
Gd-Complexes of New Arylpiperazinyl Conjugates of DTPA-Bis(amides): Synthesis, Characterization and Magnetic Relaxation Properties




Abstract
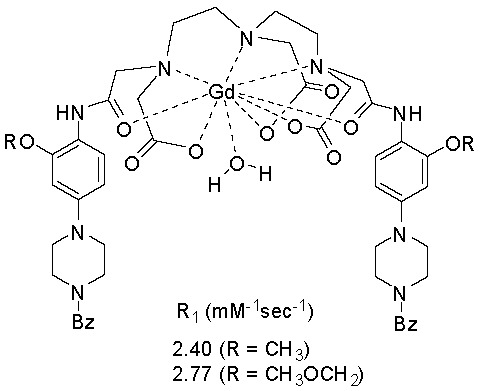
:
1. Introduction
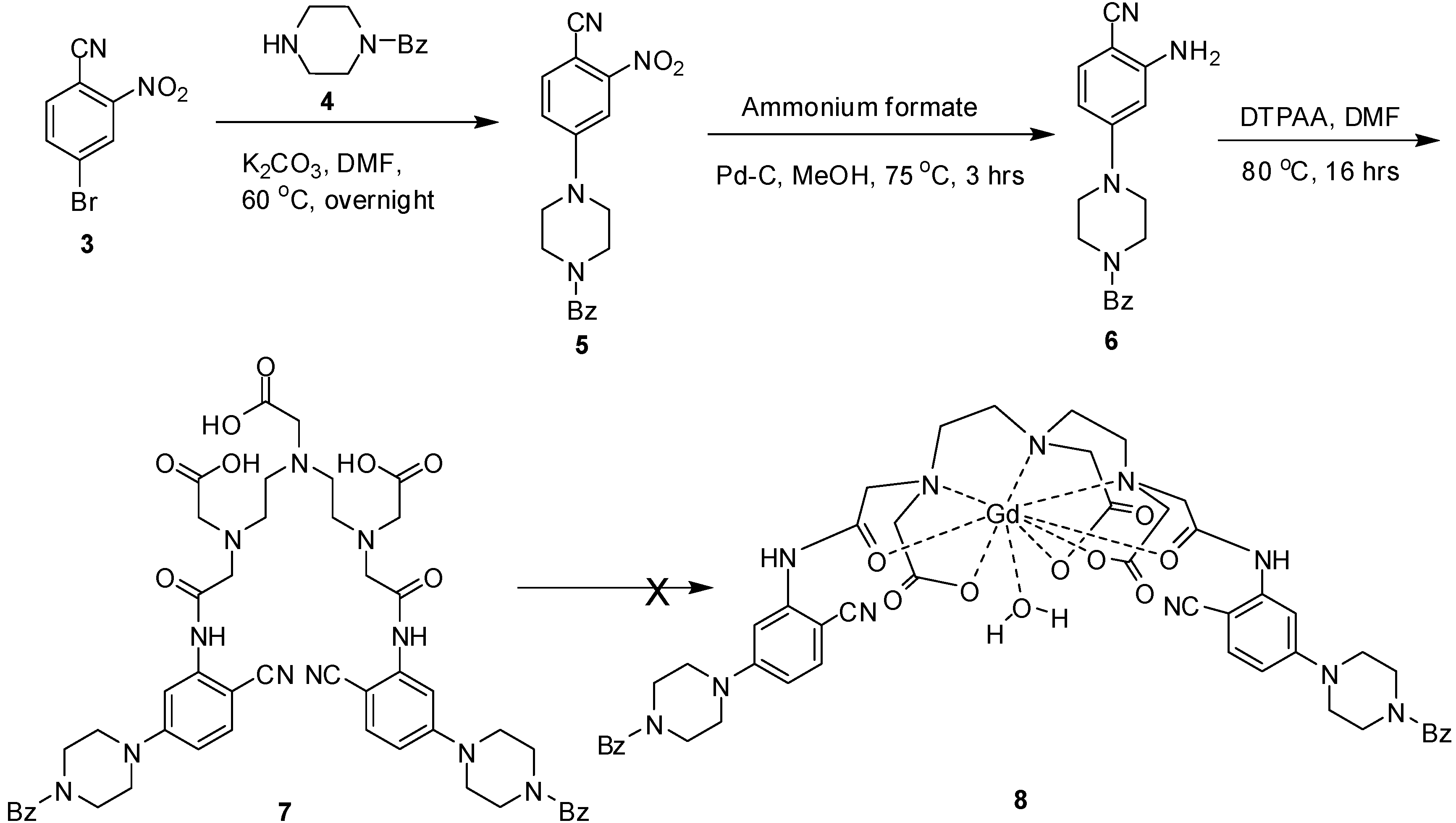
2. Results and Discussion

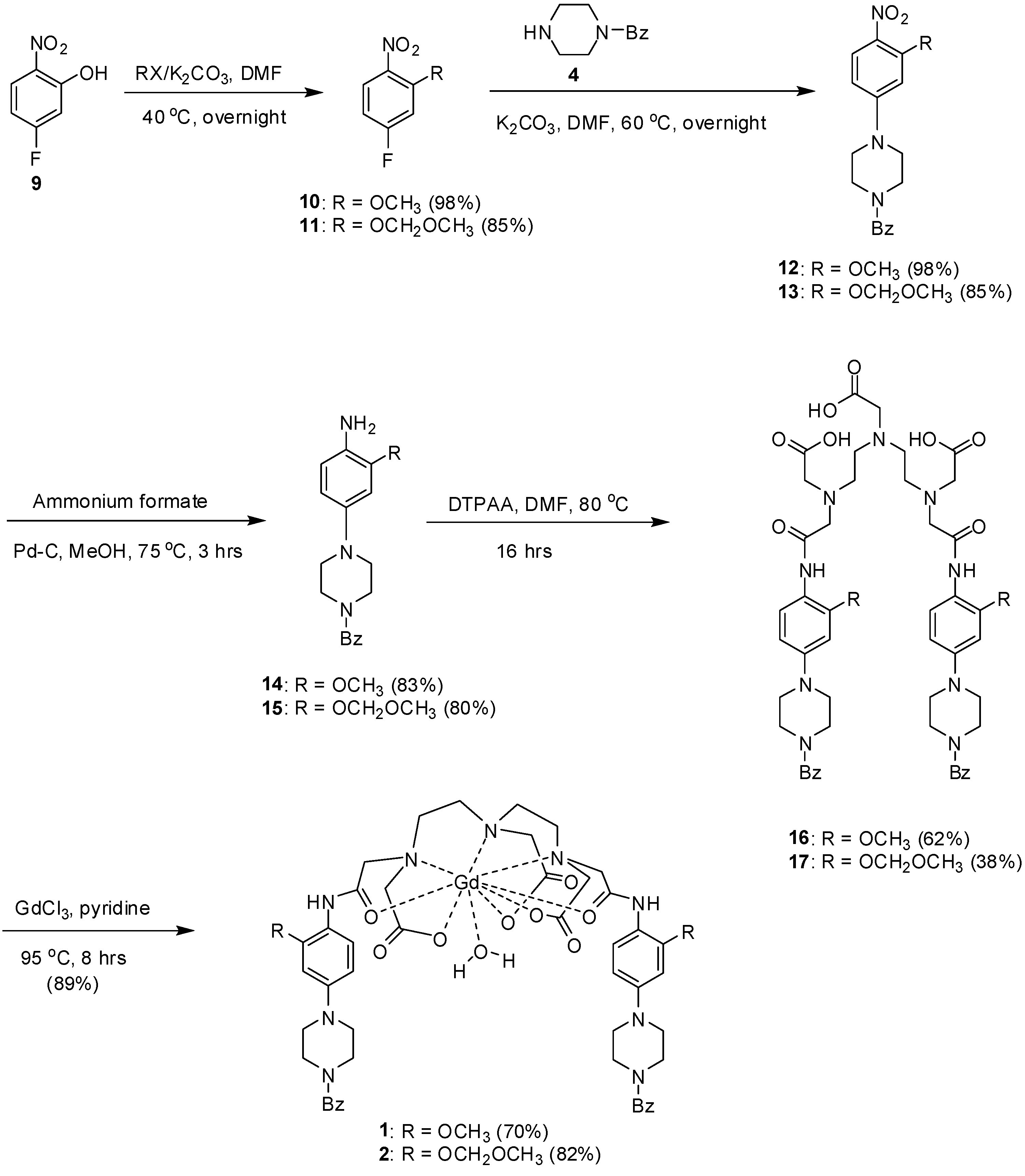

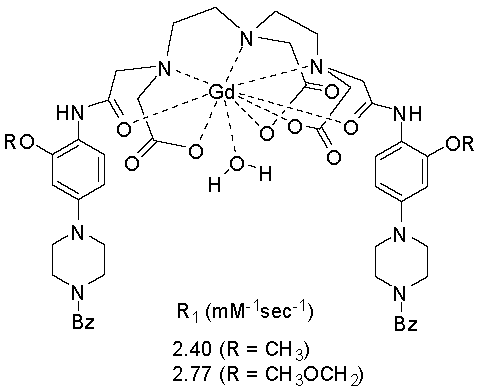{kind=link}
{kind=link}
{kind=link}
{kind=link}
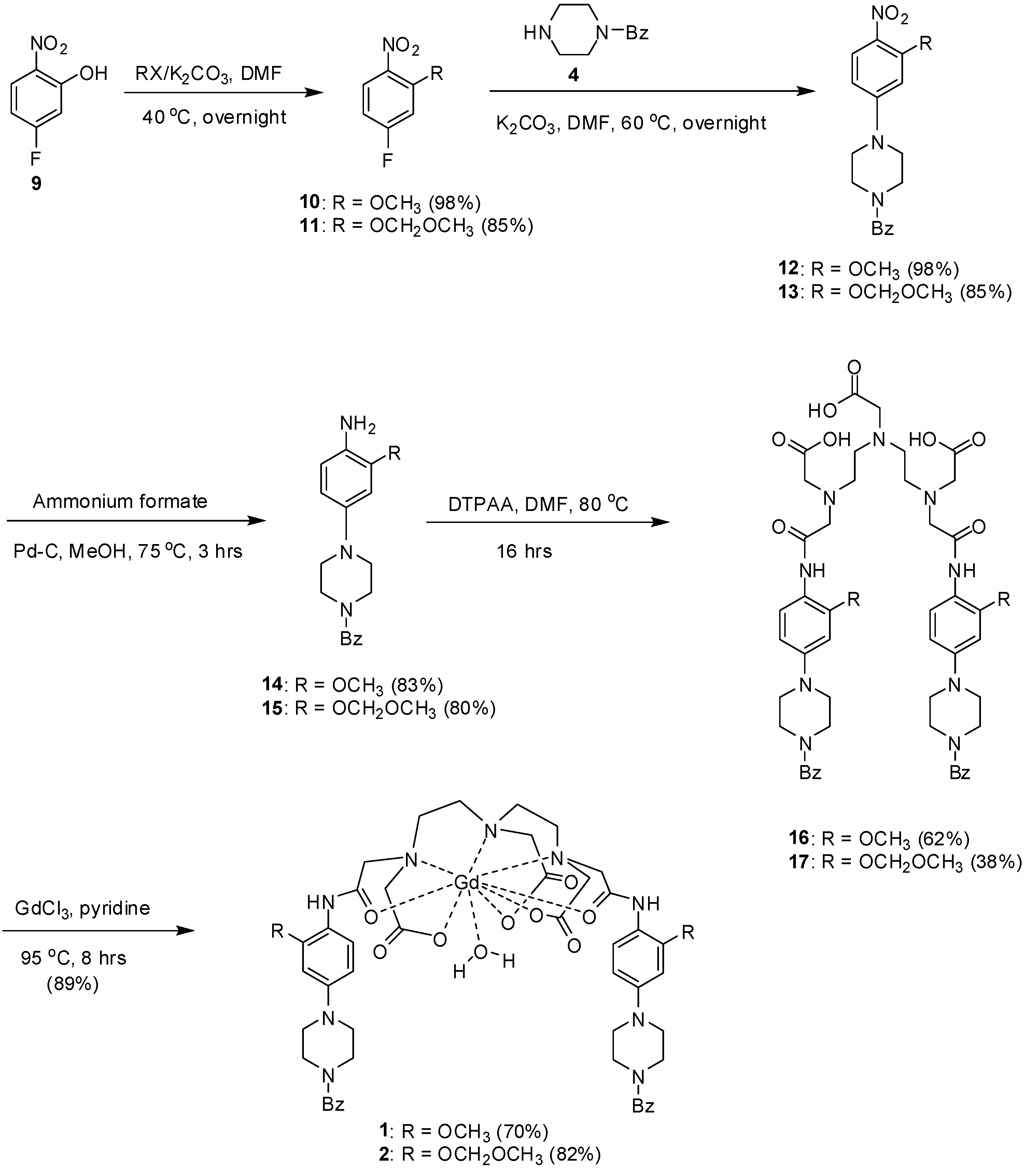{kind=link}
| CA | T1 (ms) | R1 (mM−1s−1) | In Vitro Cytotoxicity (IC50 μg/mL) | |
|---|---|---|---|---|
| 1 | 415.72 ± 2.32 | 2.40 | 1 | >50 |
| 2 | 360.93 ± 1.56 | 2.77 | 2 | >50 |
| Omniscan® a | 3.2 | Cycloheximide | 0.13 ± 0.02 | |
3. Experimental Section
3.1. General Information
3.2. Relaxivity Measurements
3.3. In Vitro Cell Toxicity
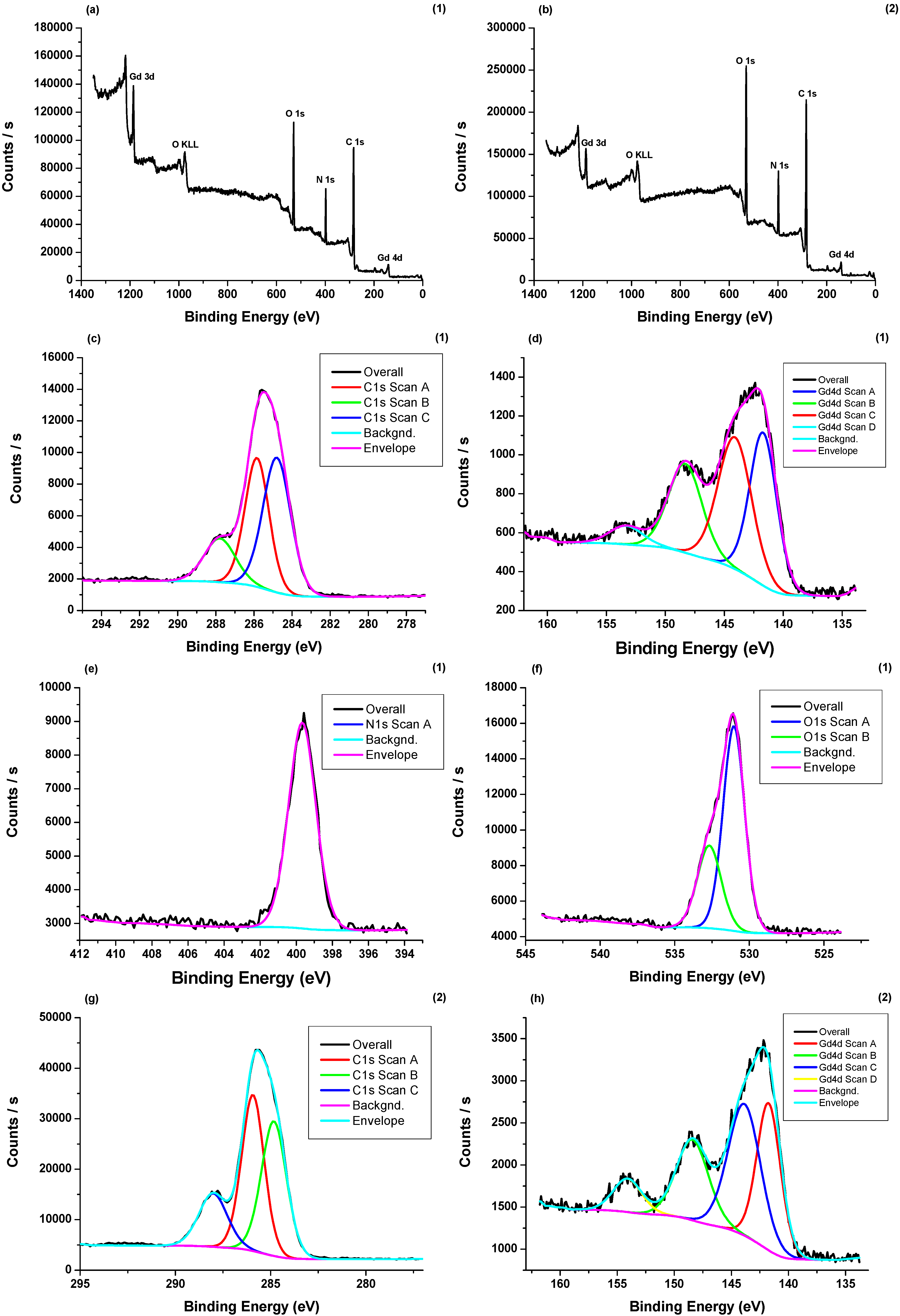
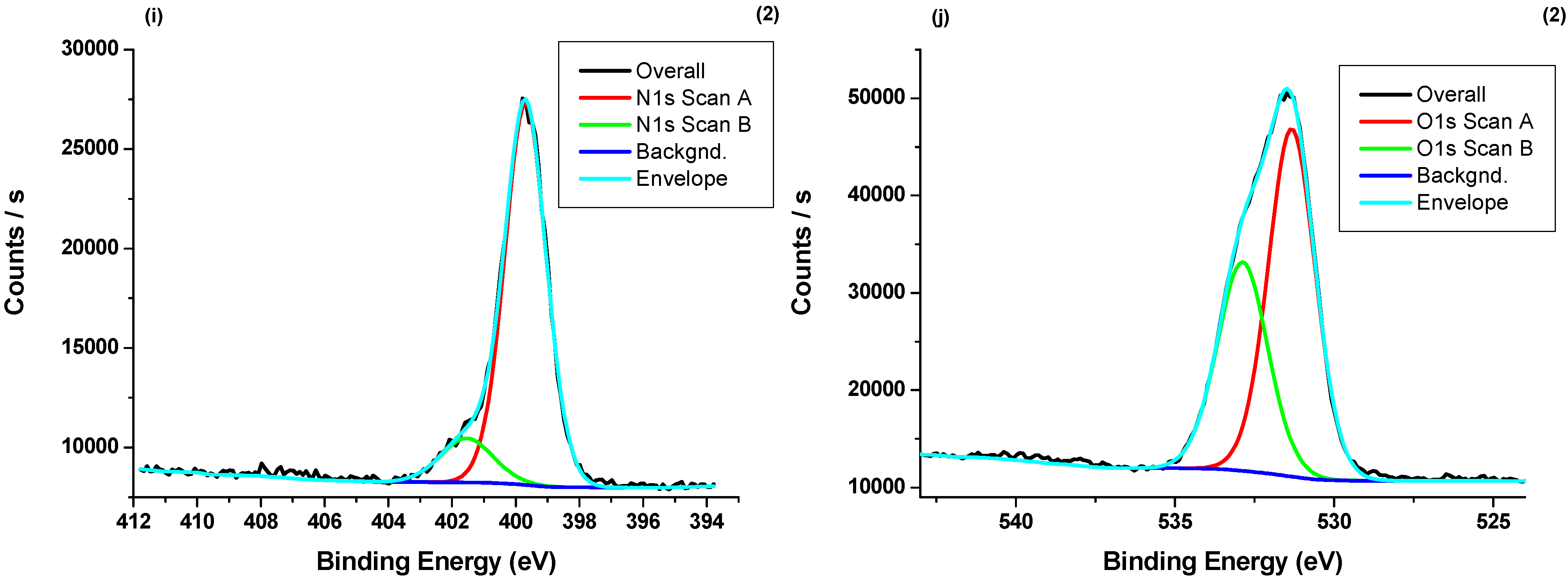
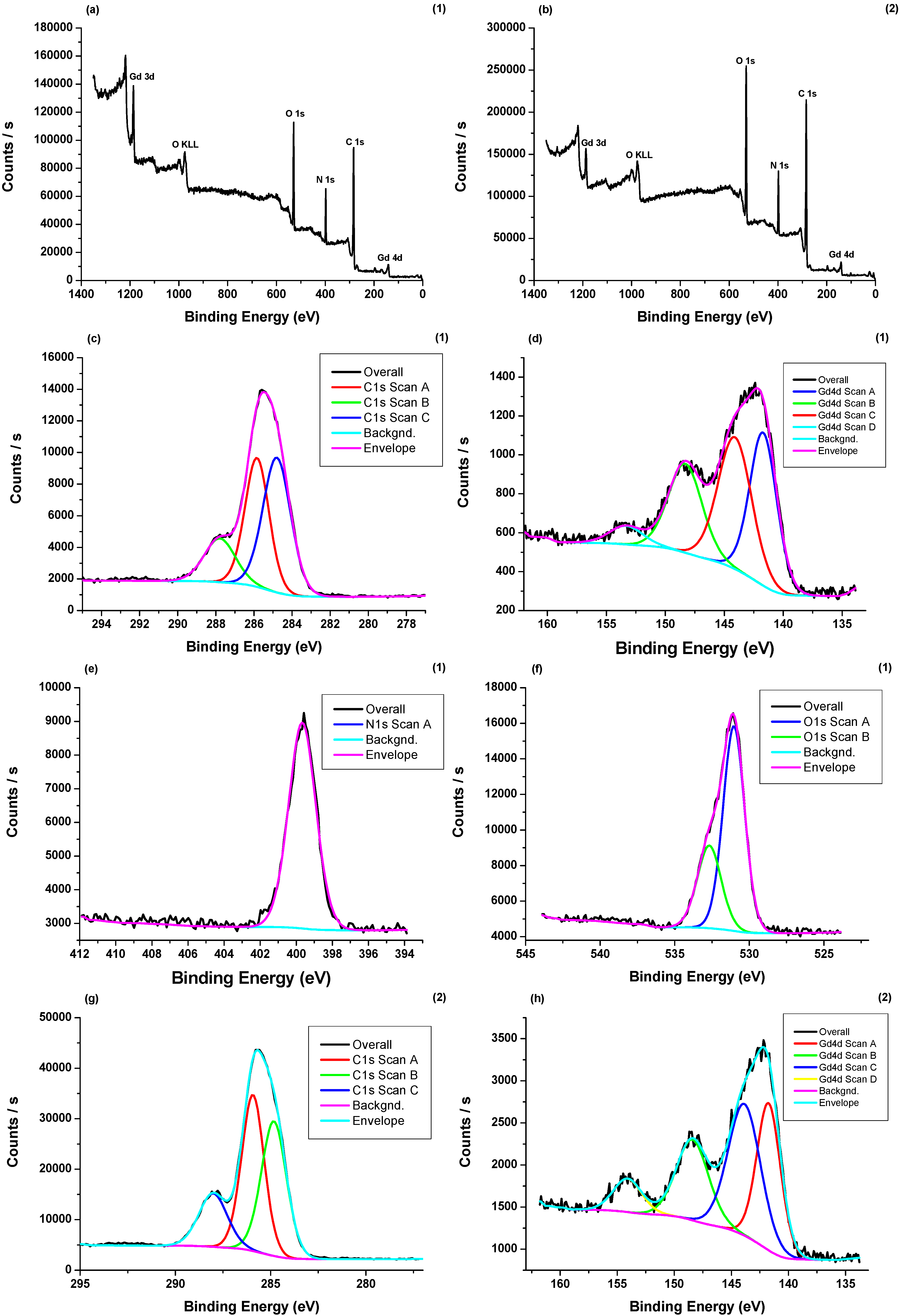
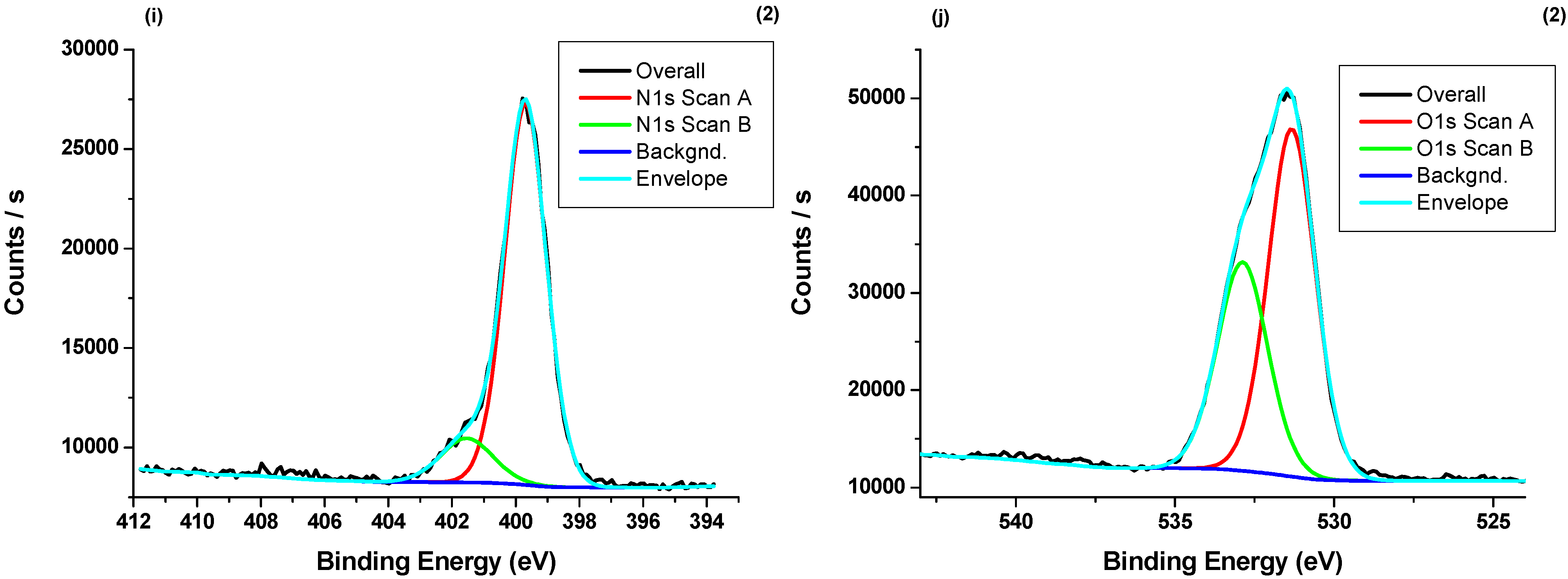
3.4. X-ray Photoelectron Spectroscopy (XPS) Measurements
