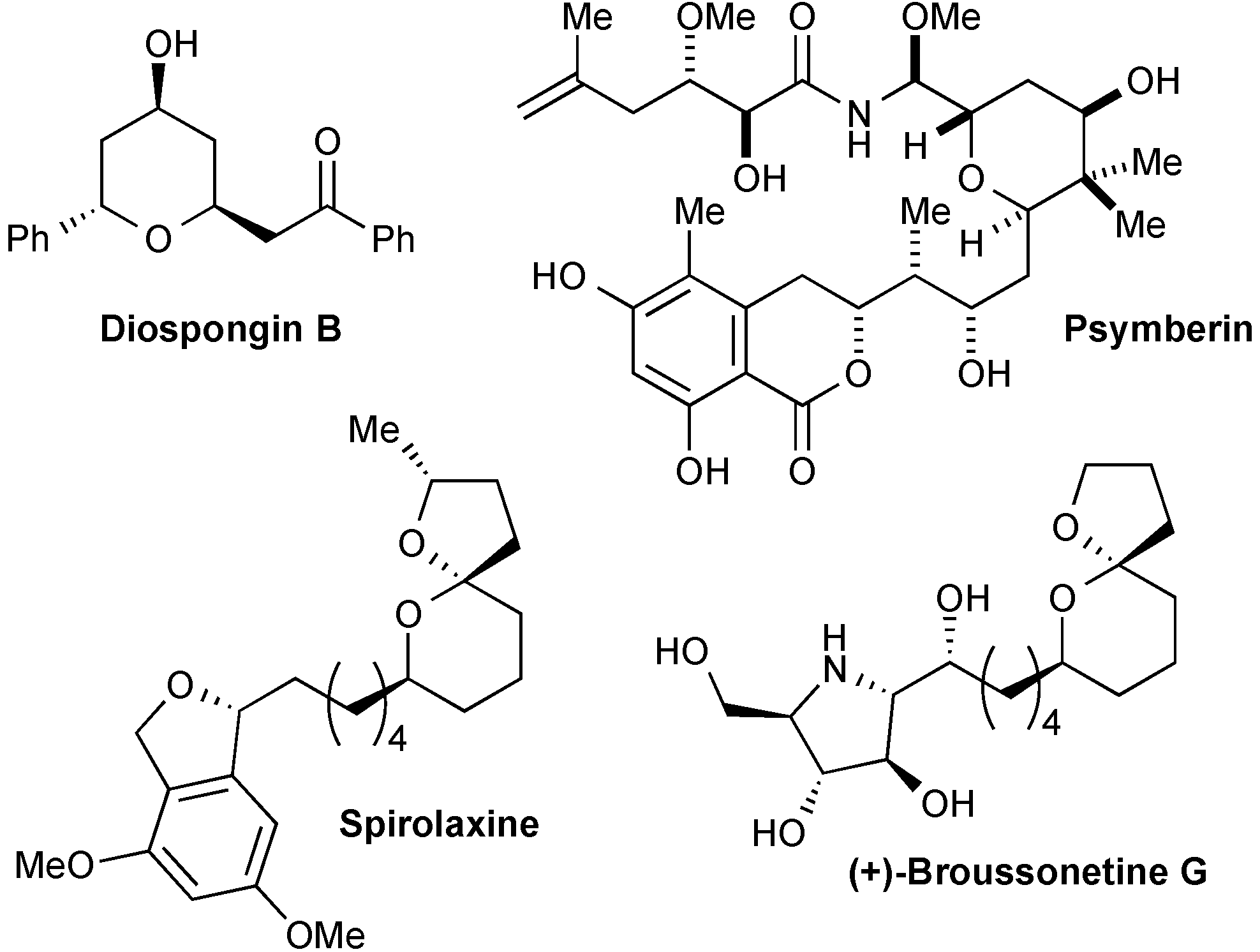
3. Experimental Section
3.2. General Procedure for the Synthesis of Racemic Dihydropyranones
To a flame dried flask under an atmosphere of argon was added ZnCl2 (39 mg, 0.28 mmol, 3 mol %) and anhydrous diethyl ether (0.4 mL, 3 mol %). Anhydrous THF (100 mL) was added followed by freshly-purified aldehyde (9.42 mmol, 1.0 eq). The reaction was stirred for 10 min before dropwise addition of Danishefsky’s Diene (1) (2.7 mL, 14.13 mmol, 1.5 eq). The reaction was stirred overnight at room temperature and then filtered through celite and concentrated. The crude product was purified by flash column chromatography to afford the respective dihydropyranones.
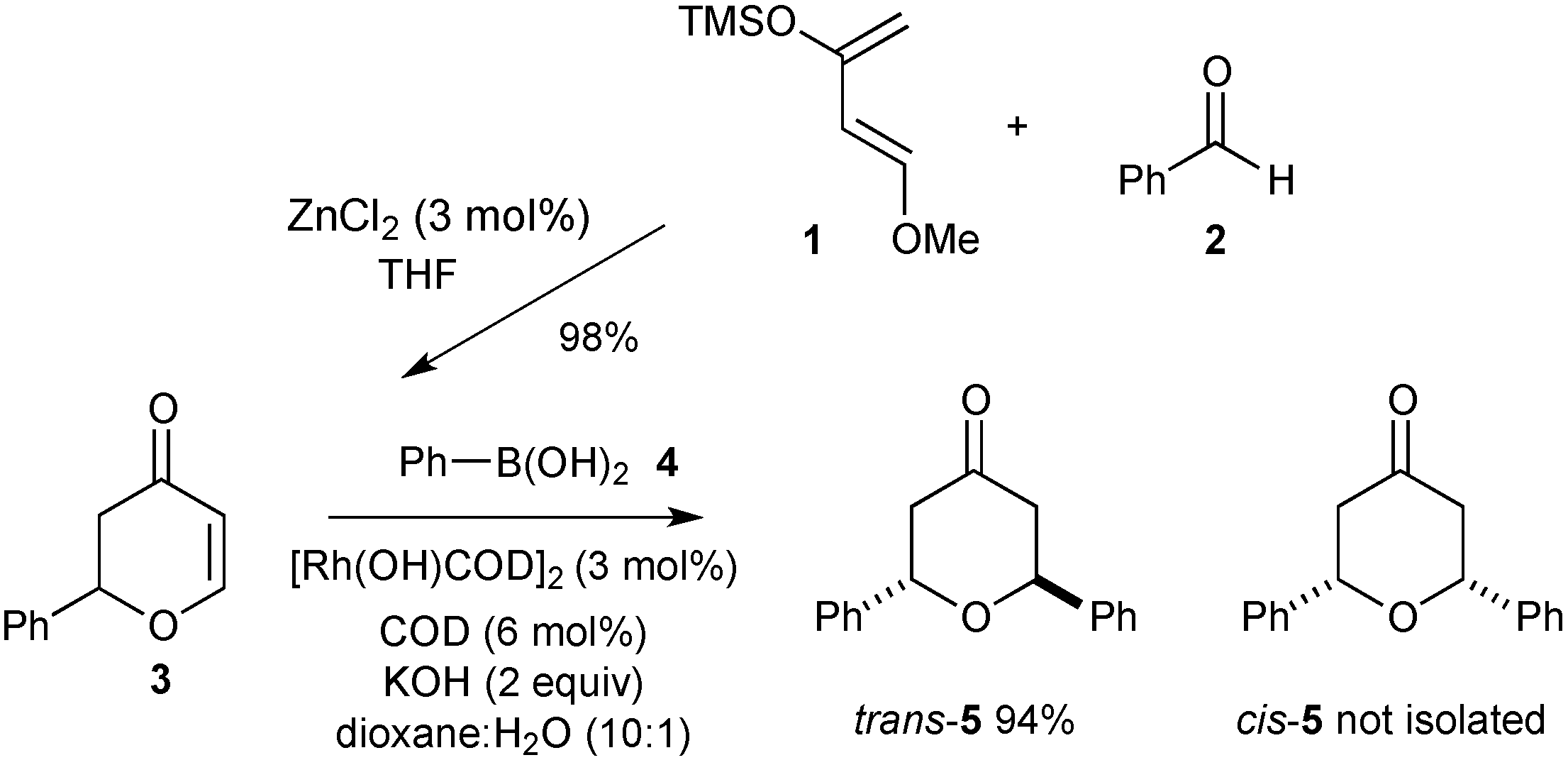
3.3. Synthesis of Racemic 2-Phenyl-2,3-dihydropyran-4-one (3)
Freshly distilled benzaldehyde (0.96 mL, 9.42 mmol) was reacted under the standard procedure and the crude product purified by flash column chromatography (eluting with petrol:ethyl acetate 8:2) to afford the title compound as a red oil (1.2 g, 73% yield).
