attenuates oxidative glutamate toxicity in PC12 cells. Planta Med. 2004, 70, 427–431. [Google Scholar] [PubMed]
- Liu, H.; Yang, X.; Tang, R.; Liu, J.; Xu, H. Effect of scutellarin on nitric oxide production in early stages of neuron damage induced by hydrogen peroxide. Pharmacol. Res. 2005, 51, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Cheng, G.; Yu, J.E.; He, Y.; An, F.; Sun, J.; Cui, F. Study on the role of hepatic first-pass elimination in the low oral bioavailability of scutellarin in rats. Die Pharm. 2005, 60, 477–478. [Google Scholar]
- Chen, X.; Cui, L.; Duan, X.; Ma, B.; Zhong, D. Pharmacokinetics and metabolism of the flavonoid scutellarin in humans after a single oral administration. Drug Metab. Dispos. 2006, 34, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, N.; Yu, Y.; Weng, W.; Huang, X. Determination of aglycone conjugated metabolites of scutellarin in rat plasma by HPLC. J. Pharm. Biomed. Anal. 2006, 40, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, W.; Dong, Z.X.; Gu, T.; Li, N.G.; Shi, Z.H.; Kai, J.; Qu, C.; Shang, G.X.; Tang, Y.P.; et al. A new and practical synthetic method for the synthesis of 6-O-methyl-scutellarein: One metabolite of scutellarin in vivo. Int. J. Mol. Sci. 2015, 16, 7587–7594. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Z.; Shi, Z.H.; Li, N.G.; Tang, H.; Shi, Q.P.; Tang, Y.P.; Yang, J.P.; Duan, J.A. Efficient synthesis of 6-O-methyl-scutellarein from scutellarin via selective methylation. Lett. Org. Chem. 2013, 10, 733–737. [Google Scholar] [CrossRef]
- Li, N.G.; Song, S.L.; Shen, M.Z.; Tang, Y.P.; Shi, Z.H.; Tang, H.; Shi, Q.P.; Fu, Y.F.; Duan, J.A. Mannich bases of scutellarein as thrombin-inhibitors: Design, synthesis, biological activity and solubility. Bioorg. Med. Chem. 2012, 20, 6919–6923. [Google Scholar] [CrossRef] [PubMed]
- Li, N.G.; Shen, M.Z.; Wang, Z.J.; Tang, Y.P.; Shi, Z.H.; Fu, Y.F.; Shi, Q.P.; Tang, H.; Duan, J.A. Design, synthesis and biological evaluation of glucose-containing scutellarein derivatives as neuroprotective agents based on metabolic mechanism of scutellarin in vivo. Bioorg. Med. Chem. Lett. 2013, 23, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Samples of the compounds 1, 2, 3, 7, 8, 9, 10 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Dong, Z.-X.; Gu, T.; Li, N.-G.; Zhang, P.-X.; Wu, W.-Y.; Yu, S.-P.; Tang, Y.-P.; Yang, J.-P.; Shi, Z.-H. A New and Efficient Synthesis of 6-O-Methylscutellarein, the Major Metabolite of the Natural Medicine Scutellarin. Molecules 2015, 20, 10184-10191. https://doi.org/10.3390/molecules200610184
Zhang W, Dong Z-X, Gu T, Li N-G, Zhang P-X, Wu W-Y, Yu S-P, Tang Y-P, Yang J-P, Shi Z-H. A New and Efficient Synthesis of 6-O-Methylscutellarein, the Major Metabolite of the Natural Medicine Scutellarin. Molecules. 2015; 20(6):10184-10191. https://doi.org/10.3390/molecules200610184
Chicago/Turabian StyleZhang, Wei, Ze-Xi Dong, Ting Gu, Nian-Guang Li, Peng-Xuan Zhang, Wen-Yu Wu, Shao-Peng Yu, Yu-Ping Tang, Jian-Ping Yang, and Zhi-Hao Shi. 2015. "A New and Efficient Synthesis of 6-O-Methylscutellarein, the Major Metabolite of the Natural Medicine Scutellarin" Molecules 20, no. 6: 10184-10191. https://doi.org/10.3390/molecules200610184