
LC-ESI-MS/MS Analysis and Pharmacokinetics of GP205, an Innovative Potent Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease in Rats

Abstract
:
1. Introduction
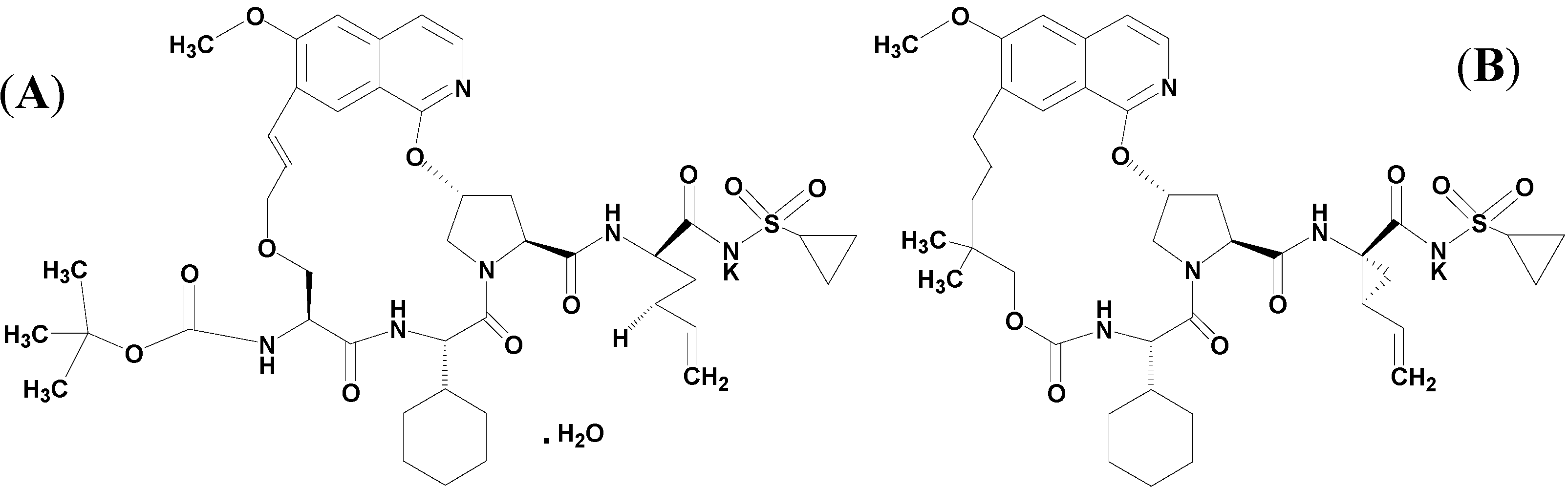
2. Results and Discussion
2.1. Method Development
2.1.1. Sample Preparation
2.1.2. LC-MS/MS Optimization

2.2. Method Validation
2.2.1. Selectivity
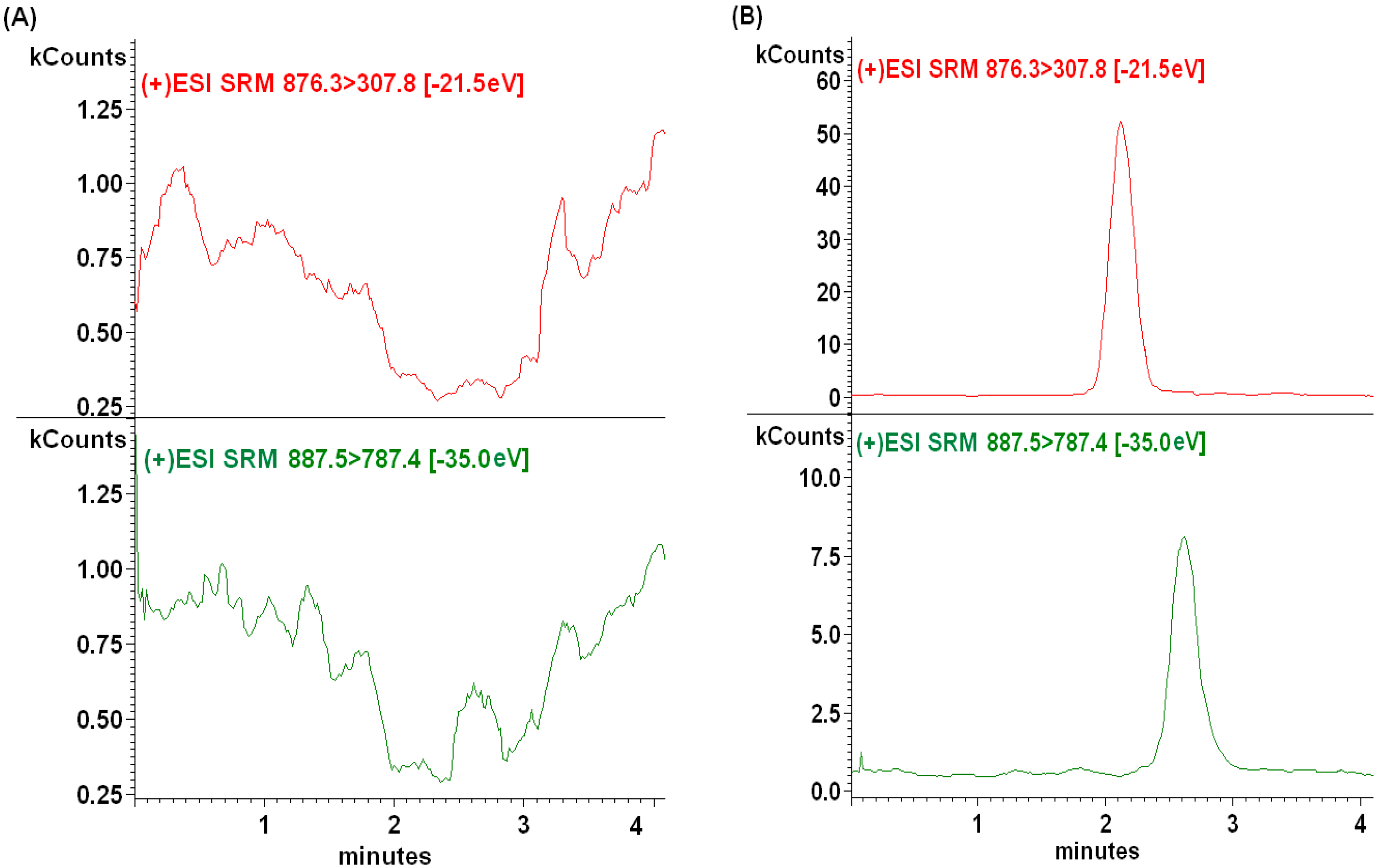

2.2.2. Sensitivity and Linearity
2.2.3. Accuracy and Precision

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 15) | ||||
|---|---|---|---|---|---|---|
| Measured Concentration (ng/mL) (Mean ± S.D.) | RSD (%) | RE (%) | Measured Concentration (ng/mL) (Mean ± S.D.) | RSD (%) | RE (%) | |
| 5 | 5.07 ± 0.36 | 7.1 | 0.8 | 5.18 ± 0.26 | 5.0 | 3.0 |
| 100 | 106.15 ± 2.99 | 2.9 | 5.5 | 103.53 ± 5.16 | 5.0 | 2.9 |
| 4000 | 4062.09 ± 93.81 | 2.4 | 1.0 | 4059.29 ± 110.69 | 2.8 | 0.9 |
2.2.4. Extraction Recovery and Matrix Effect
