
Targeting Carbonic Anhydrase IX Activity and Expression

Abstract
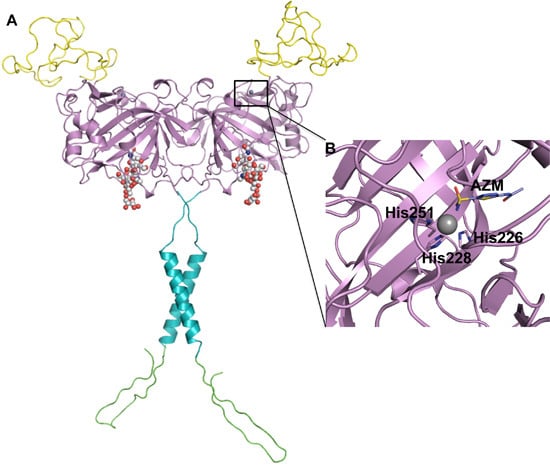
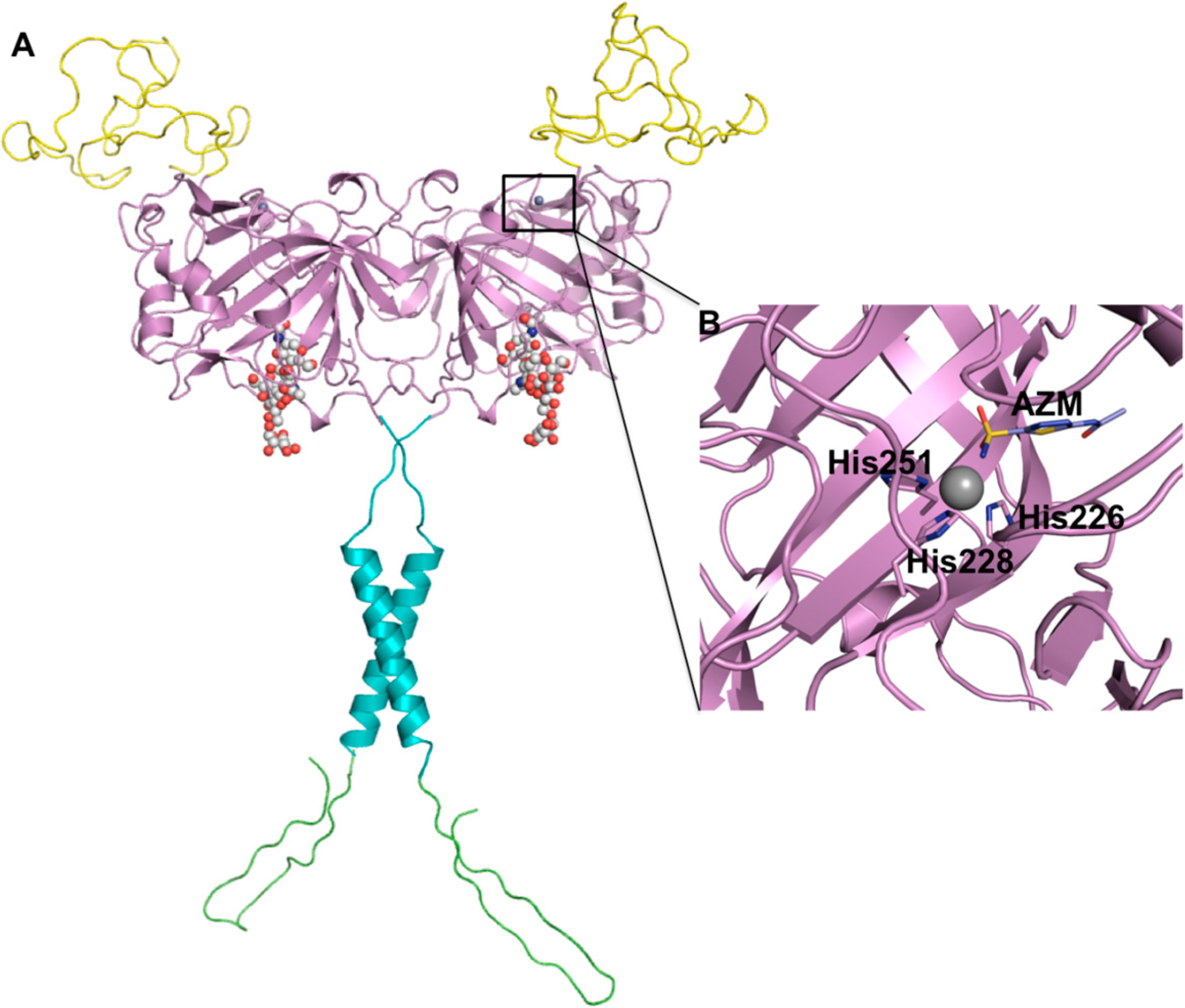
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
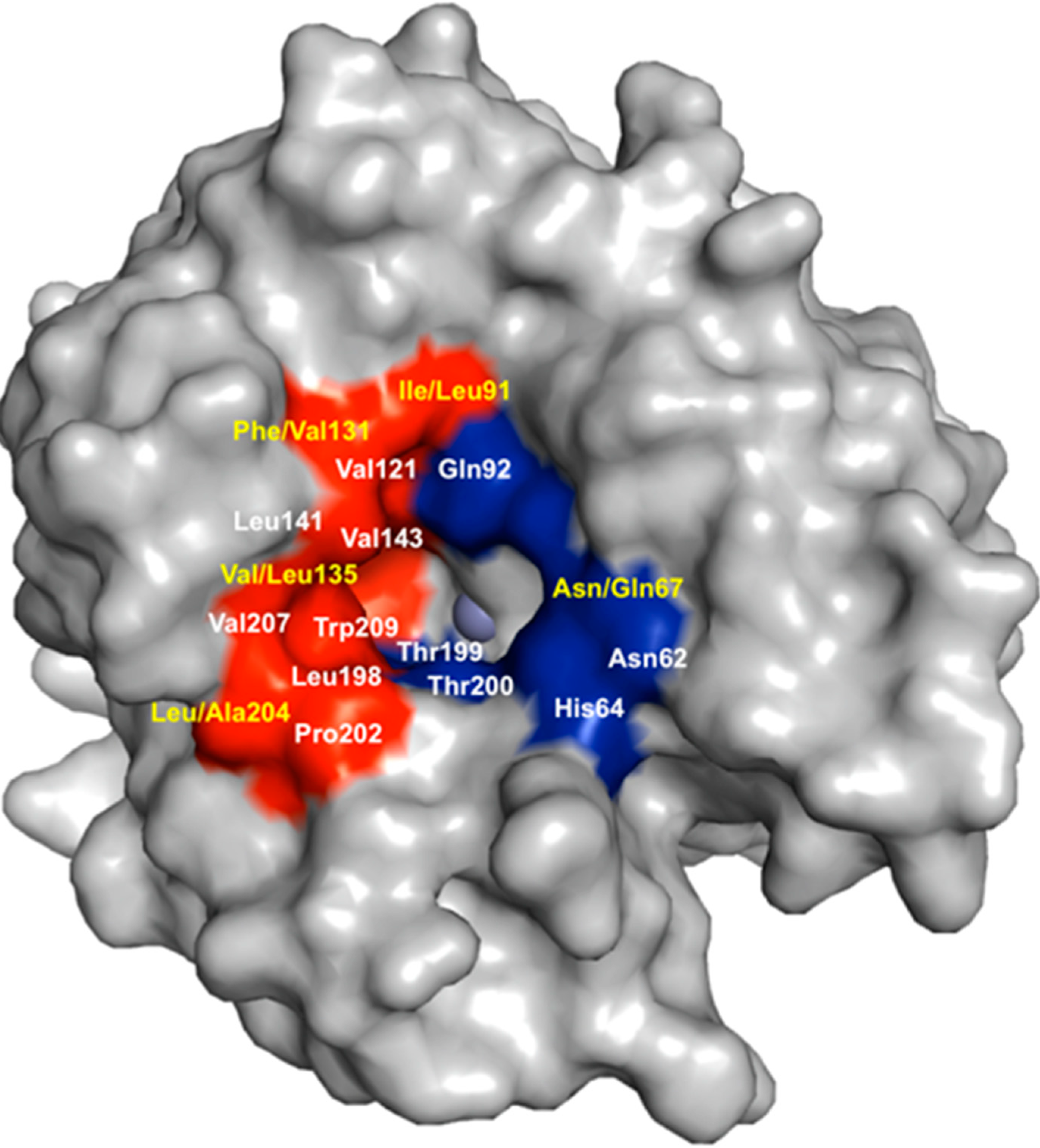{