Rf (petrol:ethyl acetate, 7:3); 0.29; νmax (CH2Cl2)/cm−1; 3063 (C-H), 1722 (C=O), 1670 (C=C), 1593, 1583 (C=C), 1268, 1037 (C-O); δH (300 MHz; CDCl3); 7.48 (1H, dd, J = 6.0, 0.5 Hz, OCH), 7.44–7.35 (5H, m, ArH), 5.52 (1H, dd, J = 6.0 Hz, 1.3 Hz, CHCO), 5.43 (1H, dd, J = 14.4, 3.5 Hz, CH2CHAr), 2.91 (1H, dd, J = 16.9, 14.3 Hz, COCHH), 2.66 (ddd, J = 16.9, 3.5, 1.3 Hz, COHH); δC (75.5 MHz; CDCl3); 192.1, 163.2, 137.9, 129.0, 128.9, 126.2, 107.4, 81.1, 43.4; HRMS (ESI+) calcd for C11H10NaO2 [M+Na+] m/z 197.0579 found: m/z 197.0590.
All data in accordance with literature values [
22].
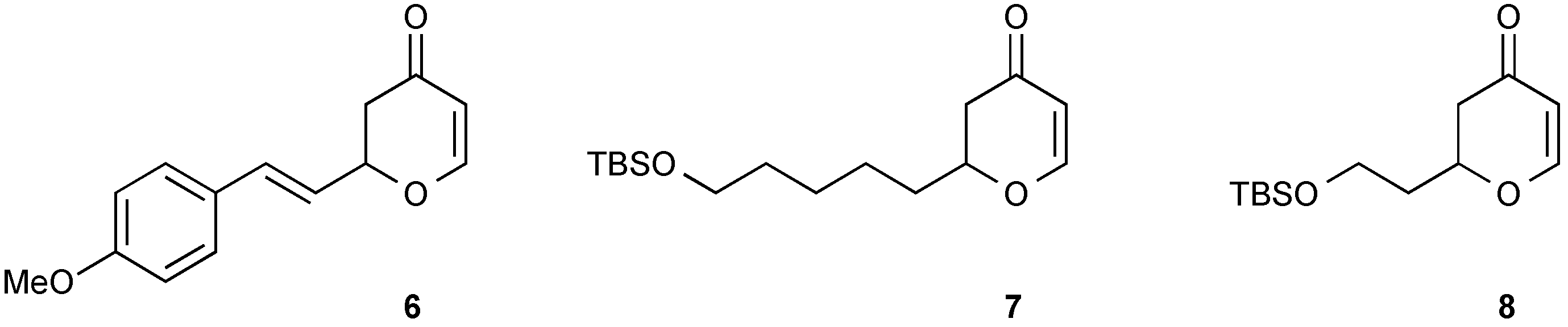
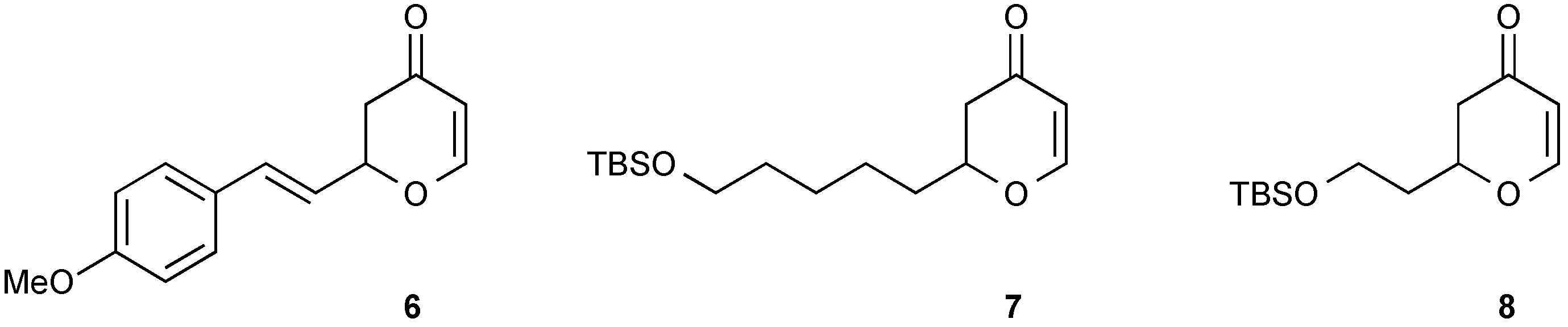
3.4. Synthesis of Racemic 2-[2-(4-Methoxyphenyl)vinyl]-2,3-dihydropyran-4-one (6)
Recrystallised 4-methoxycinnamaldehyde (1.0 g, 6.17 mmol) was reacted under the standard procedure and the crude product purified by flash column chromatography (eluting with petrol:ethyl acetate 8:2) to afford the title compound as an orange solid (0.28g, 20% yield).
Rf (petrol:ethyl acetate, 4:1); 0.29; δH (300 MHz; CDCl3); 7.40 (2H, d, J = 6.0 Hz, ArH), 7.34 (1H, d, J = 8.5 Hz, OCHCH), 6.87 (2H, d, J = 8.5 Hz, ArH), 6.70 (1H, d, J 15.9 Hz, ArCHCH), 6.16 (1H, dd, J = 15.9, 6.8 Hz, ArCHCH), 5.46 (1H, d, J = 6.0 Hz, COCH), 5.07–5.00 (1H, m, CH2CHO), 3.81 (3H, s, OCH3), 2.78–2.56 (2H, m, CH2CO); δC (75.5 MHz; CDCl3); 192.2, 163.2, 150.0, 133.6, 128.2, 128.1, 122.7, 114.1, 107.3, 80.1, 55.4, 42.1; HRMS (CI+) calcd for C14H15O3 [M+H]+ /z 231.1016 found: m/z. 231.1059.
All data in accordance with literature values [
22].
3.5. Synthesis of Racemic 2-[5-(tert-Butyldimethylsilanyloxy)pentyl]-2,3-dihydropyran-4-one (7)
6-(tert-Butyldimethylsilanyloxy)hexanal (1.0 g, 4.09 mmol) was reacted under the standard procedure and the crude product purified by flash column chromatography (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a yellow oil (0.990 g, 81% yield).