| Nominal Concentration (ng/mL) | Peak Area a (e5) (A) (Mean ± S.D.) | Peak Area b (e5) (B) (Mean ± S.D.) | Peak area c (e5) (C) (Mean ± S.D.) | Extraction Recovery d (%) (A/B) | Matrix Effect e (%) (B/C) |
|---|---|---|---|---|---|
| 5 | 0.39 ± 0.02 | 0.45 ± 0.02 | 0.47 ± 0.02 | 86.7 | 95.7 |
| 100 | 5.20 ± 0.11 | 5.91 ± 0.19 | 6.09 ± 0.16 | 88.0 | 97.0 |
| 4000 | 192.50 ± 12.01 | 206.90 ± 5.62 | 215.47 ± 5.44 | 93.0 | 96.0 |
| 100 (I.S.) | 7.27 ± 0.47 | 7.83 ± 0.46 | 8.45 ± 0.22 | 92.9 | 92.7 |
2.2.5. Stability
2.2.6. Sample Dilution
| Sample Condition | Nominal Conc. (ng/mL) | Measured Conc. (ng/mL) | Accuracy (%) |
|---|---|---|---|
| Bench top stability a | 5 | 4.84 | −3.3 |
| 100 | 98.39 | −1.6 | |
| 4000 | 3884.71 | −2.9 | |
| Auto-sampler stability b | 5 | 4.79 | −4.2 |
| 100 | 96.13 | −3.9 | |
| 4000 | 3904.44 | −2.4 | |
| Freeze-thaw stability c | 5 | 4.73 | −5.5 |
| 100 | 94.90 | −5.1 | |
| 4000 | 3925.86 | −1.9 | |
| Long time stability d | 5 | 4.61 | −7.7 |
| 100 | 92.84 | −7.2 | |