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
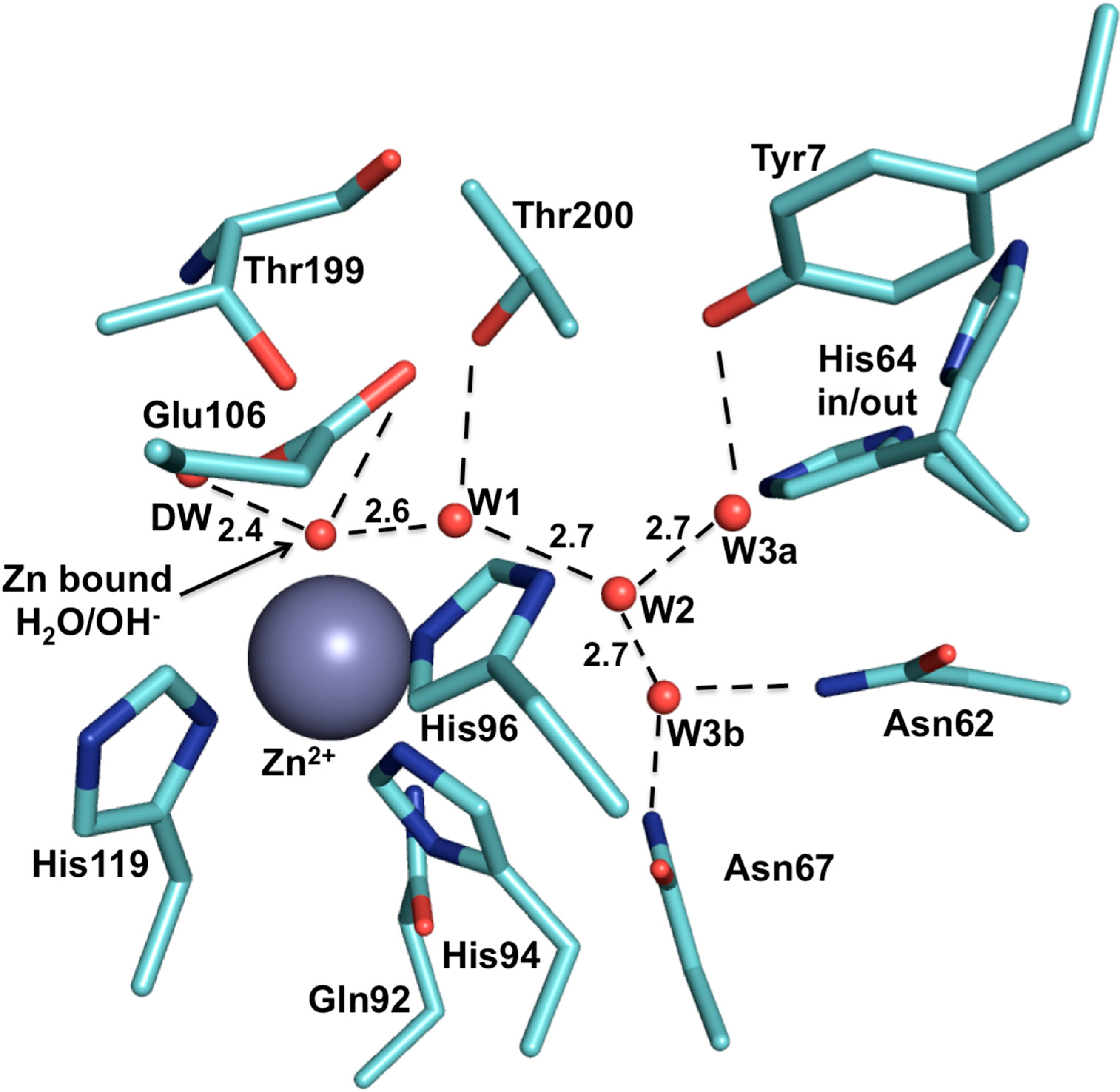
2. CA IX Structure and Function