3.5. Chemistry
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lauterbur, P.C. Image formation by induced local interactions: Examples employing nuclear magnetic resonance. Nature 1973, 242, 190–191. [Google Scholar] [CrossRef]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef] [PubMed]
- Aneta, K.; Thomas, Z. Bioorganic synthesis of some recent tactical application of bioisosteres in drug design. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Ogawa, S.; Lee, T.-M.; Nayak, A.S.; Glynn, P. Oxygenation-sensitive contrasts in magnetic resonance image of rodent brain at high magnetic fields. Magn. Reson. Med. 1990, 14, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Belliveau, J.; Rosen, B.; Kantor, H.; Rzedzian, R.; Kennedy, D.; McKinstry, R.; Vevea, J.; Cohen, M.; Pykett, I.; Brady, T. Functional cerebral imaging by susceptibility-contrast NMR. Magn. Reson. Med. 1990, 14, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.W.-Y.; Wong, W.-T. Small molecular gadolinium(III) complexes as MRI contrast agents for diagnostic imaging. Coord. Chem. Rev. 2007, 251, 2428–2451. [Google Scholar] [CrossRef]
- Vlaardingerbroek, M.T.; den Boer, J.A. Magnetic Resonance Imaging. Theory and Practice; Springer Verlag: Heidelberg, Germany, 1996. [Google Scholar]
- Yang, C.-T.; Chuang, K.-H. Gd(III) chelates for MRI contrast agents: From high relaxivity to “smart”, from blood pool to blood–brain barrier permeable. Med. Chem. Commun. 2012, 3, 552–565. [Google Scholar] [CrossRef]
- Schmitt-Willich, H.; Brehm, M.; Evers, C.L.J.; Michl, G.; Muller-Fahrnow, A.; Petrov, O.; Platzek, J.; Raduchel, B.; Sulzle, D. Synthesis and physicochemical characterization of a new gadolinium chelate: The liver-specific magnetic resonance imaging contrast agent Gd-EOB-DTPA. Inorg. Chem. 1999, 38, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, H.J.; Brash, R.C.; Press, W.R. Characteristics of gadolinium-DTPA complex: A potential NMR contrast agent. Am. Roentgenol. 1984, 142, 619–622. [Google Scholar] [CrossRef]