| 4000 | 3812.43 | −4.7 | |
| Room temperature stabilityfor stock solution e | 5 | 5.09 | 1.8 |
| 100 | 103.26 | 3.3 | |
| 4000 | 3987.91 | −0.3 | |
| At 4 °C stability for stock solution f | 5 | 5.14 | 2.8 |
| 100 | 100.63 | 0.6 | |
| 4000 | 3992.75 | −0.2 |
| Dilution Factor | Assayed Concentration (ng/mL) | Reported Concentration (ng/mL) |
|---|---|---|
| 2 | 4426.87 | 8853.74 |
| 4234.72 | 8469.44 | |
| 4347.85 | 8695.70 | |
| Mean | 8672.96 | |
| RSD (%) | 2.3 | |
| Accuracy (%) | 8.4 | |
| 5 | 4365.77 | 21,828.85 |
| 4039.57 | 20,197.85 | |
| 4104.06 | 20,520.30 | |
| Mean | 20,849.01 | |
| RSD (%) | 4.2 | |
| Accuracy (%) | 4.3 |
2.3. Pharmacokinetic Study
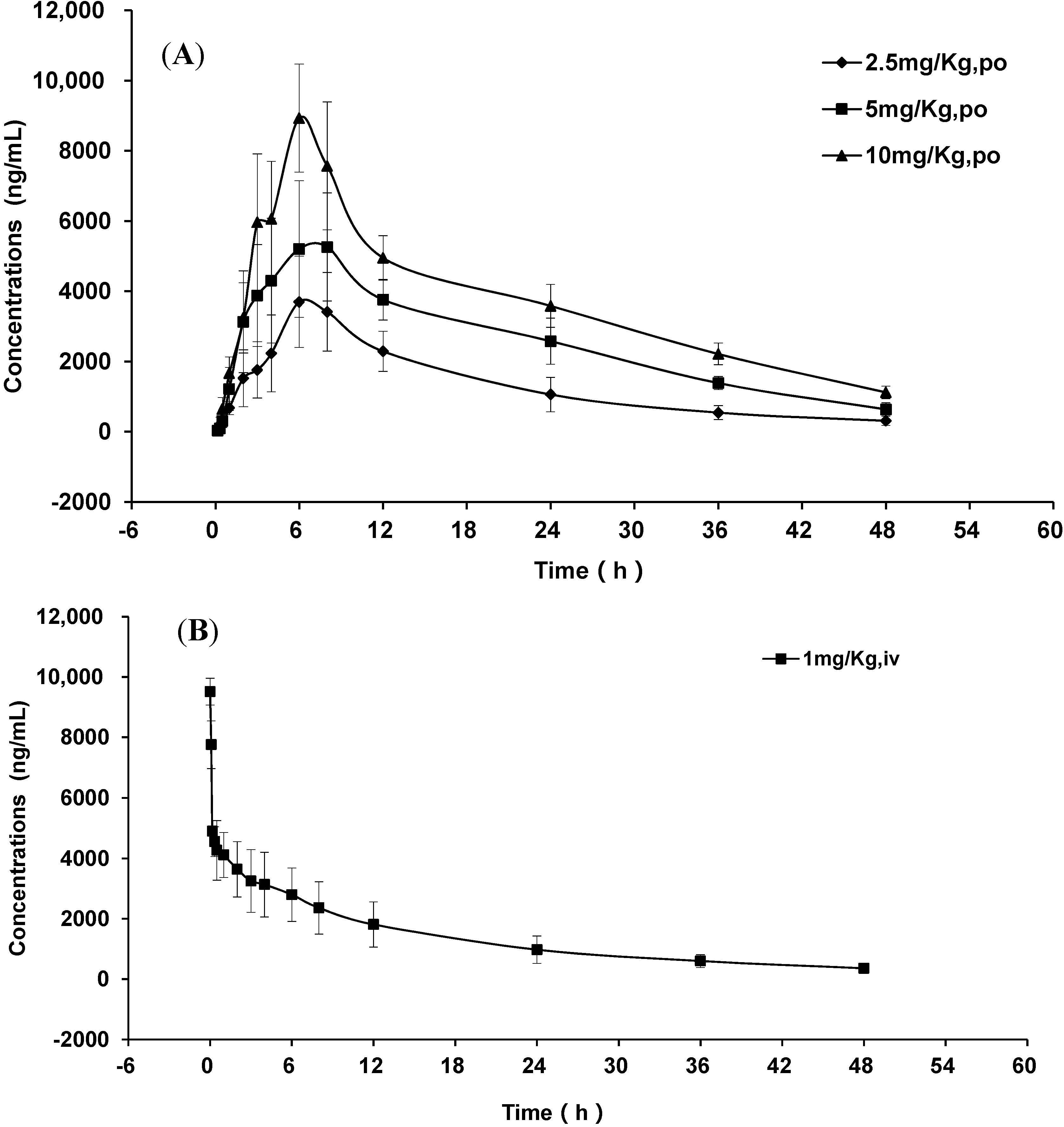
| Pharmacokinetic Parameters | GP205 | ||
|---|---|---|---|
| Oral | 2.5 mg/Kg | 5 mg/Kg | 10 mg/Kg |
| Tmax (h) | 6.30 ± 0.80 | 6.30 ± 1.50 | 6.30 ± 0.80 |
| Cmax (ng/mL) | 4260.32 ± 400.35 | 6782.15 ± 749.52 | 9406.12 ± 582.99 |