Rf (petrol:ethyl acetate, 4:1); 0.56; δH (300 MHz; CDCl3); 7.35 (1H, d, J = 5.9 Hz, CHCHO), 5.39 (1H, dd, J = 5.9, 1.0 Hz, CHCHO), 4.39 (1H, ddt, J = 12.7, 7.7, 4.6 Hz, CH2CHCH2), 3.61 (2H, t, J = 6.3 Hz, CH2OSi), 2.52 (1H, dd, J = 16.7, 12.9 Hz, COCHH), 2.42 (1H, ddd, J = 16.7, 4.3, 1.0 Hz, COCHH), 1.89–1.64 (2H, m, CHCH2CH2), 1.58–1.49 (2H, m, CH2CH2OSi), 1.48–1.33 (4H, m, (CH2)2(CH2)2OSi), 0.89 (9H, s, SiC(CH3)3), 0.04 (6H, s, Si(CH3)2); δC (75.5 MHz; CDCl3); 192.9, 163.4, 107.0, 79.6, 63.0, 41.9, 34.5, 32.7, 26.0, 25.7, 24.7, 18.4, −5.2; HRMS (ESI+) calcd for C16H30O3Si [M+H]+ m/z 299.2037 found: m/z 299.2052.
All data in accordance with literature values [
23].
3.6. Synthesis of Racemic 2-[2-(tert-Butyl-dimethyl-silanyloxy)-ethyl]-2,3-dihydropyran-4-one (8)
3-(tert-butyldimethylsilanyloxy)propionaldehyde (1.0 g, 9.336 mmol) was reacted under the standard procedure and the crude product purified by flash column chromatography (eluting with petrol:ethyl acetate 4:1) to afford the title compound as a yellow oil (0.400 g, 39% yield).
Rf (petrol:ethyl acetate, 4:1); 0.51; δH (300 MHz; CDCl3); 7.35 (1H, d, J = 6.0 Hz, OCH), 5.41 (1H, dd, J = 6.0, 1.0 Hz, CHCO), 4.67–4.57 (1H, m, OCHCH2), 3.85–3.71 (2H, m, OCH2), 2.63–2.43 (2H, m, COCH2), 2.07–1.96 (1H, m, CHCHHCH2), 1.90–1.79 (1H, m, CHCHHCH2), 0.89 (9H, s, SiC(CH3)3), 0.05 (6H, s, Si(CH3)2); δC (75.5 MHz; CDCl3); 192.7, 163.2, 107.2, 58.4, 42.1, 37.4, 26.0, 18.4, −5.32, −5.36; HRMS (ESI+) calcd for C13H25O3 [M+H]+ m/z 257.1572 found: m/z 257.1536.
All data in accordance with literature values [
23].
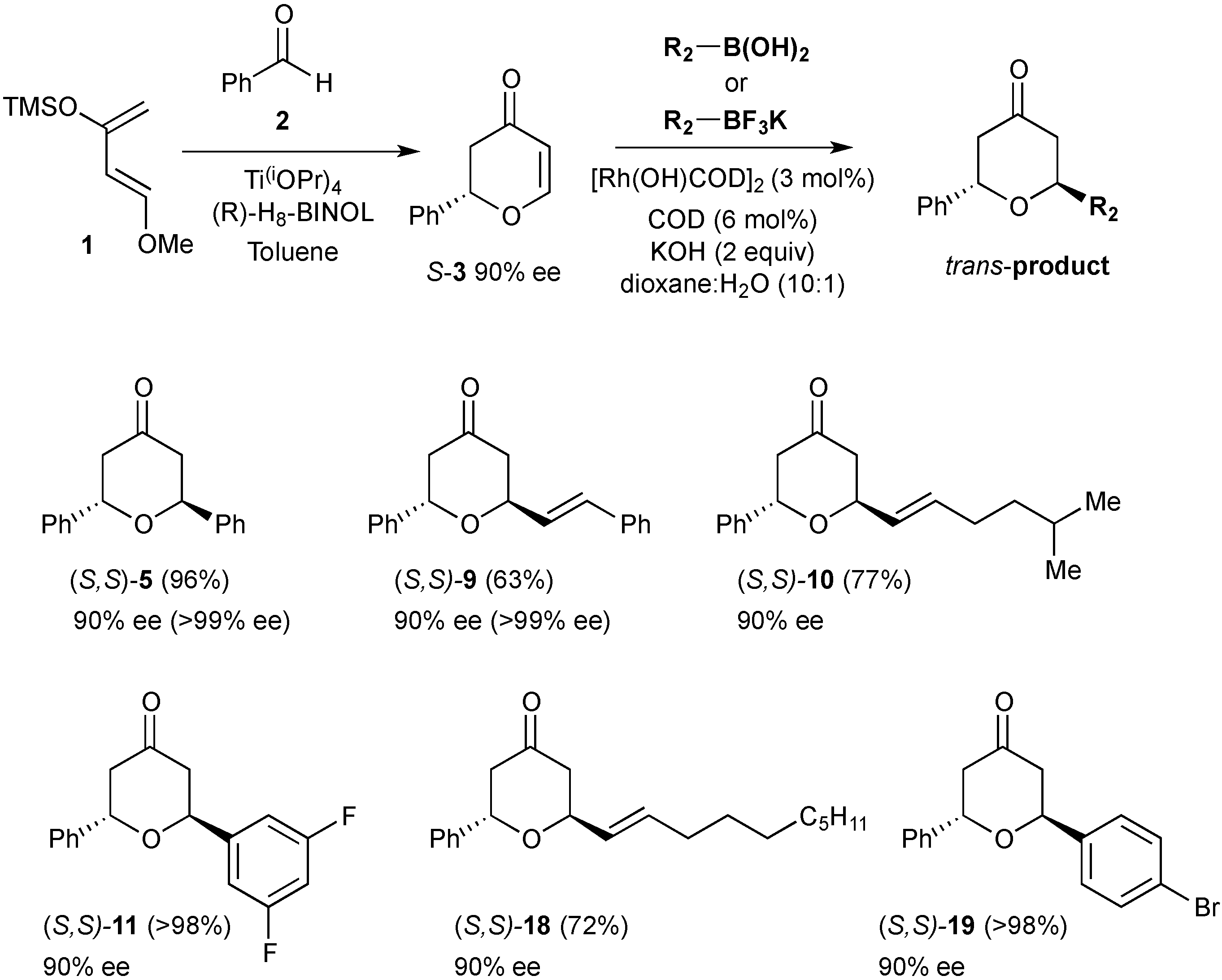
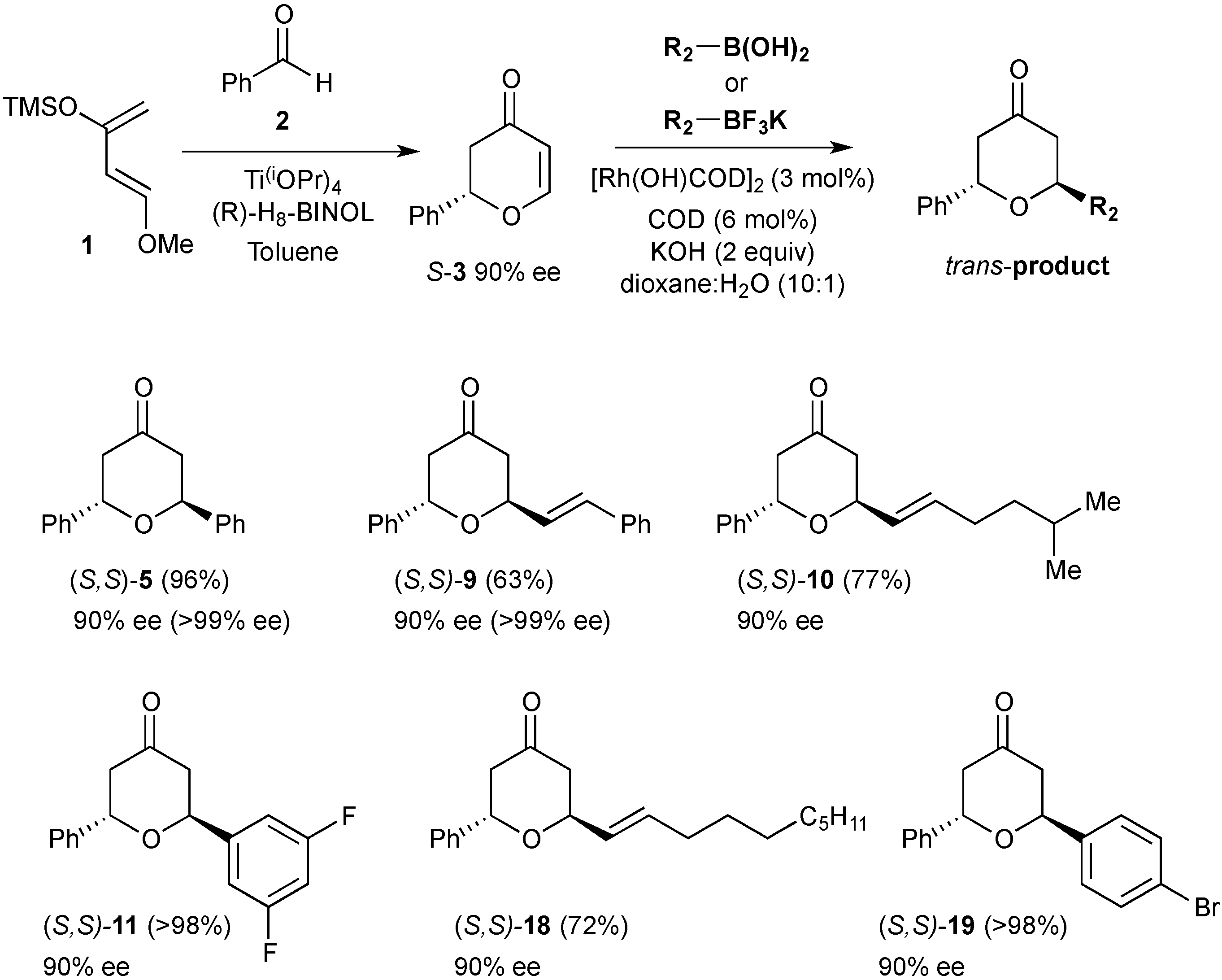
3.7. Synthesis of (S)-2-Phenyl-2,3-dihydropyran-4-one ((S)-3)