- Chang, C.A. Magnetic resonance imaging contrast agents. Design and physicochemical properties of gadodiamide. Investig. Radiol. 1993, 28 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef]
- Mark, S.K.; William, C.D.; David, B.L.; Kenneth, N.R.; Steven, C.Q.; Scott, M.R. Gadolinium complexation by a new DPTA-amide ligand. Amide oxygen coordination. J. Inorg. Chem. 1990, 29, 1488–1491. [Google Scholar] [CrossRef]
- Dutta, S.; Kim, S.-K.; Lee, E.J.; Kim, T.-J.; Kang, D.-S.; Chang, Y.; Kang, S.O.; Han, W.-S. Synthesis and magnetic relaxation properties of paramagnetic Gd-complexes of new DTPA-bis-amides. The X-ray crystal structure of [Gd(L)(H2O)]·3H2O (L = DTPA-bis(4-carboxylicphenyl)amide). Bull. Korean Chem. Soc. 2006, 27, 1038–1042. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Cheng, T.-H.; Liu, G.-C.; Sheu, R.-S. Synthesis of some N,N''-bis(amide) derivatives of diethylenetriaminepentaacetic acid and the stabilities of their complexes with Gd3+, Ca2+, Cu2+ and Zn2+. J. Chem. Soc. Dalton Trans. 1997, 5, 833–837. [Google Scholar] [CrossRef]