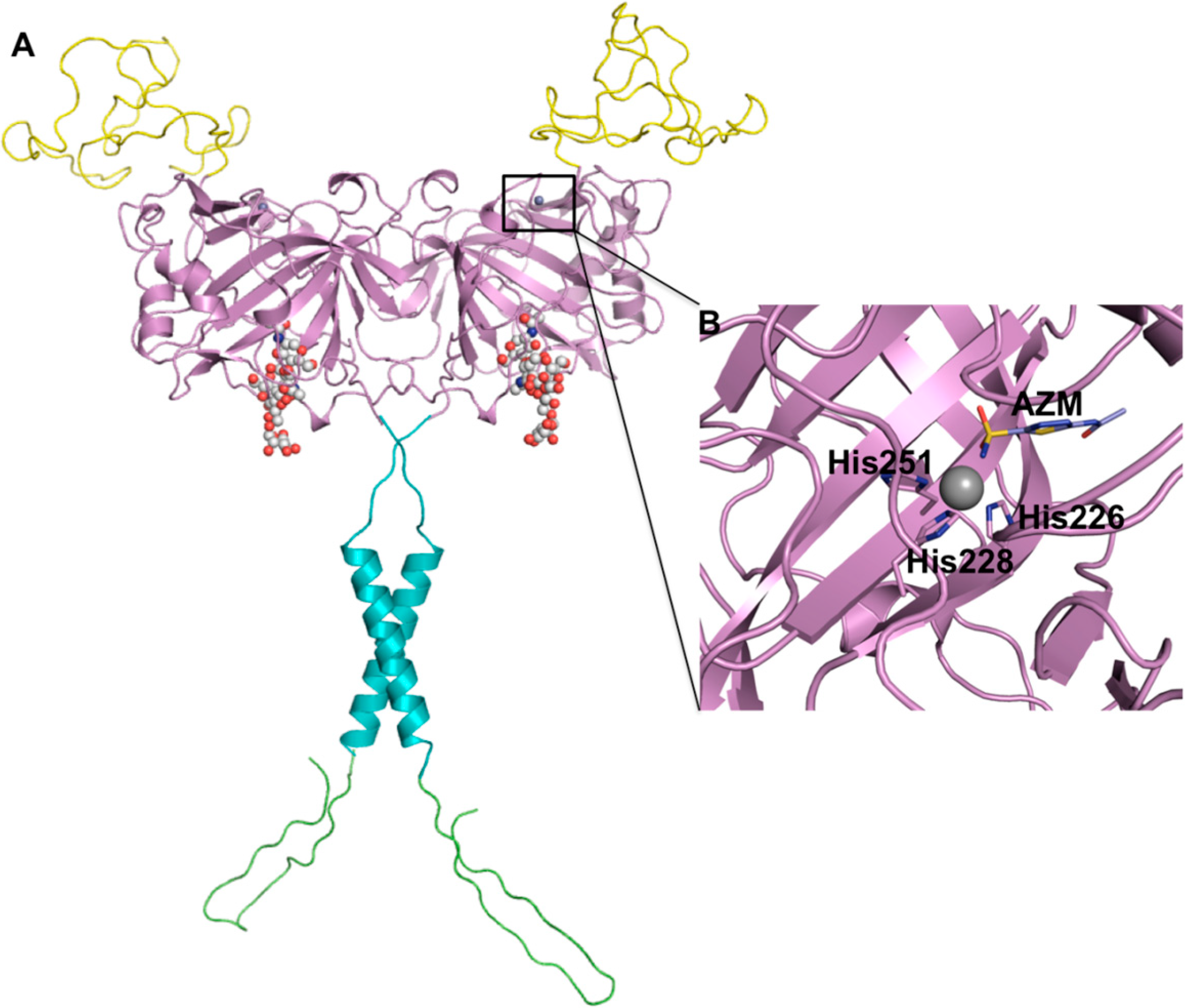
3. HIF-1 Regulates CA IX Expression
4. CA IX Expression in Normal vs. Neoplastic Tissue