| t1/2 (h) | 12.38 ± 1.51 | 13.91 ± 1.48 | 17.21 ± 2.73 |
| MRT (h) | 19.35 ± 3.22 | 22.17 ± 2.18 | 25.55 ± 3.48 |
| AUC0-τ (ng·h/mL) | 64,342.95 ± 13,770.51 | 122,230.48 ± 9225.20 | 176,357.52 ± 16,331.60 |
| AUC0-∞ (ng·h/mL) | 70,136.75 ± 16,623.71 | 135,312.81 ± 13,439.93 | 204,693.19 ± 15,949.70 |
| IV | 1 mg/Kg | ||
| C0 (ng/mL) | 9511.56 ± 442.31 | ||
| t1/2 (h) | 16.26 ± 2.99 | ||
| MRT (h) | 20.76 ± 3.41 | ||
| Cl(L/h/Kg) | 0.02 ± 0.01 | ||
| VD(L) | 0.31 ± 0.12 | ||
| AUC0-τ (ng·h/mL) | 66,640.04 ± 21,721.23 | ||
| AUC0-∞ (ng·h/mL) | 75,210.88 ± 23,751.74 | ||
3. Experimental Section
3.1. Chemicals and Reagents
3.2. LC-MS/MS Instrumentation
3.3. Liquid Chromatographic Conditions
3.4. Mass Spectrometer Conditions
3.5. Preparation of Standard and Quality Control (QC) Samples
3.6. Extraction Procedure
3.7. Pharmacokinetic Study in Rats
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hepatitis C—Global prevalence (update). Wkly. Epidemiol. Rec. 1999, 74, 425–427.