A mixture of (R)-H8-BINOL (0.610 g, 2.07 mmol) and Ti(OiPr)4 (0.56 mL, 1.884 mmol) with activated 4 Å molecular sieves (4.54 g) in anhydrous toluene (38 mL) under an inert atmosphere was heated at 35 °C for 1 h. The yellow mixture was cooled to room temperature and freshly distilled benzaldehyde (0.96 mL, 9.42 mmol, 1.0 eq) added. After stirring for 10 min the mixture was cooled to 0 °C and Danishefsky’s diene (11.3 mmol, 1.2 eq) was added. The reaction was stirred at 0 °C for 24 h and then treated with trifluoroacetic acid (0.1 mL). After stirring for a further 15 min at 0 °C, NaHCO3 (10 mL) was added and the reaction stirred for 10 min and then filtered through a plug of celite. The organic layer was then separated and the aqueous extracted with diethylether (3 × 25 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (eluting with petrol:ethyl acetate 8:2) to afford the title compound as a red oil (1.2 g, 73% yield). The chromatographed material was determined to be in 90% ee by chiral HPLC analysis (Chiralcel OD, 9:1 Hexanes: propan-2-ol, 1.0 mL·min−1, tR = 11.22 min (major) and 13.23 min (minor).
Rf (petrol:ethyl acetate, 7:3); 0.29; = +81° (c = 0.8, CHCl3); νmax (CH2Cl2)/cm−1; 3063 (C-H), 1722 (C=O), 1670 (C=C), 1593, 1583 (C=C), 1268, 1037 (C-O)δH (300 MHz; CDCl3); 7.48 (1H, dd, J = 6.0, 0.6 Hz, OCHCH), 7.44–7.38 (5H, m, ArH), 5.53 (1H, dd, J = 6.0 Hz, 1.2 Hz, CHCO), 5.43 (1H, dd, J = 14.4, 3.5 Hz, CH2CHAr), 2.91 (1H, dd, J = 17.0, 14.4 Hz, COCHH), 2.66 (ddd, J = 17.0, 3.5, 1.3 Hz, COHH); δC (75.5 MHz; CDCl3); 192.2, 163.2, 137.9, 129.0, 128.9, 126.2, 107.5, 81.2, 43.5; HRMS (ESI+) calcd for C11H10NaO2 [M+Na]+ m/z 197.0579 found: m/z 197.0590.
All data in accordance with literature values [
22].
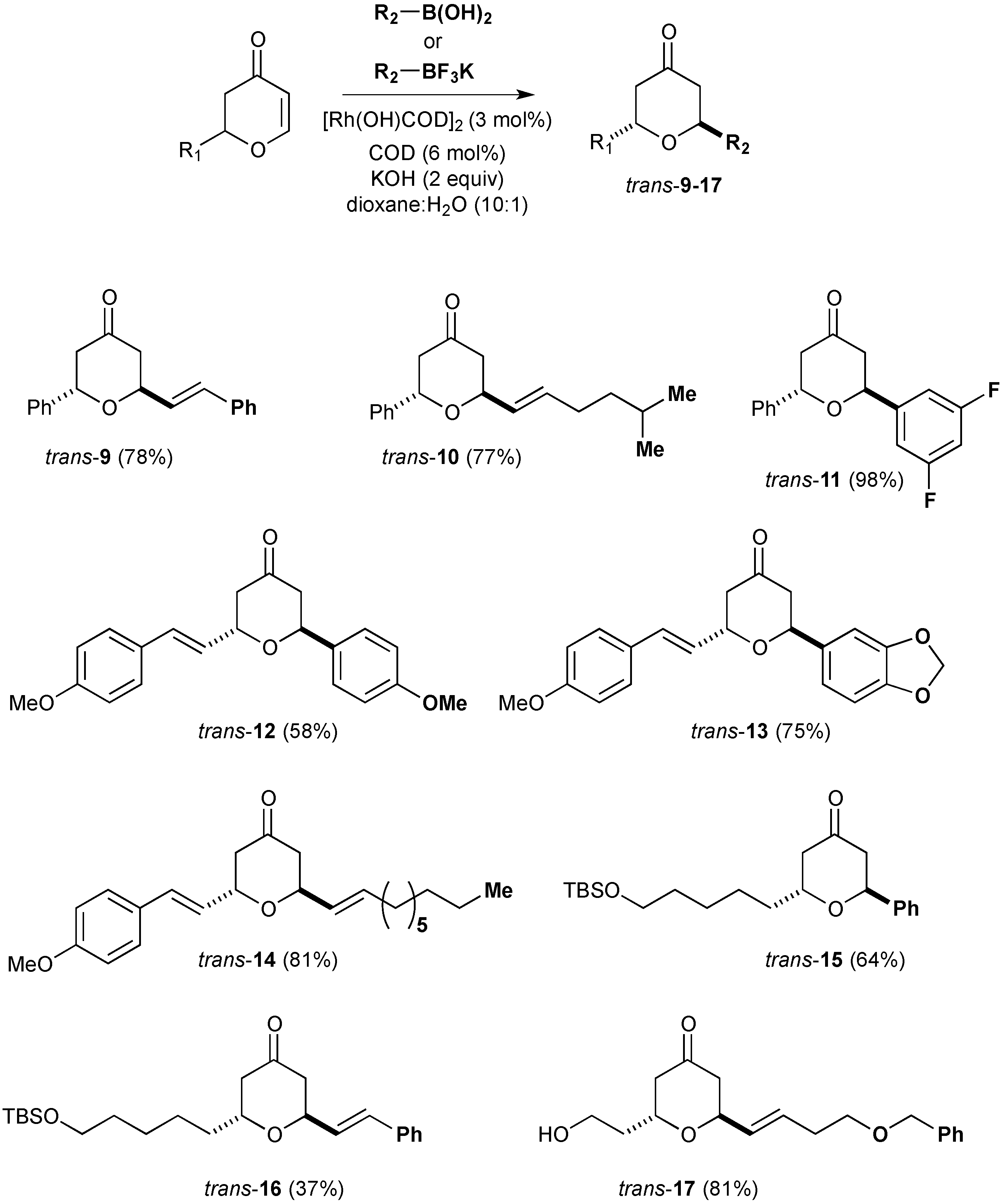
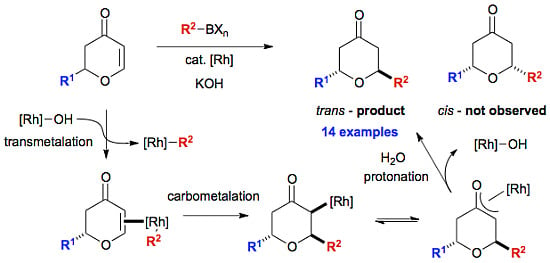
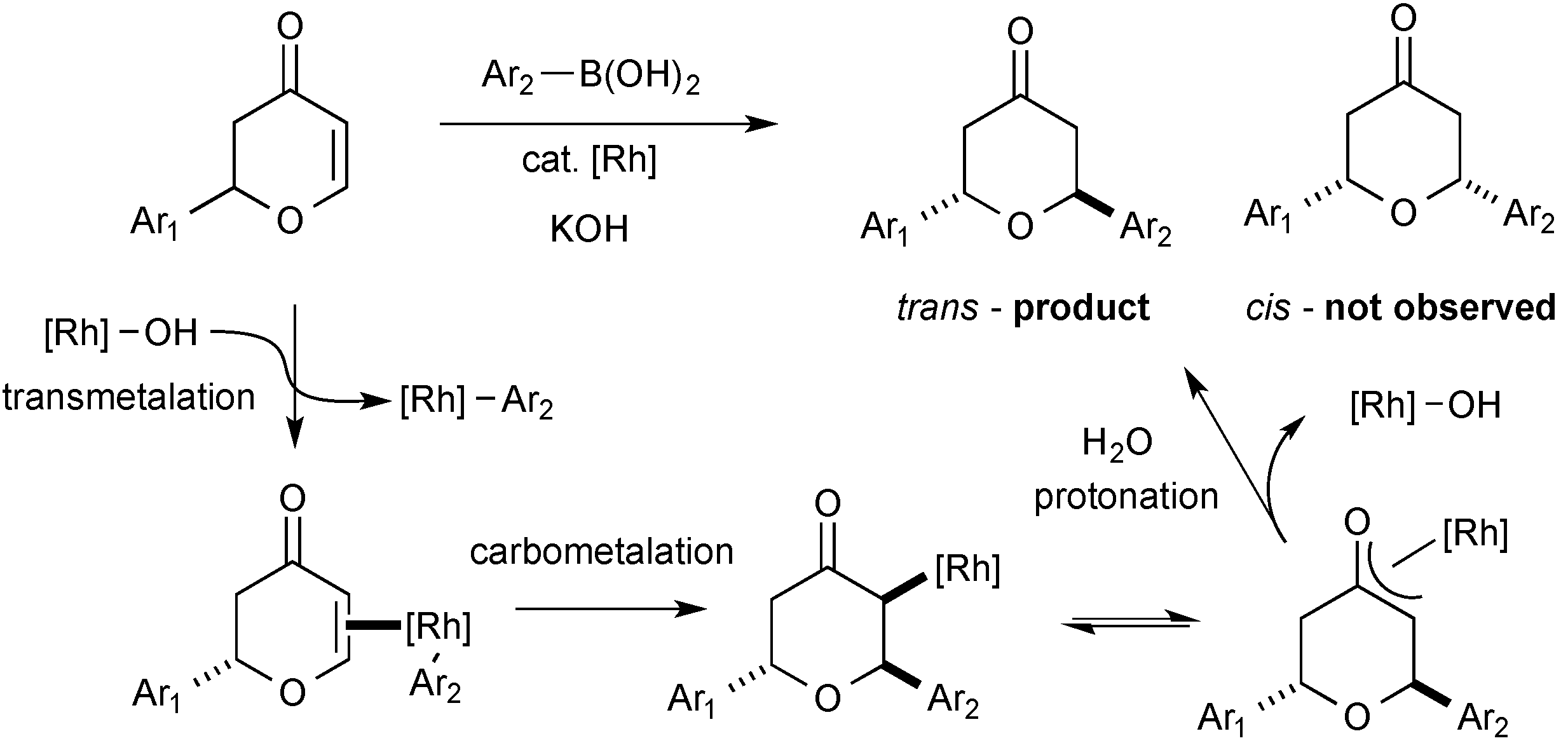