- Dutta, S.; Park, J.-A.; Jung, J.-C.; Chang, Y.; Kim, T.-J. Gd-complexes of DTPA-bis(amide) conjugates of and its with high relaxivity and stability for magnetic resonance imaging. Dalton Trans. 2008, 16, 2199–2206. [Google Scholar] [PubMed]
- Dutta, S.; Kim, S.-K.; Patel, D.B.; Kim, T.-J.; Chang, Y. Some new DTPA-N,N''-bis(amides) functionalized by alkyl carboxylates: Synthesis, complexation and stability properties. Polyhedron 2007, 26, 3799–3809. [Google Scholar] [CrossRef]
- Ullah, N. Synthesis and dual D2 and 5-HT1A receptor binding affinities of 7-piperazinyl and 7-piperidinyl-3,4-dihydroquinazolin-2(1H)-ones. Med. Chem. 2014, 10, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Hnatowich, D.J.; Layne, W.W.; Childs, R.L. The preparation and labeling of DTPA-coupled albumin. Appl. Radiat. Isot. 1982, 33, 327–332. [Google Scholar] [CrossRef]
- Han, C.; Wan, L.; Ji, H.; Ding, K.; Huang, Z.; Lai, Y.; Peng, S.; Zhang, Y. Synthesis and evaluation of 2-anilinopyrimidines bearing 3-aminopropamides as potential epidermal growth factor receptor inhibitors. Eur. J. Med. Chem. 2014, 77, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gooyit, M.; Lee, M.; Schroeder, V.A.; Ikejiri, M.; Suckow, M.A.; Mobashery, S.; Chang, M. Selective Water-Soluble Gelatinase Inhibitor Prodrugs. J. Med. Chem. 2011, 54, 6676–6690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, X.; Linb, Z.; Zhang, H.; Chen, Y.; He, S. Synthesis and relaxation properties of two non-ion complexes of gadolinium(III) and manganese(II) with derivatives from diethylene triamine pentaacetic acid and isoniazid. Inorg. Chem. Commun. 2014, 40, 66–68. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M.P. Practical Surface Analysis, 2nd ed.; Wiley: New York, NY, USA, 1990; Volume 1. [Google Scholar]