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Memon, M.I.; Memon, M.A. Hepatitis C: An epidemiological review. J. Viral Hepat. 2002, 9, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Wasley, A.; Alter, M.J. Epidemiology of hepatitis C: Geographic differences and temporal trends. Semin. Liver Dis. 2000, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J.; Heller, T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.C.; Bortotti, A.C.; Nunes, N.N.; Al Bacha, I.; Parise, E.R. Association between age at diagnosis and degree of liver injury in hepatitis C. Braz. J. Infect. Dis. 2014, 18, 507–511. [Google Scholar]
- Kondo, Y.; Ninomiya, M.; Kimura, O.; Machida, K.; Funayama, R.; Nagashima, T.; Kobayashi, K.; Kakazu, E.; Kato, T.; Nakayama, K.; et al. HCV infection enhances Th17 commitment, which could affect the pathogenesis of autoimmune diseases. PLoS One 2014, 9, e98521. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C virus: Standard-of-care treatment. Adv. Pharmacol. 2013, 67, 169–215. [Google Scholar] [PubMed]
- Hadziyannis, S.J.; Sette, H., Jr.; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer, H., Jr.; Bernstein, D.; Rizzetto, M.; et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W. Side effects of therapy of hepatitis C and their management. Hepatology 2002, 36, S237–S244. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.L.; Suter, F.; Bacon, B.R.; Nelson, D.; Harley, H.; Sola, R.; Shafran, S.D.; Barange, K.; Lin, A.; Soman, A.; et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N. Engl. J. Med. 2007, 357, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Foster, G.R.; Rockstroh, J.K.; Zeuzem, S.; Zoulim, F.; Houghton, M. The way forward in HCV treatment-finding the right path. Nat. Rev. Drug Discov. 2007, 6, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.P.; Keller, P.A. Control of hepatitis C: A medicinal chemistry perspective. J. Med. Chem. 2005, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, R.; Migliaccio, G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005, 436, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Reesink, H.W.; Zeuzem, S.; Weegink, C.J.; Forestier, N.; van Vliet, A.; van de Wetering de Rooij, J.; McNair, L.; Purdy, S.; Kauffman, R.; Alam, J.; et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: A phase Ib, placebo-controlled, randomized study. Gastroenterology 2006, 131, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Tsantrizos, Y.S. Peptidomimetic therapeutic agents targeting the protease enzyme of the human immunodeficiency virus and hepatitis C virus. Acc. Chem. Res. 2008, 41, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.M.; Ascione, A. Future directions in therapy for chronic hepatitis C. Antivir. Ther. 2008, 13, 31–36. [Google Scholar] [PubMed]
- Kneteman, N.M.; Howe, A.Y.; Gao, T.; Lewis, J.; Pevear, D.; Lund, G.; Douglas, D.; Mercer, D.F.; Tyrrell, D.L.; Immermann, F.; et al. HCV796: A selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 2009, 49, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Narjes, F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr. Top. Med. Chem. 2007, 7, 1302–1329. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, Y.; Hou, X.; Zhao, Y.; Fabrycki, J.; Chen, D.; Wang, X.; Agarwal, A.; Phadke, A.; Deshpande, M.; et al. ACH-806, an NS4A antagonist, inhibits hepatitis C virus replication by altering the composition of viral replication complexes. Antimicrob. Agents Chemother. 2013, 57, 3168–3177. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Seko, Y.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saito, S.; Arase, Y.; et al. Prevalence of hepatitis C virus variants resistant to NS3 protease inhibitors or the NS5A inhibitor (BMS-790052) in hepatitis patients with genotype 1b. J. Clin. Virol. 2012, 54, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Perni, R.B. Hepatitis C virus NS3–4A protease inhibitors: Countering viral subversion in vitro and showing promise in the clinic. Curr. Opin. Drug Discov. Dev. 2006, 9, 606–617. [Google Scholar]