3.8. General Procedure for the Rhodium-Catalysed Conjugate Additions to Dihydropyranones
An oven dried, 24 mL screw-capped vial equipped with a rubber septum was charged with organoboron reagent (0.228 mmol, 2.0 eq), [Rh(OH)(cod)]2 (0.0016 g, 0.00342 mmol, 3 mol %), cyclooctadiene (0.007 g, 0.00684 mmol) and potassium hydroxide (0.009 g, 0.228 mmol). The reaction vessel was purged with argon and dioxane (0.5 mL) and water (0.05 mL) were subsequently added by syringe. The red solution was stirred for 15 minutes at room temperature, before the addition of dihydropyranone (0.114 mmol, 1.0 eq). The reaction was transferred to a preheated hotplate at 80 °C for 20 h. Upon completion, the crude reaction mixture was taken up in diethyl ether (5 mL) and filtered through a short plug of silica (elution; diethyl ether) and the solvent removed in vacuo. The crude residue was purified by flash column chromatography on silica gel to afford the desired compounds.
3.9. Synthesis (2S,6S)-Diphenyltetrahydropyran-4-one (5)
Phenylboronic acid (0.210 g, 1.72 mmol) was treated with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.150 g, 0.86 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a white solid (0.207 g, 96% yield).
Rf (petrol:ethyl acetate, 4:1); 0.47; = −16° (c = 1, CHCl3); νmax (CH2Cl2)/cm−1; 3067, 3066, 2974, 2886 (C-H), 1714 (C=O), 1601 (C=C aryl), 1133 (C-O); δH (300 MHz; CDCl3); 7.27–7.26 (5H, m, ArH), 7.05 (2H, m, CHOCH), 6.87 (2H, dd, J = 14.6, 6.6 Hz, CHHCOCHH), 6.81 (2H, dd, J = 15.0, 5.0 Hz, CHHCOCHH); δC (75.5 MHz; CDCl3); 206.8, 139.9, 128.8, 128.2, 126.8, 73.6, 46.4; HRMS (ESI+) calcd for C17H16NaO2 [M+Na]+ m/z 275.1048 found: m/z. 275.1029; HPLC (Chiralcel ODH, 97:3 Hexanes:propan-2-ol, 0.5 mL·min−1, tR = 11.07 min (major) and 13.11 min (minor).
