
Development and Validation of a Docking-Based Virtual Screening Platform for the Identification of New Lactate Dehydrogenase Inhibitors

Abstract
:
1. Introduction
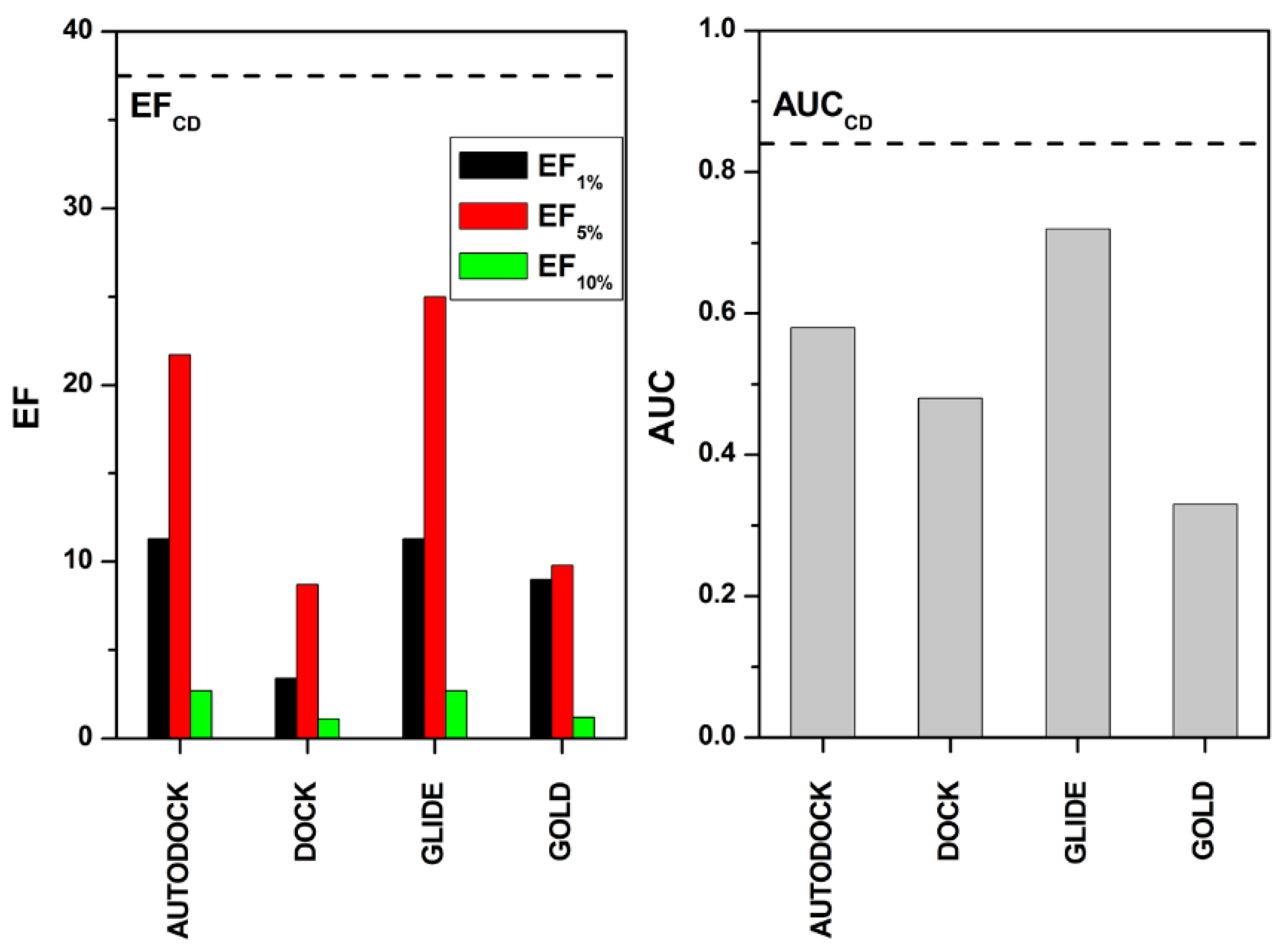
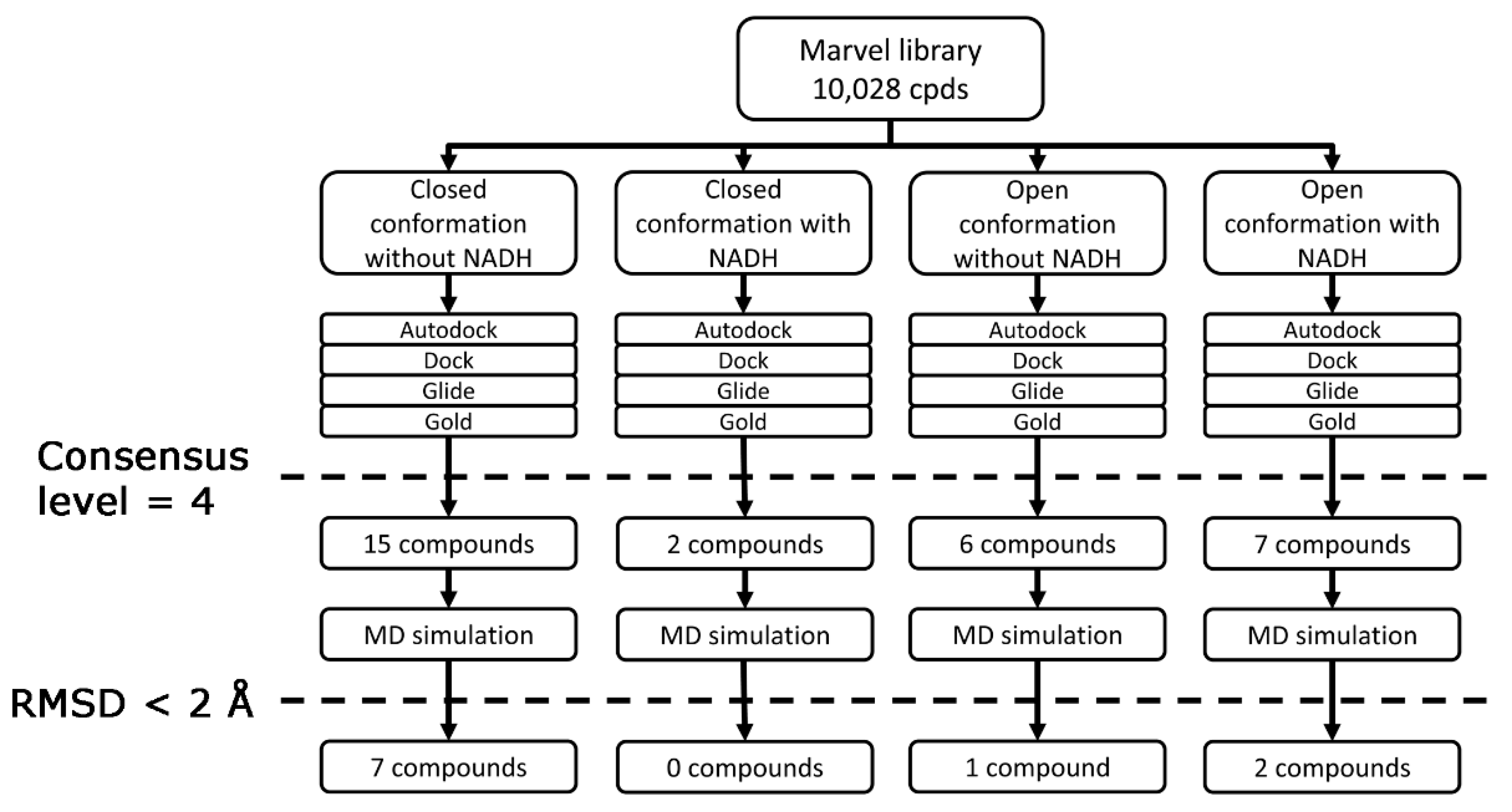
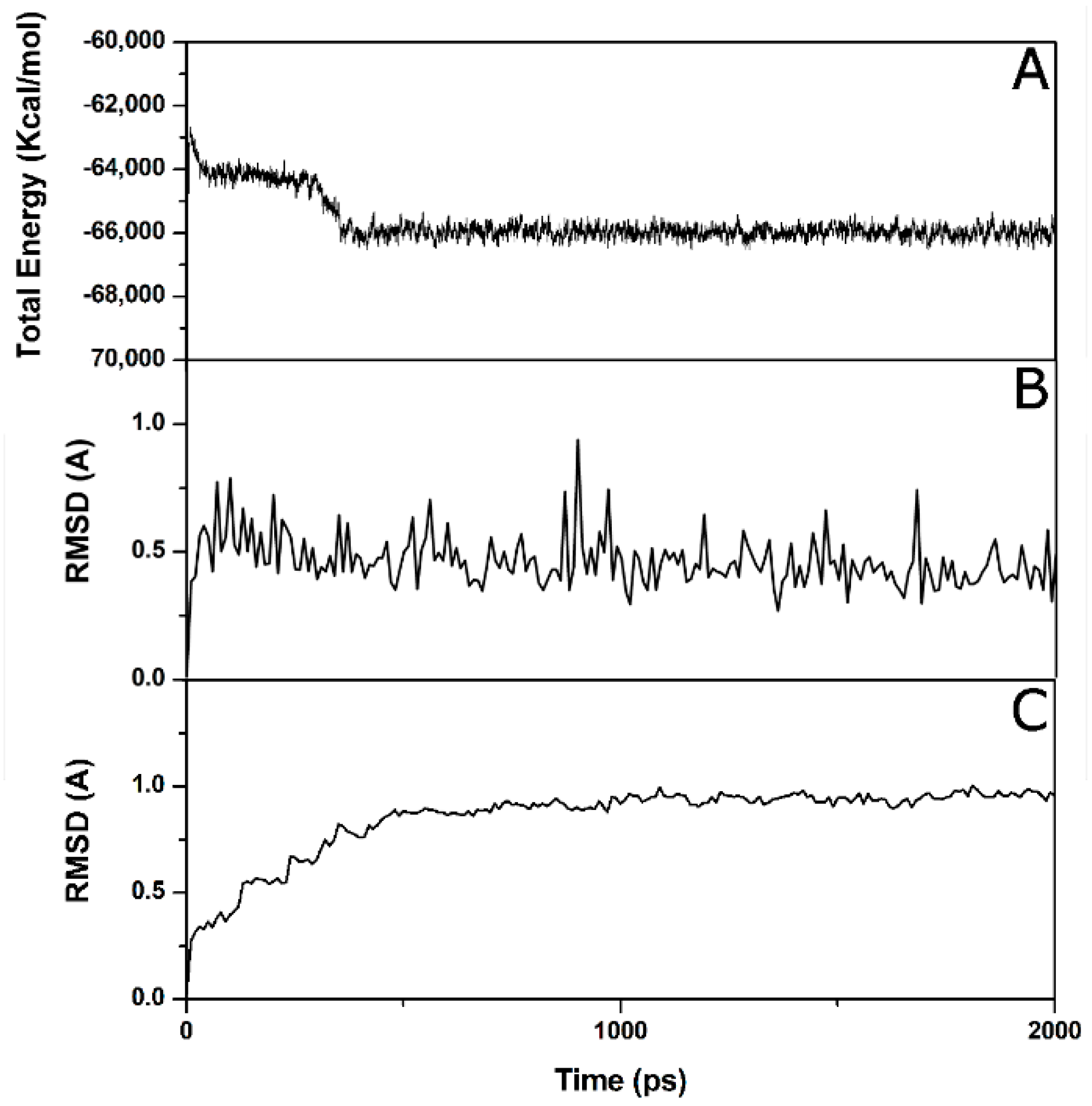
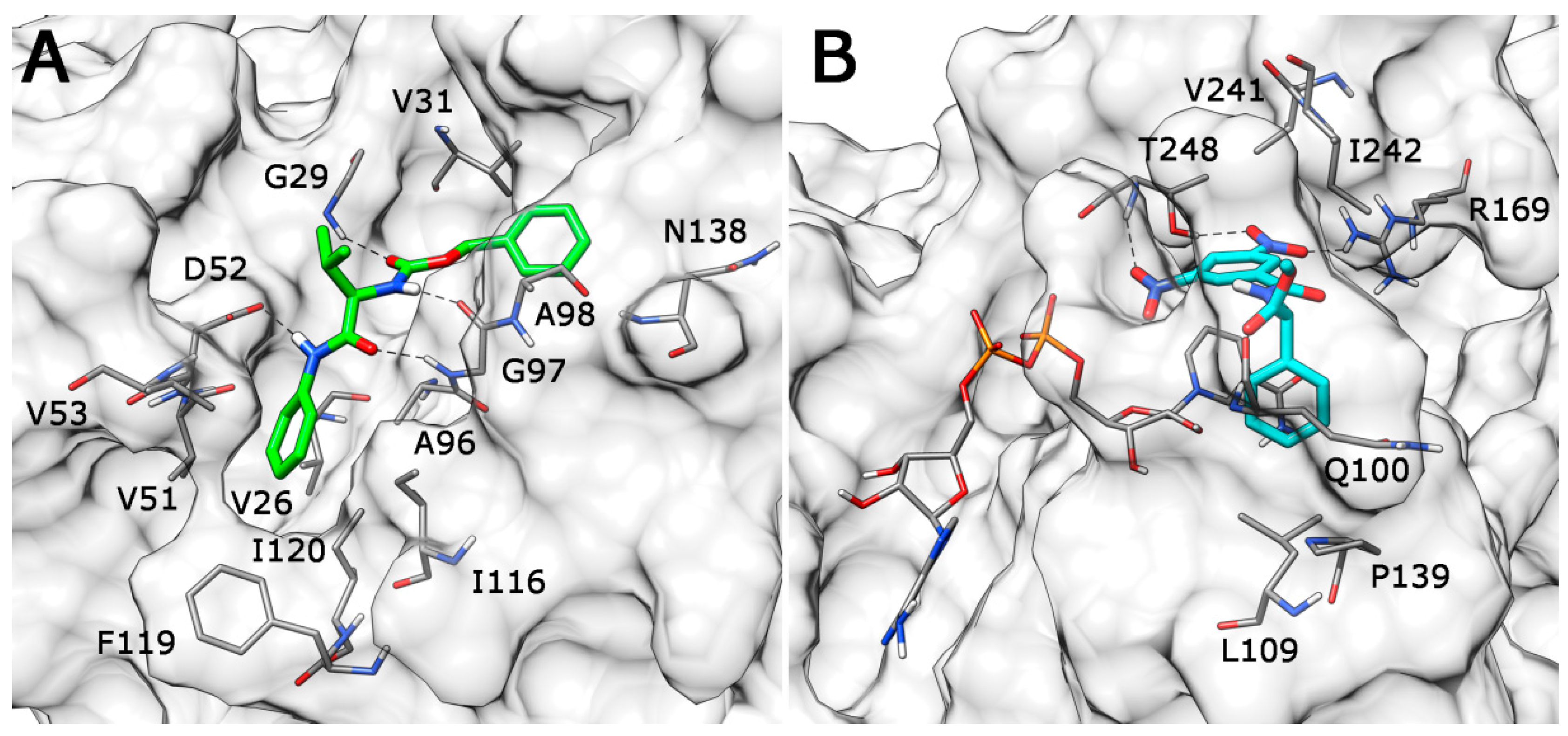
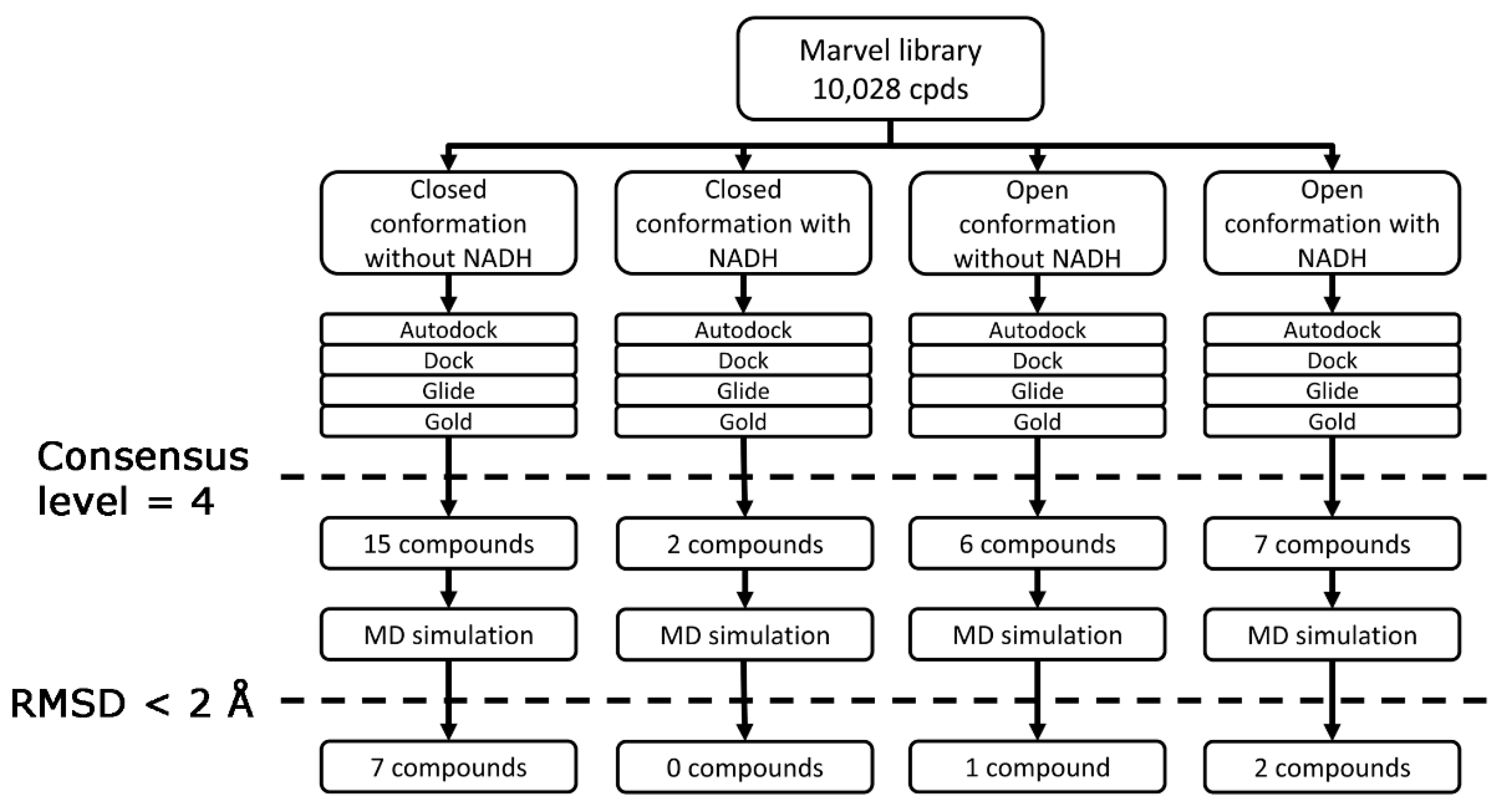
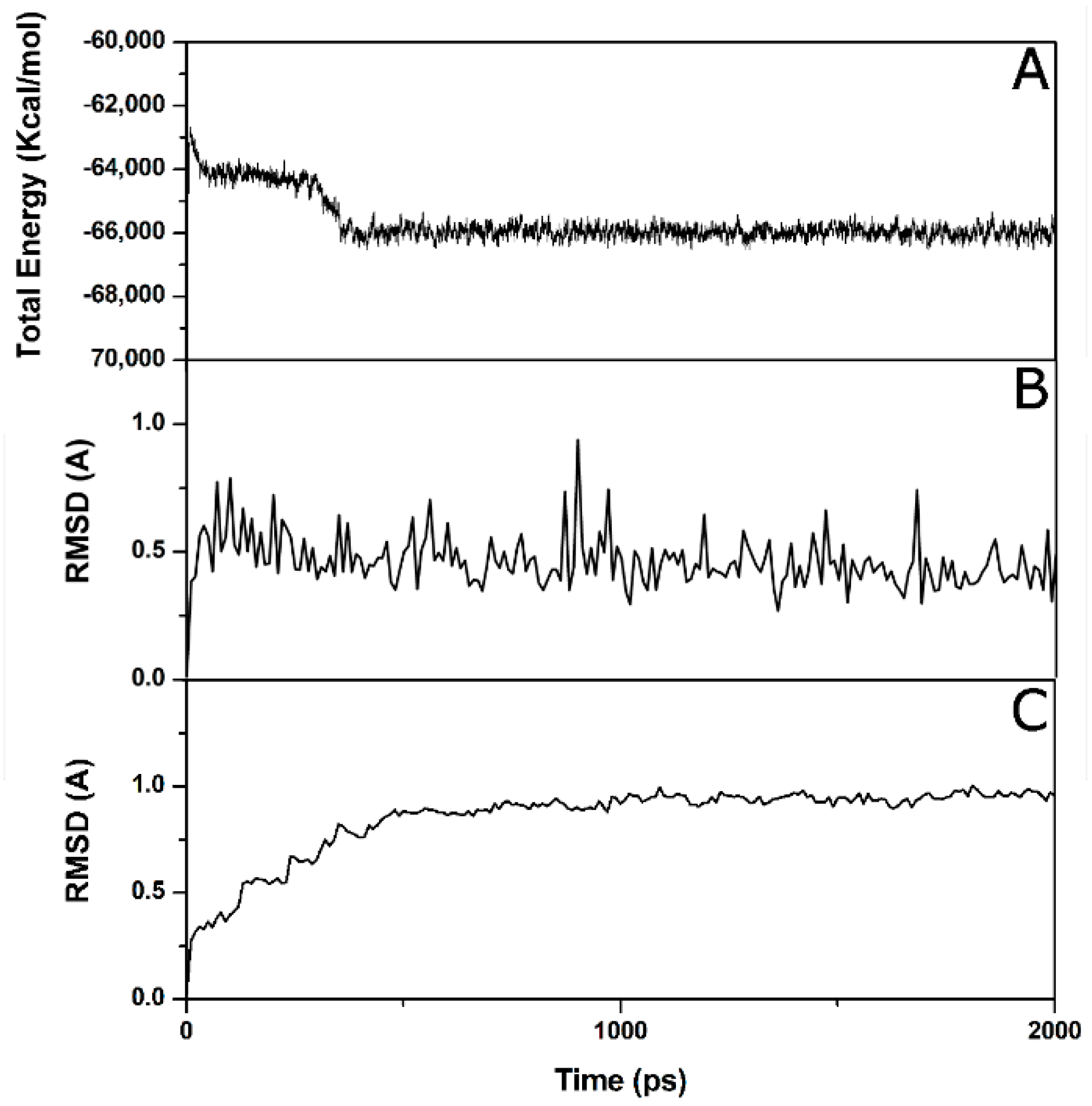
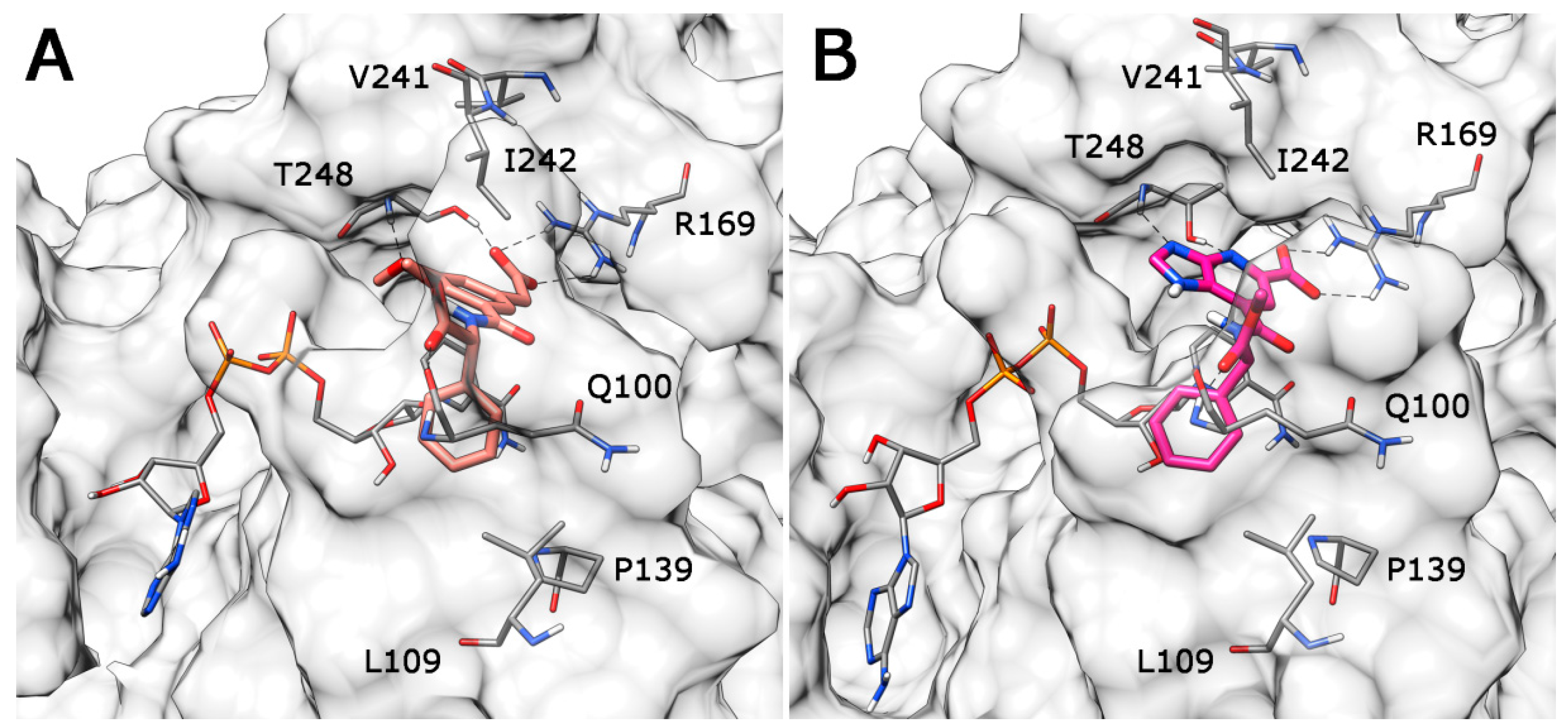
2. Results and Discussion

| Consensus Level | Actives | Decoys | EF |
|---|---|---|---|
| 1 | 93 | 10,000 | 1.0 |
| 2 | 75 | 1952 | 3.7 |
| 3 | 43 | 177 | 19.5 |
| 4 | 12 | 20 | 40.7 |
| AUC | 0.84 |
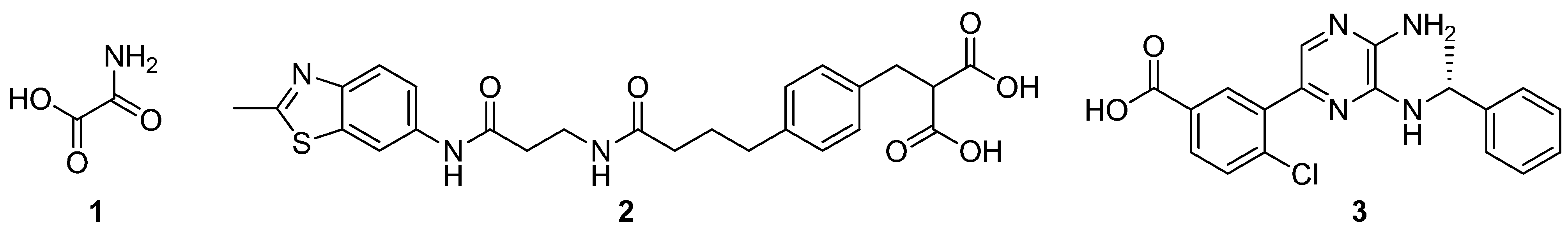
| Structure | hLDH5, IC50 (µM) | |
|---|---|---|
| VS1 | 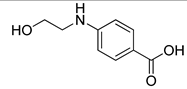 | >500 |
| VS2 |  | >500 |
| VS3 | 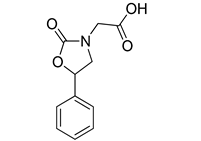 | >500 |
| VS4 | 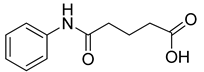 | >500 |
| VS5 | 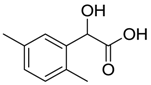 | >500 |
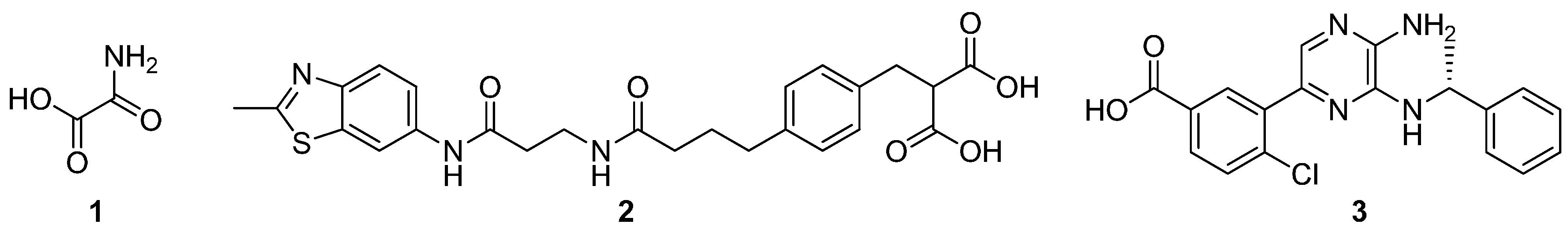
| VS6 |  | 245.7 ± 10.8 |
| VS7 | 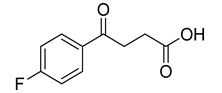 | >500 |