- Kolykhalov, A.A.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3' nontranslated region are essential for virus replication in vivo. J. Virol. 2000, 74, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Cosset, F.L.; Lohmann, V. Hepatitis C virus replication cycle. J. Hepatol. 2010, 53, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Narjes, F.; Koch, U.; Steinkuhler, C. Recent developments in the discovery of hepatitis C virus serine protease inhibitors—Towards a new class of antiviral agents? Expert Opin. Investig. Drugs 2003, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Llinas-Brunet, M.; Bailey, M.D.; Bolger, G.; Brochu, C.; Faucher, A.M.; Ferland, J.M.; Garneau, M.; Ghiro, E.; Gorys, V.; Grand-Maitre, C.; et al. Structure-activity study on a novel series of macrocyclic inhibitors of the hepatitis C virus NS3 protease leading to the discovery of BILN 2061. J. Med. Chem. 2004, 47, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Perni, R.B.; Farmer, L.J.; Cottrell, K.M.; Court, J.J.; Courtney, L.F.; Deininger, D.D.; Gates, C.A.; Harbeson, S.L.; Kim, J.L.; Lin, C.; et al. Inhibitors of hepatitis C virus NS3.4A protease. Part 3: P2 proline variants. Bioorg. Med. Chem. Lett. 2004, 14, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.; Victor, F.; Snyder, N.; Johnson, R.B.; Wang, Q.M.; Glass, J.I.; Chen, S.H. Novel P4 truncated tripeptidyl alpha-ketoamides as HCV protease inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Nizi, E.; Koch, U.; Ontoria, J.M.; Marchetti, A.; Narjes, F.; Malancona, S.; Matassa, V.G.; Gardelli, C. Capped dipeptide phenethylamide inhibitors of the HCV NS3 protease. Bioorg. Med. Chem. Lett. 2004, 14, 2151–2154. [Google Scholar] [CrossRef] [PubMed]
- Priestley, E.S.; de Lucca, I.; Ghavimi, B.; Erickson-Viitanen, S.; Decicco, C.P. P1 Phenethyl peptide boronic acid inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 2002, 12, 3199–3202. [Google Scholar] [CrossRef] [PubMed]
- Arasappan, A.; Njoroge, F.G.; Parekh, T.N.; Yang, X.; Pichardo, J.; Butkiewicz, N.; Prongay, A.; Yao, N.; Girijavallabhan, V. Novel 2-oxoimidazolidine-4-carboxylic acid derivatives as hepatitis C virus NS3–4A serine protease inhibitors: Synthesis, activity, and X-ray crystal structure of an enzyme inhibitor complex. Bioorg. Med. Chem. Lett. 2004, 14, 5751–5755. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.S.; DiMuzio, J.; McHale, C.; Burlein, C.; Olsen, D.; Carroll, S.S. A time-resolved, internally quenched fluorescence assay to characterize inhibition of hepatitis C virus nonstructural protein 3–4A protease at low enzyme concentrations. Anal. Biochem. 2008, 373, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mathy, J.E.; Ma, S.; Compton, T.; Lin, K. Combinations of cyclophilin inhibitor NIM811 with hepatitis C Virus NS3–4A Protease or NS5B polymerase inhibitors enhance antiviral activity and suppress the emergence of resistance. Antimicrob. Agents Chemother. 2008, 52, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Lamarre, D.; Anderson, P.C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bos, M.; Cameron, D.R.; Cartier, M.; Cordingley, M.G.; et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Rodriguez-Torres, M.; Muir, A.J.; Kieffer, T.L.; McNair, L.; Khunvichai, A.; McHutchison, J.G. Antiviral effects and safety of telaprevir, peginterferon alfa-2a, and ribavirin for 28 days in hepatitis C patients. J. Hepatol. 2008, 49, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Rouzier, R.; Wagner, F.; Forestier, N.; Larrey, D.; Gupta, S.K.; Hussain, M.; Shah, A.; Cutler, D.; Zhang, J.; et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology 2007, 132, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Forestier, N.; Larrey, D.; Guyader, D.; Marcellin, P.; Rouzier, R.; Patat, A.; Smith, P.; Bradford, W.; Porter, S.; Blatt, L.; et al. Treatment of chronic hepatitis C patients with the NS3/4A protease inhibitor danoprevir (ITMN-191/RG7227) leads to robust reductions in viral RNA: A phase 1b multiple ascending dose study. J. Hepatol. 2011, 54, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Reiser, M.; Hinrichsen, H.; Benhamou, Y.; Reesink, H.W.; Wedemeyer, H.; Avendano, C.; Riba, N.; Yong, C.L.; Nehmiz, G.; Steinmann, G.G. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology 2005, 41, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Shankar, H.; Bichoupan, K.; Dieterich, D.T. The pharmacokinetic evaluation of boceprevir for treatment of hepatitis C virus. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, S.D.; Andrews, S.W.; Jiang, Y.; Serebryany, V.; Tan, H.; Kossen, K.; Rajagopalan, P.T.; Misialek, S.; Stevens, S.K.; Stoycheva, A.; et al. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Hezode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J.P.; Bourliere, M.; Gharakhanian, S.; Bengtsson, L.; et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009, 360, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Andrews, S.W.; Condroski, K.R.; Buckman, B.; Serebryany, V.; Wenglowsky, S.; Kennedy, A.L.; Madduru, M.R.; Wang, B.; Lyon, M.; et al. Discovery of danoprevir (ITMN-191/R7227), a highly selective and potent inhibitor of hepatitis C virus (HCV) NS3/4A protease. J. Med. Chem. 2014, 57, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Raboisson, P.; de Kock, H.; Rosenquist, A.; Nilsson, M.; Salvador-Oden, L.; Lin, T.I.; Roue, N.; Ivanov, V.; Wahling, H.; Wickstrom, K.; et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- White, P.W.; Llinas-Brunet, M.; Amad, M.; Bethell, R.C.; Bolger, G.; Cordingley, M.G.; Duan, J.; Garneau, M.; Lagace, L.; Thibeault, D.; et al. Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob. Agents Chemother. 2010, 54, 4611–4618. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.A.; McIntyre, C.J.; Rudd, M.T.; Nguyen, K.T.; Romano, J.J.; Butcher, J.W.; Gilbert, K.F.; Bush, K.J.; Holloway, M.K.; Swestock, J.; et al. Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor. J. Med. Chem. 2010, 53, 2443–2463. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.J.; Holloway, M.K.; McCauley, J.A.; Rudd, M.T.; Butcher, J.W.; Carroll, S.S.; DiMuzio, J.; Fandozzi, C.; Gilbert, K.F.; Mao, S.S.; et al. Molecular modeling based approach to potent P2-P4 macrocyclic inhibitors of hepatitis C NS3/4A protease. J. Am. Chem. Soc. 2008, 130, 4607–4609. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.J.; Carroll, S.S.; Dimuzio, J.; Fandozzi, C.; Graham, D.J.; Hazuda, D.; Holloway, M.K.; Ludmerer, S.W.; McCauley, J.A.; McIntyre, C.J.; et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 2010, 54, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.T.; McCauley, J.A.; Butcher, J.W.; Romano, J.J.; McIntyre, C.J.; Nguyen, K.T.; Gilbert, K.F.; Bush, K.J.; Holloway, M.K.; Swestock, J.; et al. Discovery of MK-1220: A Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease with Improved Preclinical Plasma Exposure. ACS Med. Chem. Lett. 2011, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.D.; Liu, M.Y.; Yu, W.L.; Li, J.Q.; Peng, M.; Dai, Q.; Liu, X.; Zhou, Z.Q. Hepatitis C virus infections and genotypes in China. Hepatobiliary Pancreat. Dis. Int. 2002, 1, 194–201. [Google Scholar] [PubMed]
- Xia, X.; Lu, L.; Tee, K.K.; Zhao, W.; Wu, J.; Yu, J.; Li, X.; Lin, Y.; Mukhtar, M.M.; Hagedorn, C.H.; et al. The unique HCV genotype distribution and the discovery of a novel subtype 6u among IDUs co-infected with HIV-1 in Yunnan, China. J. Med. Virol. 2008, 80, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, L.; Zhai, P.; He, S.; Jiang, T.; Gan, L.; Xiao, B.; Wen, X.; Sun, L. Macrocyclic Compounds for Suppressing Replication of Hepatitis C Virus. China Patent ZL 201210034872.0, 31 December 2014. [Google Scholar]
- Li, B.; Chen, L.; Zhai, P.; Jiang, T. Macrocyclic Compounds for Suppressing Replication of Hepatitis C Virus. PTC WO/2013/120371, 22 August 2013. [Google Scholar]
- Kiser, J.J.; Burton, J.R.; Anderson, P.L.; Everson, G.T. Review and management of drug interactions with boceprevir and telaprevir. Hepatology 2012, 55, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds GP205 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, N.; Sun, Q.; Xu, Z.; Wang, X.; Zhao, X.; Cao, Y.; Chen, L.; Fan, G. LC-ESI-MS/MS Analysis and Pharmacokinetics of GP205, an Innovative Potent Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease in Rats. Molecules 2015, 20, 4319-4336. https://doi.org/10.3390/molecules20034319
Yang N, Sun Q, Xu Z, Wang X, Zhao X, Cao Y, Chen L, Fan G. LC-ESI-MS/MS Analysis and Pharmacokinetics of GP205, an Innovative Potent Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease in Rats. Molecules. 2015; 20(3):4319-4336. https://doi.org/10.3390/molecules20034319
Chicago/Turabian StyleYang, Nan, Qiushi Sun, Zihua Xu, Xiuyun Wang, Xin Zhao, Yuqing Cao, Li Chen, and Guorong Fan. 2015. "LC-ESI-MS/MS Analysis and Pharmacokinetics of GP205, an Innovative Potent Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease in Rats" Molecules 20, no. 3: 4319-4336. https://doi.org/10.3390/molecules20034319
APA StyleYang, N., Sun, Q., Xu, Z., Wang, X., Zhao, X., Cao, Y., Chen, L., & Fan, G. (2015). LC-ESI-MS/MS Analysis and Pharmacokinetics of GP205, an Innovative Potent Macrocyclic Inhibitor of Hepatitis C Virus NS3/4A Protease in Rats. Molecules, 20(3), 4319-4336. https://doi.org/10.3390/molecules20034319