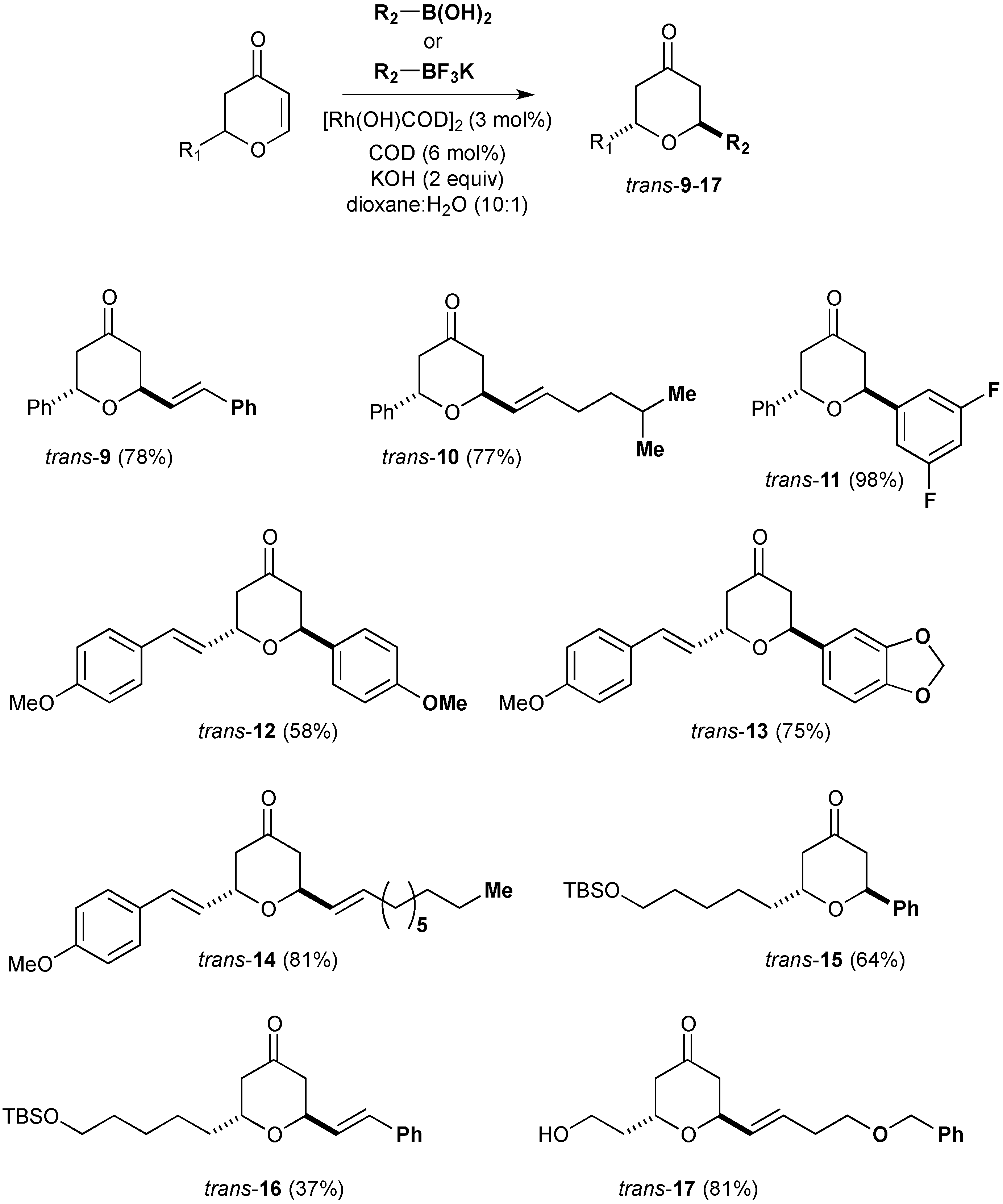
3.10. Synthesis of (2S,6S)-2-Phenyl-6-styryltetrahydropyran-4-one (9)
Potassium (E)-styryltrifluoroborate (0.907 g, 4.32 mmol) was reacted with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.20 g, 1.148 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a white solid (0.20 g, 63% yield).
Rf (petrol:ethyl acetate, 4:1); 0.5; = −77° (c = 1, CHCl3), νmax (neat)/cm−1; 3035, 2979, 2882 (C-H), 1720 (C=O), 1658 (C=C), 1600, 1579 (C=C aryl), 1231, 1048 (C-O); δH (300 MHz; CDCl3); 7.34–7.17 (10H, m, ArH), 6.53 (1H, dd, J = 16.3, 1.4 Hz, ArCH), 6.23 (1H, dd, J = 16.3, 5.0, ArCHCH), 5.12 (1H, dd, J = 7.4, 5.0 Hz, ArCHO), 4.83 (1H, ddd, J = 10.7, 5.2, 1.4 Hz, CHCHO), 2.79–2.63 (4H, m, CH2COCH2); δC (75.5 MHz; CDCl3); 206.5, 140.3, 136.0, 133.5, 128.8, 128.7, 128.3, 128.2, 127.8, 126.7, 126.5, 73.6, 72.9, 47.8, 45.4; HRMS (ESI+) calcd for C19H18NaO2 [M+Na]+ m/z 301.1204 found: m/z. 301.1177; HPLC (Chiralcel ODH; 95.5 Hexanes:propan-2-ol, 1.0 mL·min−1, tR = 13.37 min (major) and 21.93 min (minor).
3.11. Synthesis of (2S, 6S)-2-(5-Methylhex-1-enyl)-6-phenyltetrahydropyran-4-one (10)
Potassium (E)-trifluoro(5-methyl-hex-1-enyl)borate (0.047 g, 0.23 mmol) was reacted with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.020 g, 0.115 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a yellow oil (0.024 g, 77% yield).
Rf (petrol:ethyl acetate, 4:1); 0.78; νmax (neat)/cm−1; 3068, 2955, 2870 (C-H), 1706 (C=O), 1648 (C=C), 1602 (C=C aryl), 1268, 1069 (C-O); δH (300 MHz; CDCl3); 7.38–7.27 (5H, m, ArH), 5.69 (1H, dtd, J = 15.7, 6.3, 0.9 Hz, CH2CHCH), 5.57 (1H, ddt, J = 15.7, 5.0, 1.0 Hz, CH2CHCH), 5.11 (1H, dd, J = 7.4, 5.3 Hz, ArCHO), 4.70 (1H, dd, J = 9.7, 4.8 Hz, CHCHO), 2.76 (1H, dd, J = 14.4, 5.7, CHHCOCHH), 2.70 (2H, d, J = 5.4 Hz, CHHCOCHH), 2.60 (1H, ddd, J = 14.4, 4.6, 1.0 Hz, CHHCOCHH), 2.10–2.03 (2H, m, CH2CH2CH), 1.53 (1H, nonet, J = 6.6 Hz, (CH3)2CH), 1.29–1.24 (2H, m, (CH3)2CHCH2), 0.88 (6H, d, J = 6.6 Hz, (CH3)2CH); δC (75.5 MHz; CDCl3); 206.9, 140.5, 136.1, 128.1, 126.4, 73.1, 72.9, 48.0, 45.4, 38.1, 30.4, 27.6, 22.5; HPLC (Chiralcel AD; 98:2 Hexanes:propan-2-ol, 1.0 mL·min−1, tR = 6.85 min (major) and 15.91 min (minor).
3.12. Synthesis of (2S,6S)-2-(3,5-Difluorophenyl)-6-phenyltetrahydropyran-4-one (11)
3,5-difluorophenylboronic acid (0.045 g, 0.287 mmol) was reacted with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.025 g, 0.144 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a yellow solid (0.038 g, 98% yield).