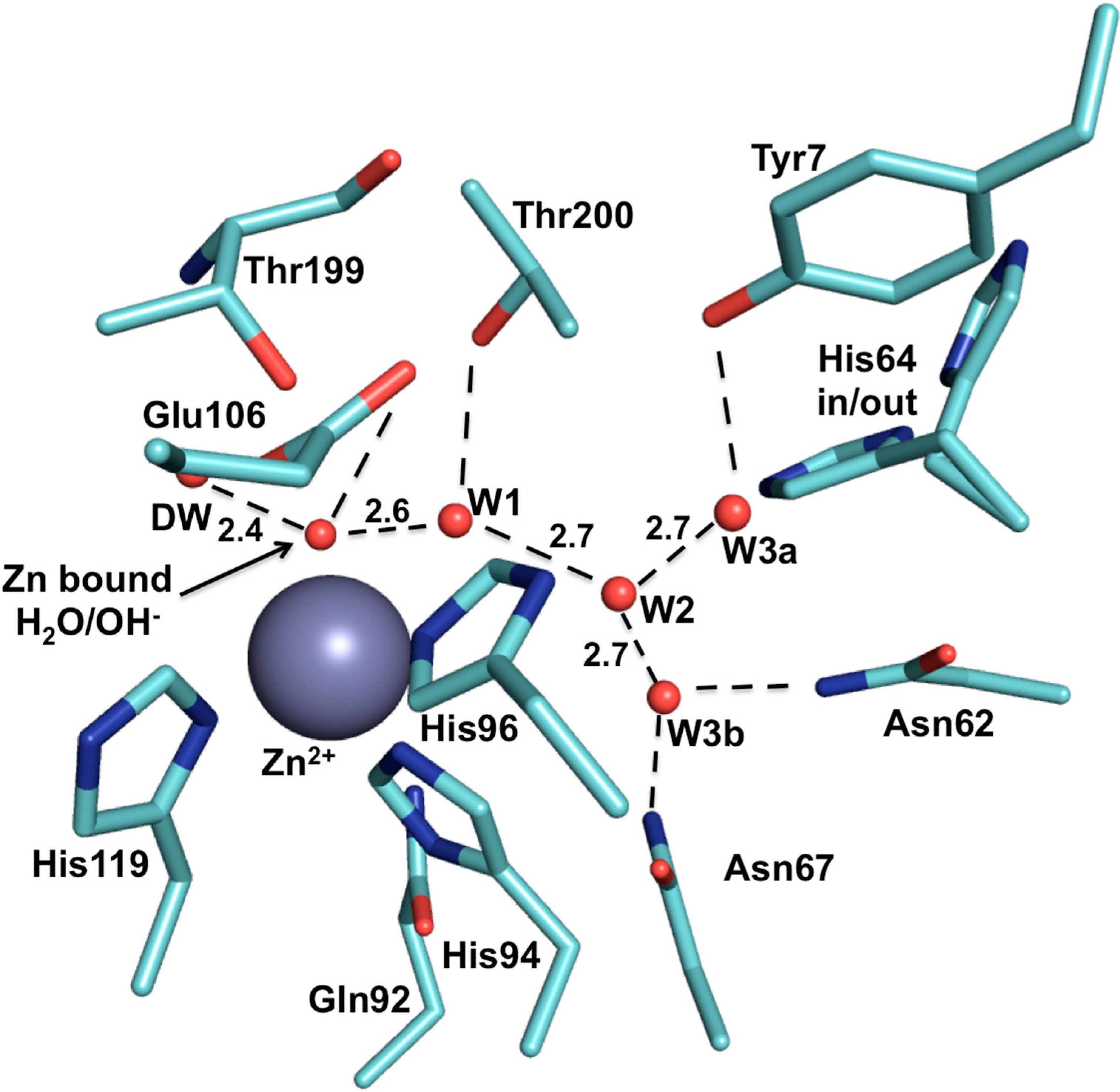
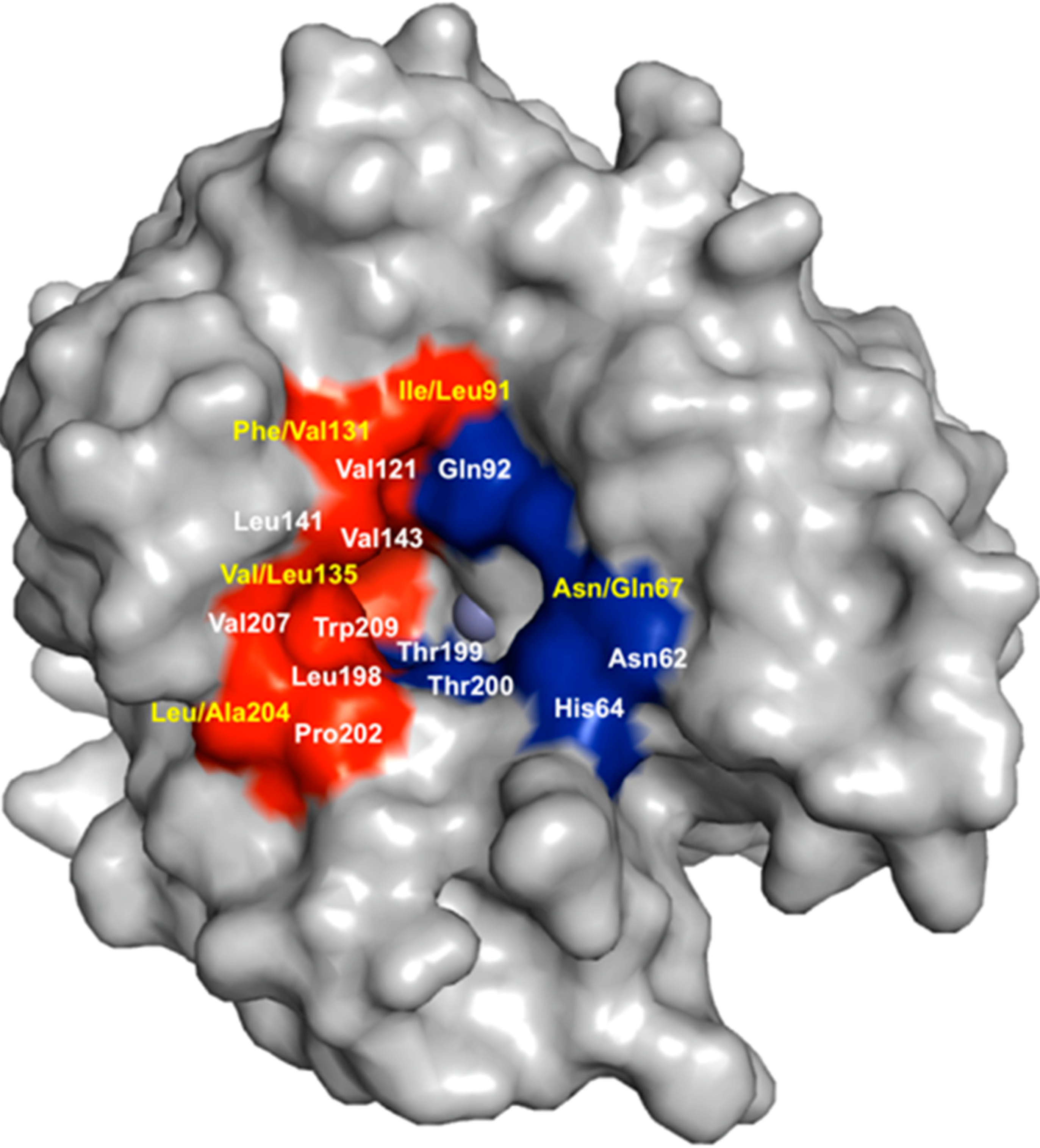
5. Structural Homology among Human CAs