| VS8 | 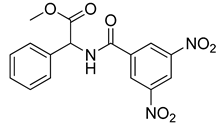 | 268.6 ± 58.1 |
| VS9 | 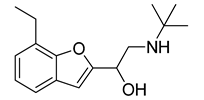 | >500 |
| VS10 | 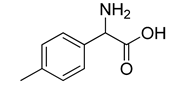 | >500 |
| galloflavin | 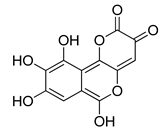 | 91.7 ± 10.7 |
| oxamic acid | 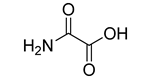 | 97.7 ± 11.2 |
| NHI-1 | 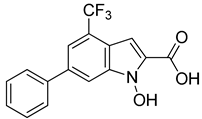 | 51.7 ± 4.2 |
| NHI-2 |  | 10.8 ± 3.5 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
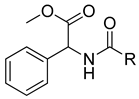
| R | ||
|---|---|---|
| VS8a | VS8b | VS8c |
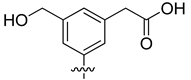 | 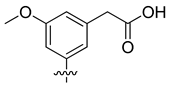 | 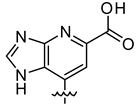 |
3. Experimental Section
3.1. Molecular Modeling
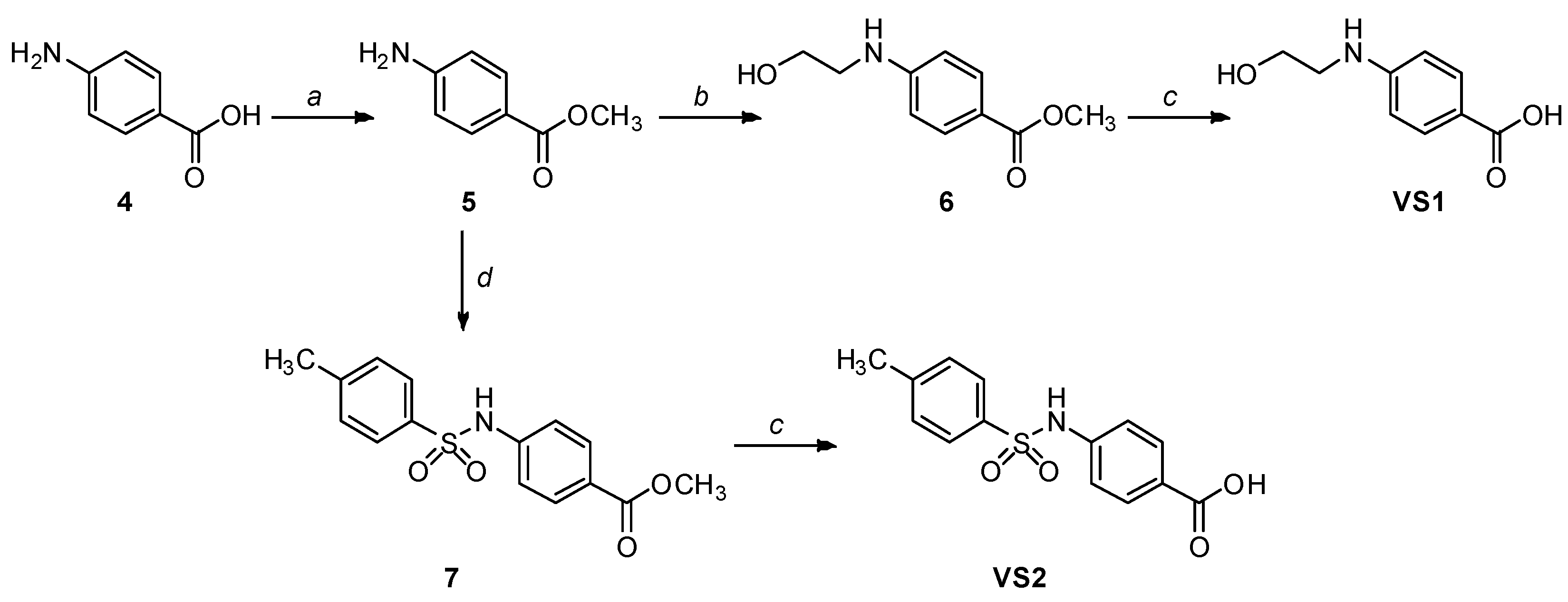
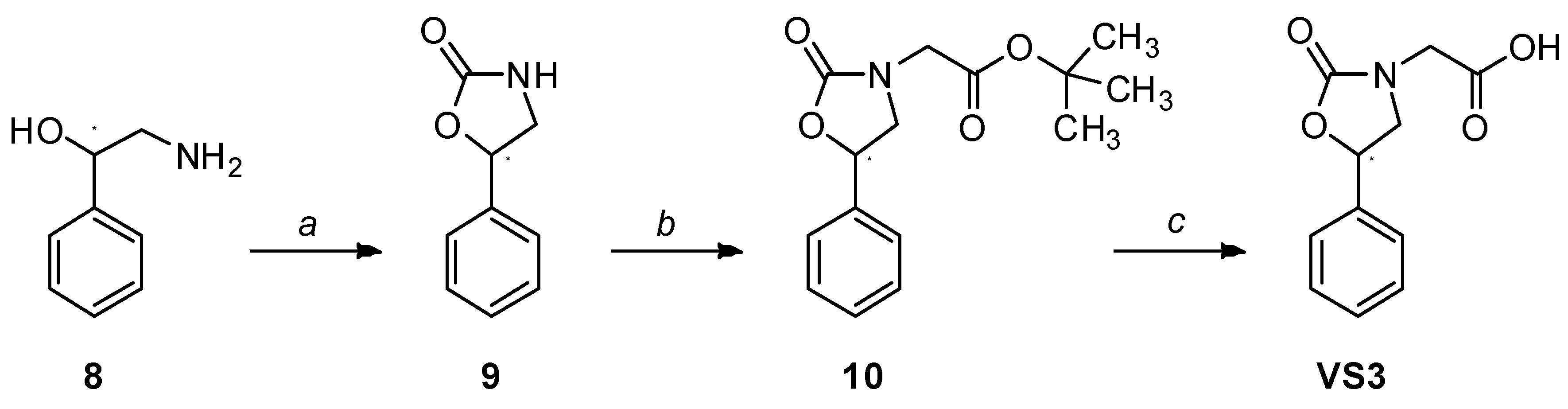
3.2. Chemistry
3.2.1. General
3.2.2. Synthetic Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Granchi, C.; Bertini, S.; Macchia, M.; Minutolo, F. Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials. Curr. Med. Chem. 2010, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Minutolo, F. Anticancer agents that counteract tumor glycolysis. ChemMedChem 2012, 7, 1318–1350. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Fancelli, D.; Minutolo, F. An update on therapeutic opportunities offered by cancer glycolytic metabolism. Bioorg. Med. Chem. Lett. 2014, 24, 4915–4925. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-molecule inhibitors of human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; De Simone, A.; Salvetti, I.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Lanza, M.; Betti, L.; Giannaccini, G.; et al. N-Hydroxyindole-based inhibitors of lactate dehydrogenase against cancer cell proliferation. Eur. J. Med. Chem. 2011, 46, 5398–5407. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Mottinelli, M.; Nardini, E.; Campinoti, F.; Tuccinardi, T.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Synthesis of sulfonamide-containing N-hydroxyindole-2-carboxylates as inhibitors of human lactate dehydrogenase-isoform 5. Bioorgainc Med. Chem. Lett. 2011, 21, 7331–7336. [Google Scholar] [CrossRef]
- Zhang, M.; Peh, J.; Hergenrother, P.J.; Cunningham, B.T. Detection of protein-small molecule binding using a self-referencing external cavity laser biosensor. J. Am. Chem. Soc. 2014, 136, 5840–5843. [Google Scholar] [CrossRef] [PubMed]
- Calvaresi, E.C.; Granchi, C.; Tuccinardi, T.; Di Bussolo, V.; Huigens, R.W., 3rd; Lee, H.Y.; Palchaudhuri, R.; Macchia, M.; Martinelli, A.; Minutolo, F.; et al. Dual targeting of the warburg effect with a glucose-conjugated lactate dehydrogenase inhibitor. ChemBioChem 2013, 14, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Di Bussolo, V.; Calvaresi, E.C.; Granchi, C.; Del Bino, L.; Frau, I.; Dasso Lang, M.C.; Tuccinardi, T.; Macchia, M.; Martinelli, A.; Hergenrother, P.J.; et al. Synthesis and biological evaluation of non-glucose glycoconjugated N-hydroyxindole class LDH inhibitors as anticancer agents. RSC Adv. 2015, 5, 19944–19954. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. Docking-based virtual screening: Recent developments. Comb. Chem. High Throughput Screen. 