Rf (petrol:ethyl acetate, 4:1); 0.6; δH (300 MHz; CDCl3); 7.42–7.30 (5H, m, ArH), 6.90 (2H, ddd, J = 8.2, 2.2, 0.7 Hz, FCCHCCHCF), 6.75 (1H, tt, J = 8.8, 2.3 Hz, CFCHCF), 5.34 (1H, t, J = 5.7 Hz, OCH), 4.97 (1H, dd, J = 6.82, 5.64 Hz, OCH), 2.93 (2H, ddd, J = 14.5, 5.7, 0.9 Hz, CHHCOCHH), 2.85–2.72 (2H, m, CHHCOCHH); δC (75.5 MHz; CDCl3); 205.7, 144.2, 139.2, 128.9, 128.5, 127.0, 109.7, 109.3, 103.5, 74.2, 72.3, 46.8, 45.8; HPLC (Chiralcel AD: 99:1 Hexanes:propan-2-ol, 0.5 mL·min−1, tR = 37.30 min (major) and 48.68 min (minor).
3.13. Synthesis of trans-2-(4-Methoxyphenyl)-6-[2-(4-methoxyphenyl)-vinyl]-tetrahydropyran-4-one (12)
4-methoxyphenylboronic acid (0.026 g, 0.174 mmol) was reacted with 2-[2-(4-Methoxyphenyl)-vinyl]-2,3-dihydropyran-4-one (6) (0.020 g, 0.0869 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 8:2) to afford the title compound as a yellow oil (0.017 g, 58% yield).
Rf (petrol:ethyl acetate, 4:1); 0.26; δH (300 MHz; CDCl3); 7.32 (4H, dd, J 8.8, 2.2 Hz, ArH, 6.88 (4H, dd, J = 11.6, 8.8 Hz, ArH), 6.52 (1H, d, J = 16.2, 5.3 Hz, ArCH), 6.15 (1H, d, J = 16.2 Hz, 5.3 Hz, ArCHCH), 5.17 (1H, dd, J = 7.2, 4.9, ArCHO), 4.81 (1H, ddd, J = 9.4, 5.3, 1.4, CHCHO), 3.81 (6H, s, OCH3, OCH3), 2.87–2.63 (4H, m, CH2COCH2); HRMS (ESI+) calcd for C21H22NaO4 [M+Na]+ m/z 361.1416 found: m/z. 361.1404.
3.14. Synthesis of trans-2-Benzo[1,3]dioxol-5-yl-6-[2-(4-methoxyphenyl)vinyl]-tetrahydropyran-4-one (13)
Benzo-[1,3]dioxol-5-ylboronic acid (0.029 g, 0.174 mmol) was reacted with 2-[2-(4-methoxyphenyl)-vinyl]-2,3-dihydropyran-4-one (6) (0.020 g, 0.0869 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 8:2) to afford the title compound as a yellow oil (0.032 g, 75% yield).
Rf (petrol:ethyl acetate, 4:1); 0.38; νmax (neat)/cm−1; 2906, 2862 (C-H), 1715 (C=O), 1641 (C=C), 1606, 1577, 1511 (C=C aryl), 1246, 1033 (C-O); δH (300 MHz; CDCl3); 7.33 (3H, d, J 8.8 Hz, ArH), 6.89 (2H, d, J = 11.7 Hz, ArH), 6.82 (2H, d, J = 11.5 Hz, ArH), 6.53 (1H, d, J = 16.2 Hz, ArCHCH), 6.14 (1H, dd, J = 16.4, 5.5 Hz, ArCHCH), 5.96 (2H, s, OCH2O), 5.10 (1H, dd, J = 7.0, 5.4 Hz, ArCHO), 4.84 (1H, ddd, J = 10.6, 5.3, 1.2 Hz, CHCHO), 3.81 (3H, s, ArOCH3), 2.83–2.65 (4H, m, CH2COCH2); HRMS (ESI+) calcd for C21H20NaO5 [M+H]+ m/z 375.1208 found: m/z. 375.1192.
3.15. Synthesis of trans-2-Dec-1-enyl-6-[2-(4-methoxyphenyl)vinyl]-tetrahydropyran-4-one (14)
Potassium decenyl trifluoroborate (0.043 g, 0.174 mmol) was reated with 2-[2-(4-methoxyphenyl)-vinyl]-2,3-dihydropyran-4-one (6) (0.020 g, 0.0869 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 8:2) to afford the title compound as a yellow oil (0.026 g, 81% yield).
Rf (petrol:ethyl acetate, 4:1); 0.5; νmax (CH2Cl2)/cm−1; 2925, 2855 (C-H), 1718 (C=O), 1610, 1514 (C=C aryl), 1250 (C-O); δH (300 MHz; CDCl3); 7.32 (2H, d, J = 8.7 Hz, ArH), 6.85 (2H, d, J = 8.7 Hz, ArH), 6.53 (1H, d, J = 16.1 Hz, ArCHCH), 6.12 (1H, dd, J = 16.1, 5.6 Hz, ArCHCH), 5.71 (1H, dt, J = 16.0, 6.6 Hz, CH2CHCH), 5.55 (1H, dd, J = 15.7, 5.5 Hz, CH2CHCH), 4.80 (1H, dd, J = 10.7, 5.1 Hz, CHO), 4.67 (1H, dd, J = 10.7, 5.6 Hz, CHO), 3.08 (3H, s, OCH3), 2.69–2.47 (4H, m, CH2COCH2), 2.05 (2H, q, J = 6.9 Hz, CH2CHCH), 1.37–1.26 (12H, m, CH3(CH2)6), 0.88 (3H, t, J = 6.6 Hz, CH3(CH2)6); δC (75.5 MHz; CDCl3); 206.7, 159.9, 135.4, 132.4, 128.6, 127.9, 126.0, 125.7, 114.1, 72.6, 72.5, 55.4, 46.3, 32.5, 32.0, 29.5, 29.3, 29.3, 29.0, 22.8, 14.2; HRMS (ESI+) calcd for C24H35O3 [M+H]+ m/z 371.2586 found: m/z 371.2588.
3.16. Synthesis of trans-2-[5-(tert-Butyl-dimethylsilanyloxy)-pentyl]-6-phenyltetrahydropyran-4-one (15)
Phenylboronic acid (0.021 g, 0.168 mmol) was reacted with (S)-2-[5-(tert-Butyl-dimethylsilanyloxy)-pentyl]-2,3-dihydropyran-4-one (7) (0.025 g, 0.0838 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a colourless oil (0.020 g, 64% yield).
Rf (petrol:ethyl acetate, 4:1); 0.72; δH (300 MHz; CDCl3); 7.41–7.28 (5H, m, ArH), 5.21 (1H, t, J = 5.7 Hz, ArCHO), 3.98–3.90 (1H, m, OCHCH2), 3.57 (2H, t, J = 1.56 Hz, CH2OSi), 2.88–2.74 (2H, m, COCH2), 2.57 (1H, ddd, J = 14.4, 4.5, 1.1 Hz, COCHH), 2.34 (1H, dd, J = 14.4, 7.3, 1.1 Hz, COCHH), 1.54–1.40 (4H, m, CH2CH2), 1.37–1.27 (4H, m, CH2CH2), 0.88 (9H, s, C(CH3)3), 0.03 (6H, s, Si(CH3)2); δC (75.5 MHz; CDCl3); 207.3, 140.2, 128.6, 128.0, 126.8, 63.1, 47.2, 46.2, 34.55, 32.72, 25.9, 25.6, 25.1, 18.4, −5.26.