6. Improving Classic CAIs
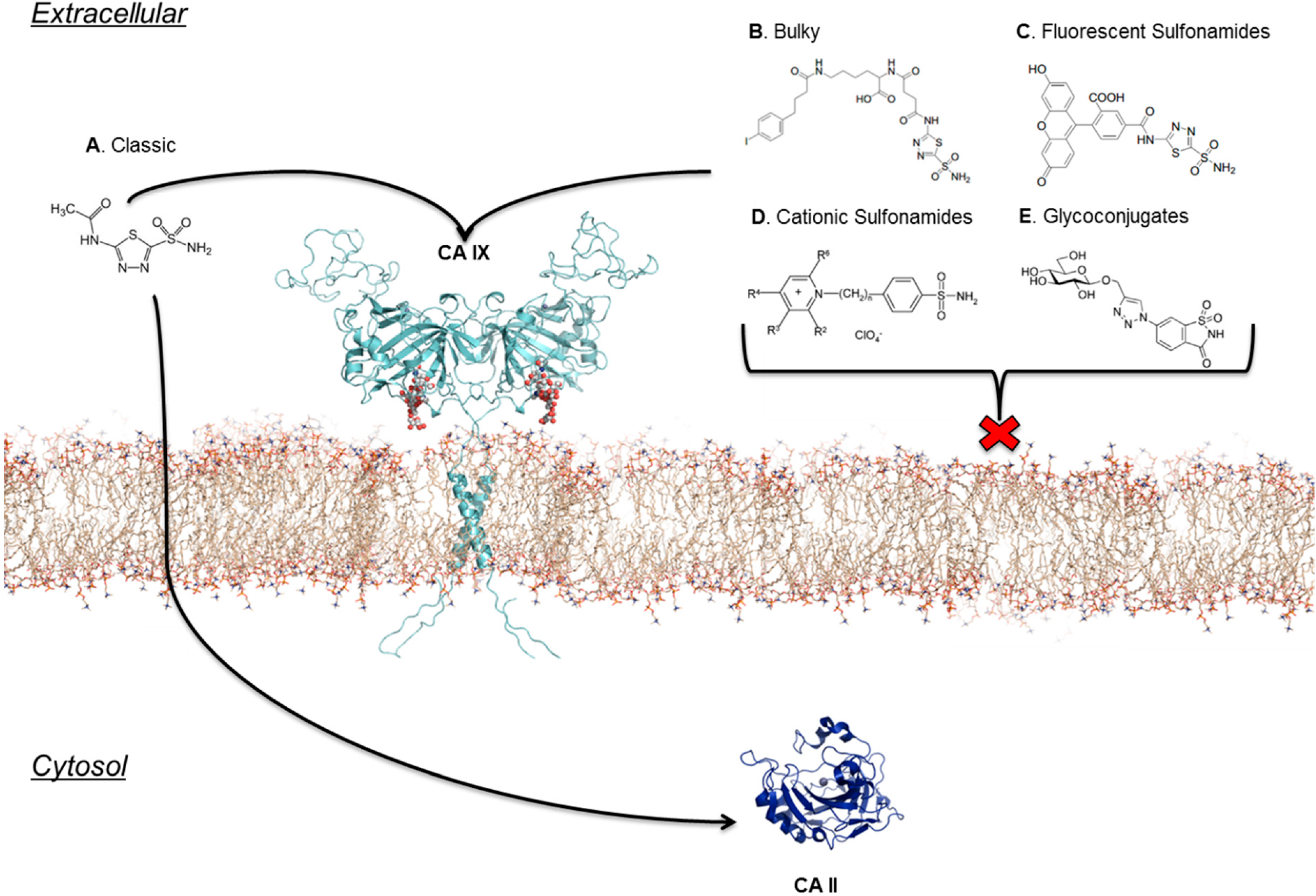
7. Location Specific Small-Molecule Inhibitors Target CA IX