- Jung, K.-H.; Kim, H.-K.; Lee, G.H.; Kang, D.-S.; Park, J.-A.; Kim, K.M.; Chang, Y.; Kim, T.-J. Gd Complexes of macrocyclic diethylenetriamine-pentaacetic acid (DTPA) biphenyl-2,20-bisamides as strong blood-pool magnetic resonance imaging contrast agents. J. Med. Chem. 2011, 54, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, M.; Bauer, H.; Mintorovitch, J.; Requardt, M.; Weinmann, H.-J. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Investig. Radiol. 2005, 40, 715–724. [Google Scholar] [CrossRef]
- Mesaik, M.A.; Haq, Z.; Murad, S.; Ismail, Z.; Abdullah, N.R.; Gill, H.K.; Rahman, A.; Yousaf, M.; Siddiqui, R.A.; Ahmad, A.; et al. Biological and molecular docking studies on coagulin-H: Human IL-2 novel natural inhibitor. Mol. Immunol. 2006, 43, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Samples of the compounds 12–17 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ba-Salem, A.O.; Ullah, N.; Shaikh, M.N.; Faiz, M.; Ul-Haq, Z. Gd-Complexes of New Arylpiperazinyl Conjugates of DTPA-Bis(amides): Synthesis, Characterization and Magnetic Relaxation Properties. Molecules 2015, 20, 7807-7819. https://doi.org/10.3390/molecules20057807
Ba-Salem AO, Ullah N, Shaikh MN, Faiz M, Ul-Haq Z. Gd-Complexes of New Arylpiperazinyl Conjugates of DTPA-Bis(amides): Synthesis, Characterization and Magnetic Relaxation Properties. Molecules. 2015; 20(5):7807-7819. https://doi.org/10.3390/molecules20057807
Chicago/Turabian StyleBa-Salem, Abdullah O., Nisar Ullah, M. Nasiruzzaman Shaikh, Mohamed Faiz, and Zaheer Ul-Haq. 2015. "Gd-Complexes of New Arylpiperazinyl Conjugates of DTPA-Bis(amides): Synthesis, Characterization and Magnetic Relaxation Properties" Molecules 20, no. 5: 7807-7819. https://doi.org/10.3390/molecules20057807
APA StyleBa-Salem, A. O., Ullah, N., Shaikh, M. N., Faiz, M., & Ul-Haq, Z. (2015). Gd-Complexes of New Arylpiperazinyl Conjugates of DTPA-Bis(amides): Synthesis, Characterization and Magnetic Relaxation Properties. Molecules, 20(5), 7807-7819. https://doi.org/10.3390/molecules20057807