3.17. Synthesis of trans-2-[5-(tert-Butyldimethylsilanyloxy)pentyl]-6-styryltetrahydropyran-4-one (16)
Potassium (E)-styryl trifluoroborate (0.028 g, 0.13 mmol) was reacted with (S)-2-[5-(tert-Butyl-dimethyl-silanyloxy)-pentyl]-2,3-dihydropyran-4-one (7) (0.020 g, 0.067 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethyl acetate 9:1) to afford the title compound as a colourless oil (0.010 g, 37% yield).
Rf (petrol:ethyl acetate, 4:1); 0.62; νmax (neat)/cm−1; 2931, 2858 (C-H), 1713 (C=O), 1623 (C=C), 1579, 1569 (C=C aryl), 1251, 1049 (C-O); δH (300 MHz; CDCl3); 7.40–7.27 (5H, ArH), 6.56 (1H, dd, J = 16.1, 1.2, ArCH), 6.23 (1H, dd, J = 11.2, 5.2, ArCHCH), 4.87 (1H, dd, J = 9.6, 4.5 Hz, CHCHO), 4.12–4.03 (1H, m, OCHCH2), 3.59 (2H, t, J = 6.4 Hz, CH2OTBS), 2.68 (2H, qd, J = 14.3, 5.4 Hz, COCH2), 2.49 (1H, ddd, J = 14.2, 4.0, 1.2 Hz, COCHH), 2.29 (1H, dd, J = 13.9, 8.2Hz, COCHH), 1.55–1.46 (4H, m, CH2CH2), 1.43–1.34 (4H, m, CH2CH2), 0.88 (9H, s, C(CH3)3), 0.03 (6H, s, Si(CH3)2).
3.18. Synthesis of trans-2-(4-Benzyloxybut-1-enyl)-6-(2-hydroxyethyl)tetrahydropyran-4-one (17)
Potassium (E)-(4-(-benzyloxy)but-1-en-1-yl)trifluoroborate (0.105 g, 0.39 mmol) was reated with 2-[2-(tert-Butyldimethylsilanyloxy)ethyl]-2,3-dihydropyran-4-one (8) (0.050 g, 0.195 mmol) under the standard conditions. The crude residue was treated with TBAF (0.43 mL, 1M in THF) in THF (2 mL). After stirring for 1 h, a saturated solution of NH4Cl was added and the mixture extracted with Et2O (3 × 10 mL). Combined organic extracts were dried (MgSO4) and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (eluting with CH2Cl2:methanol 9:1) to afford the title compound as a yellow oil (0.048 g, 81% yield).
Rf (CH2Cl2:methanol 9:1); 0.46; νmax (neat)/cm−1; 3342 (O-H), 2930, 2858 (C-H), 1472, 1463 (C=C), 1254, 1094 (C-O); δH (300 MHz; CDCl3); 7.37–7.27 (5H, m, ArH), 5.71 (1H, dt, J = 15.8, 6.4 Hz, CH2CHCH), 5.58 (1H, dd, J = 15.8, 4.7 Hz, CH2CHCH), 4.75 (1H, dd, J = 9.7, 4.7 Hz, CHCHO), 4.49 (2H, s, ArCH2O), 4.26 (1H, m, OCHCH2), 3.73 (1H, br.s, OH), 3.50 (2H, t, J = 6.6 Hz, OCH2CH2), 2.67 (1H, dd, J = 14.5, 6.2 Hz, CHHCOCHH), 2.52 (1H, ddd, J = 14.6, 3.8, 1.4 Hz, CHHCOCHH), 2.43–2.28 (4H, m, CHHCOCHH, CH2CHCH), 1.91–1.62 (2H, m, OCHCH2); δC (75.5 MHz; CDCl3); 206.5, 138.3, 132.2, 130.3, 128.4, 127.7, 127.6, 73.0, 72.9, 70.5, 69.2, 60.3, 47.6, 44.9, 37.5, 32.9.
3.19. Synthesis of (2S,6S)-2-Dec-1-enyl-6-phenyltetrahydropyran-4-one (18)
Potassium decenyl trifluoroborate salt (0.057 g, 0.23 mmol) was reacted with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.020 g, 0.115 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethylacetate 9:1) to afford the title compound as a yellow oil (0.027 g, 72% yield).
Rf (petrol:ethyl acetate, 4:1); 0.69; νmax (CH2Cl2)/cm−1; 3037, 2924, 2854 (C-H), 1720 (C=O), 1667 (C=C), 1603 (C=C aryl), 1249, 1052 (C-O); δH (300 MHz; CDCl3); 7.38–7.29 (5H, m ArH), 7.00 (1H, dt, J = 15.7, 6.3 Hz, CH2CH), 5.57 (1H, dd, J = 15.7, 4.9 Hz, CH2CHCH), 5.11 (1H, dd, J = 7.4 Hz, 5.4 Hz, ArCHO, 5.71 (1H, dd, J = 9.6, 4.7 Hz, CHCHO), 2.74 (1H, dd, J = 14.4, 5.9 Hz, CHHCOCHH), 2.70 (2H, d, J = 6.6 Hz, CHHCOCHH), 2.60 (1H, dd, J = 14.4, 4.6 Hz, CHHCOCHH), 2.06 (2H, q, J = 6.9 Hz, CH2CH), 1.35 (2H, dd, J = 13.0, 5.9 Hz, CH2CH2CH), 1.30–1.22 (10H, m, CH3(CH2)8), 0.87 (3H, t, J = 6.6 Hz, CH3); δC (75.5 MHz; CDCl3); 206.9, 140.0, 136.0, 128.8, 128.3, 128.1, 126.5, 73.1, 72.9, 48.0, 45.4, 32.5, 31.9, 29.5, 29.4, 29.2, 29.0, 22.8, 14.2; HPLC (Chiralcel ODH: 98:2 Hexanes:propan-2-ol, 1.0 mL·min−1, tR = 19.43 min (minor) and 21.42 min (major).
3.20. Synthesis of (2S,6S)-2-(4-Bromophenyl)-6-phenyltetrahydropyran-4-one (19)
4-Bromophenylboronic acid (0.058 g, 0.287 mmol) was reacted with (S)-2-phenyl-2,3-dihydropyran-4-one ((S)-3) (0.025 g, 0.144 mmol) under the standard conditions. The crude residue was purified by flash column chromatography on silica gel (eluting with petrol:ethylacetate 9:1) to afford the title compound as a colourless oil (0.044 g, 93% yield).
Rf (petrol:ethyl acetate, 4:1); 0.61; νmax (neat)/cm−1; 2983, 2896 (C-H), 1719 (C=O), 1596, 1494 (C=C aryl), 1245, 1231 (C-O); δH (300 MHz; CDCl3); 7.41 (2H, d, J = 7.9 Hz, ArH), 7.30–7.24 (5H, m, ArH), 7.17 (2H, d, J = 8.3 Hz, ArH), 5.04 (1H, t, J = 5.8 Hz, ArCHO), 4.98 (1H, t, J = 5.9 Hz, ArCHO), 2.85 (1H, dd, J = 14.8, 6.5 Hz, CHHCOCHH), 2.76 (1H, dd, J = 14.8, 5.8 Hz, CHHCOCHH), 2.75 (2H, J = 6.8 Hz, CHHCOCHH); δC (75.5 MHz; CDCl3); 206.3, 139.6, 139.0, 131.9, 128.8, 128.5, 128.3, 126.8, 122.2, 73.8, 72.9, 46.9, 46.3; HRMS (ESI+) calcd for C17H15BrNaO2 [M+Na]+ m/z 353.0153 found: m/z 252.0124; HPLC (Chiralcel OJ, 9:1 Hexanes:propan-2-ol, 1.0 mL·min−1, tR = 20.36 min (minor) and 28.61 min (major).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}