2009, 12, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A.; Brassington, C.; Breeze, A.L.; Caputo, A.; Critchlow, S.; Davies, G.; Goodwin, L.; Hassall, G.; Greenwood, R.; Holdgate, G.A.; et al. Design and synthesis of novel lactate dehydrogenase a inhibitors by fragment-based lead generation. J. Med. Chem. 2012, 55, 3285–3306. [Google Scholar] [CrossRef] [PubMed]
- Fauber, B.P.; Dragovich, P.S.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y.; et al. Identification of 2-amino-5-aryl-pyrazines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 5533–5539. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Fauber, B.P.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y.; et al. Identification of substituted 2-thio-6-oxo-1,6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 3186–3194. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Fauber, B.P.; Boggs, J.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; et al. Identification of substituted 3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2014, 24, 3764–3771. [Google Scholar] [CrossRef] [PubMed]
- Fauber, B.P.; Dragovich, P.S.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Labadie, S.; Malek, S.; Peterson, D.; Purkey, H.E.; et al. Identification of 3,6-disubstituted dihydropyrones as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2014, 24, 5683–5687. [Google Scholar] [CrossRef] [PubMed]
- Labadie, S.; Dragovich, P.S.; Chen, J.; Fauber, B.P.; Boggs, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Ho, Q.; et al. Optimization of 5-(2,6-dichlorophenyl)-3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2015, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on pubchem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Giuntini, N.; Martinelli, A.; Tuccinardi, T. Application of a flap-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J. Chem. Inf. Model. 2015, 55, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Manerba, M.; Vettraino, M.; Fiume, L.; Di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (cas 568–80–9): A novel inhibitor of lactate dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Calvaresi, E.C.; Tuccinardi, T.; Paterni, I.; Macchia, M.; Martinelli, A.; Hergenrother, P.J.; Minutolo, F. Assessing the differential action on cancer cells of LDH-A inhibitors based on the N-hydroxyindole-2-carboxylate (NHI) and malonic (Mal) scaffolds. Org. Biomol. Chem. 2013, 11, 6588–6596. [Google Scholar] [CrossRef] [PubMed]
- Brood, version 2.0.0; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: http://www.eyesopen.com (accessed on 4 April 2015).
- Ligprep, version 2.5; Schrödinger Inc: Portland, OR, USA, 2011.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Dock, version 6.5; Molecular Design Institute, University of California: San Francisco, CA, USA, 1998.
- Glide, version 5.0; Schrödinger Inc: Portland, OR, USA, 2009.
- Verdonk, M.L.; Mortenson, P.N.; Hall, R.J.; Hartshorn, M.J.; Murray, C.W. Protein-ligand docking against non-native protein conformers. J. Chem. Inf. Model. 2008, 48, 2214–2225. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Tuccinardi, T.; Rizzolio, F.; Caligiuri, I.; Botta, L.; Granchi, C.; Ortore, G.; Minutolo, F.; Schenone, S.; Martinelli, A. Identification of new fyn kinase inhibitors using a flap-based approach. J. Chem. Inf. Model. 2013, 53, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber, version 11; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals—A comparison of the ewald and truncated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granchi, C.; Capecchi, A.; Del Frate, G.; Martinelli, A.; Macchia, M.; Minutolo, F.; Tuccinardi, T. Development and Validation of a Docking-Based Virtual Screening Platform for the Identification of New Lactate Dehydrogenase Inhibitors. Molecules 2015, 20, 8772-8790. https://doi.org/10.3390/molecules20058772
Granchi C, Capecchi A, Del Frate G, Martinelli A, Macchia M, Minutolo F, Tuccinardi T. Development and Validation of a Docking-Based Virtual Screening Platform for the Identification of New Lactate Dehydrogenase Inhibitors. Molecules. 2015; 20(5):8772-8790. https://doi.org/10.3390/molecules20058772
Chicago/Turabian StyleGranchi, Carlotta, Alice Capecchi, Gianluca Del Frate, Adriano Martinelli, Marco Macchia, Filippo Minutolo, and Tiziano Tuccinardi. 2015. "Development and Validation of a Docking-Based Virtual Screening Platform for the Identification of New Lactate Dehydrogenase Inhibitors" Molecules 20, no. 5: 8772-8790. https://doi.org/10.3390/molecules20058772