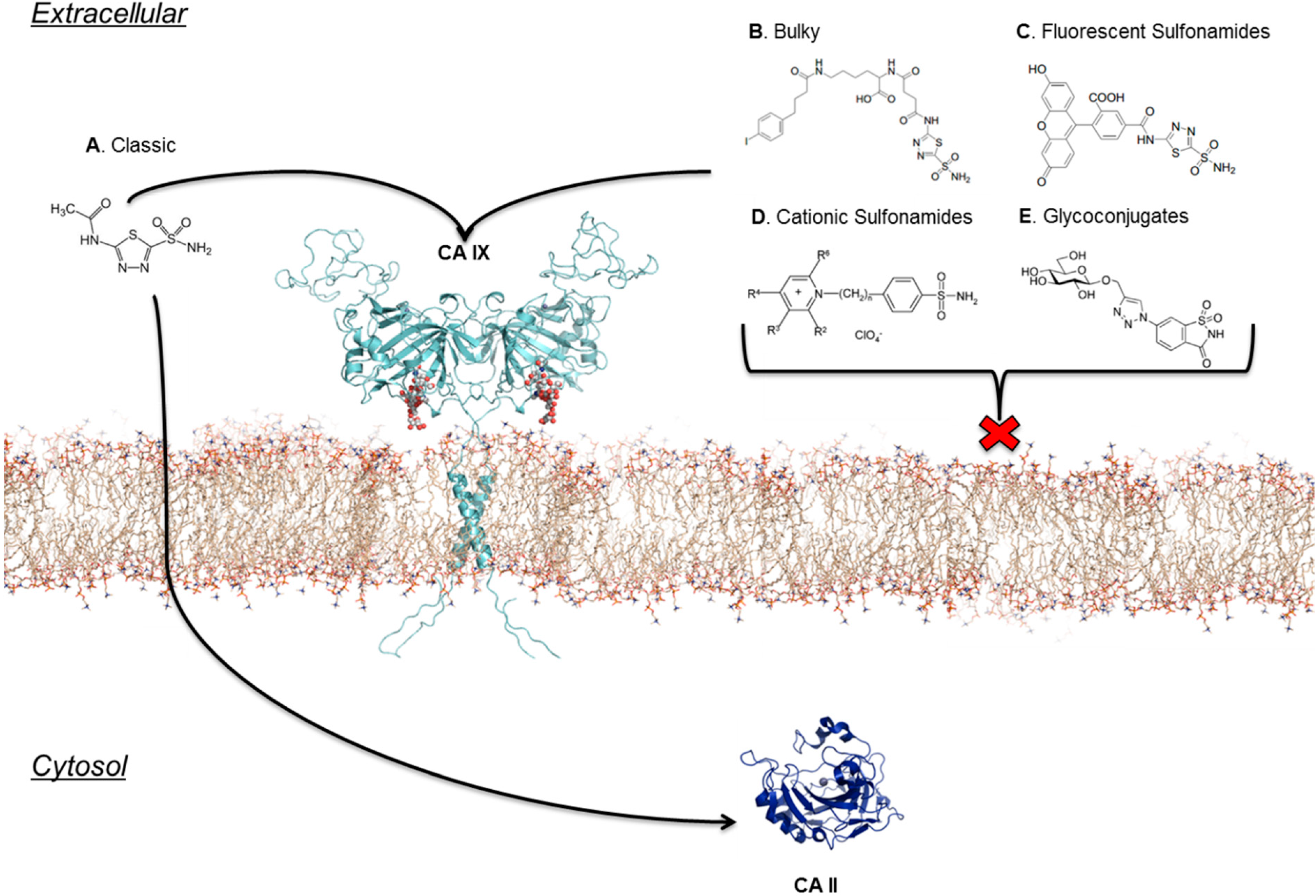
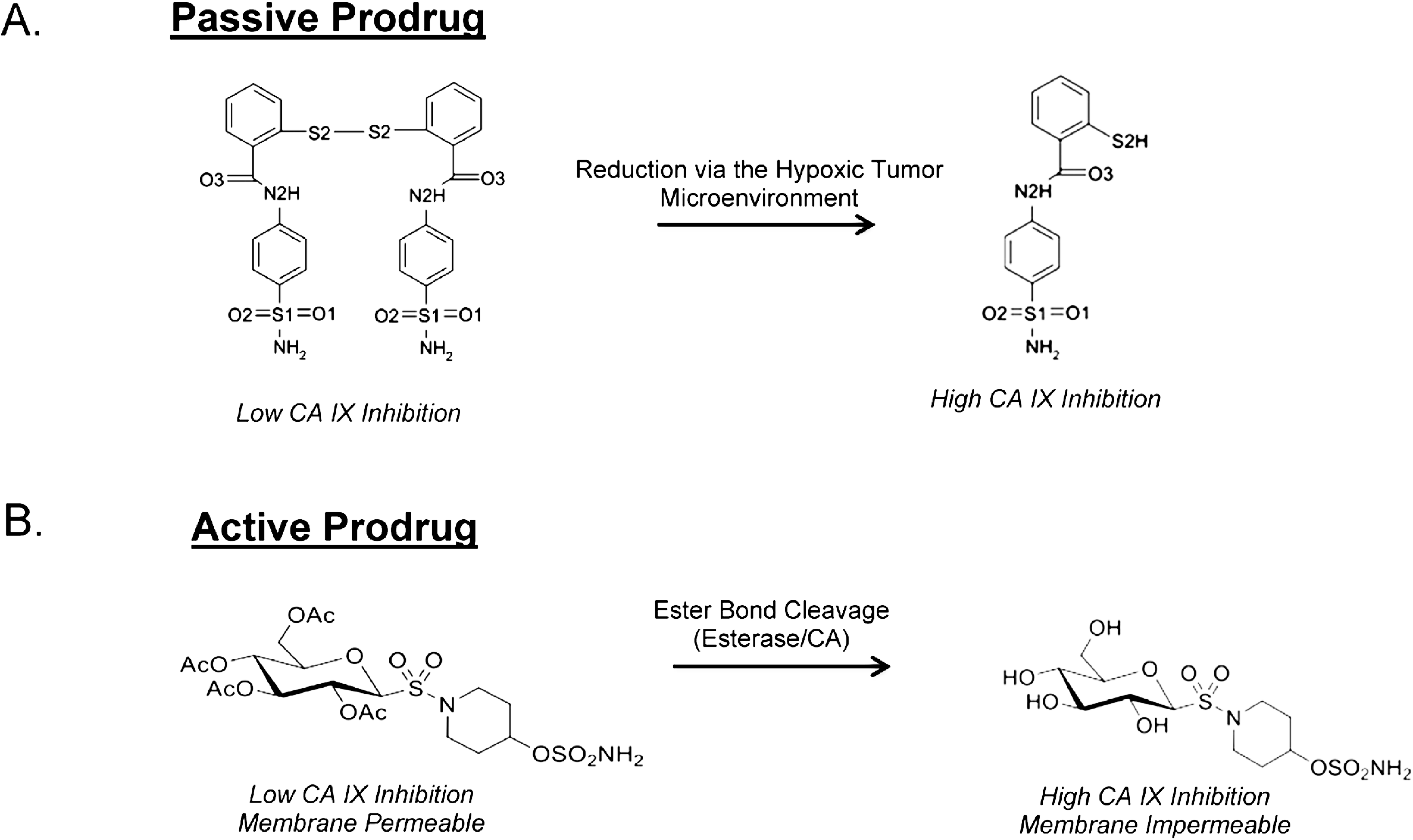
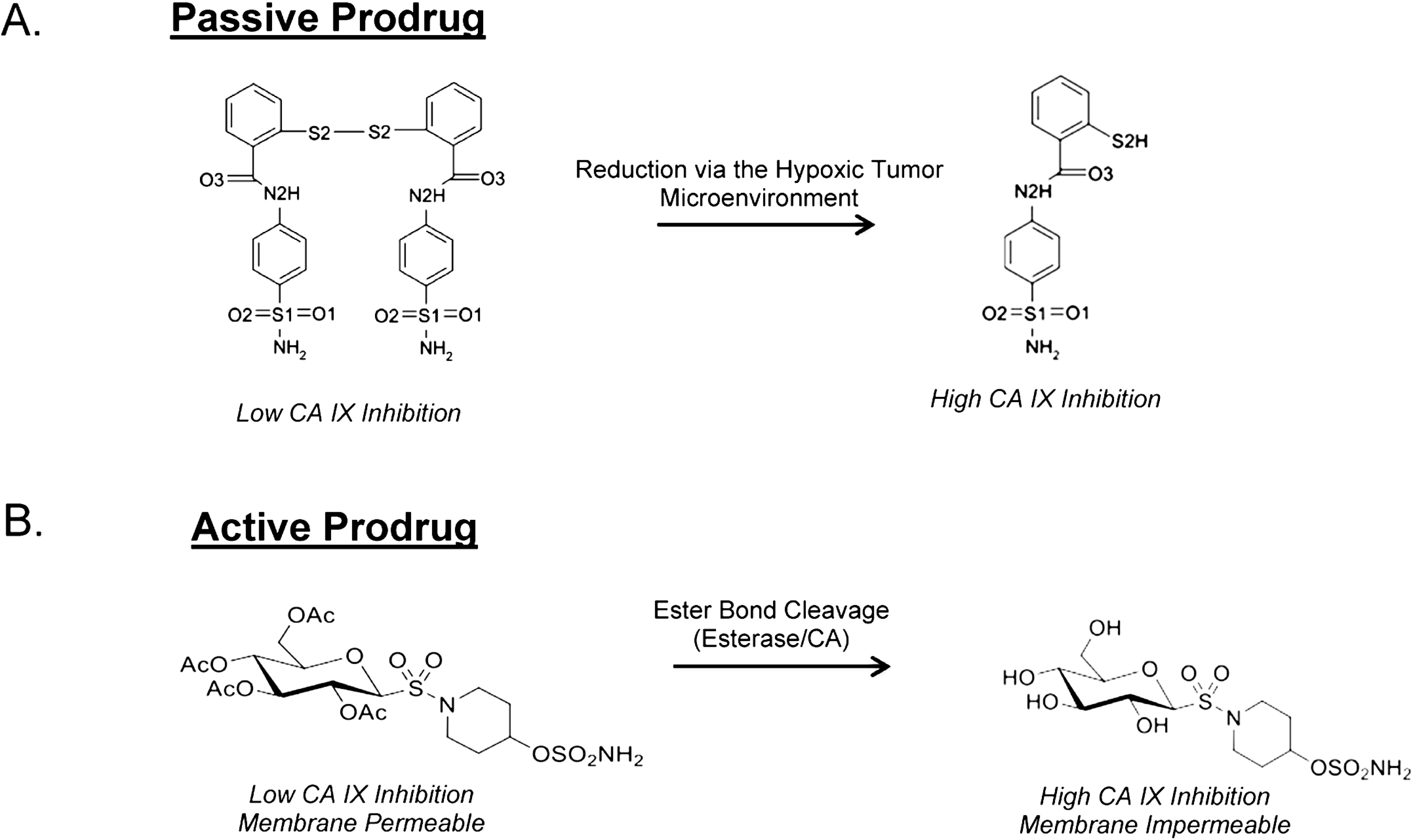
8. Taking Advantage of “Prodrug” Properties to Inhibit CA IX
9. Using CA IX as a Cell-Surface Receptor to Deliver Anti-Cancer Therapeutics
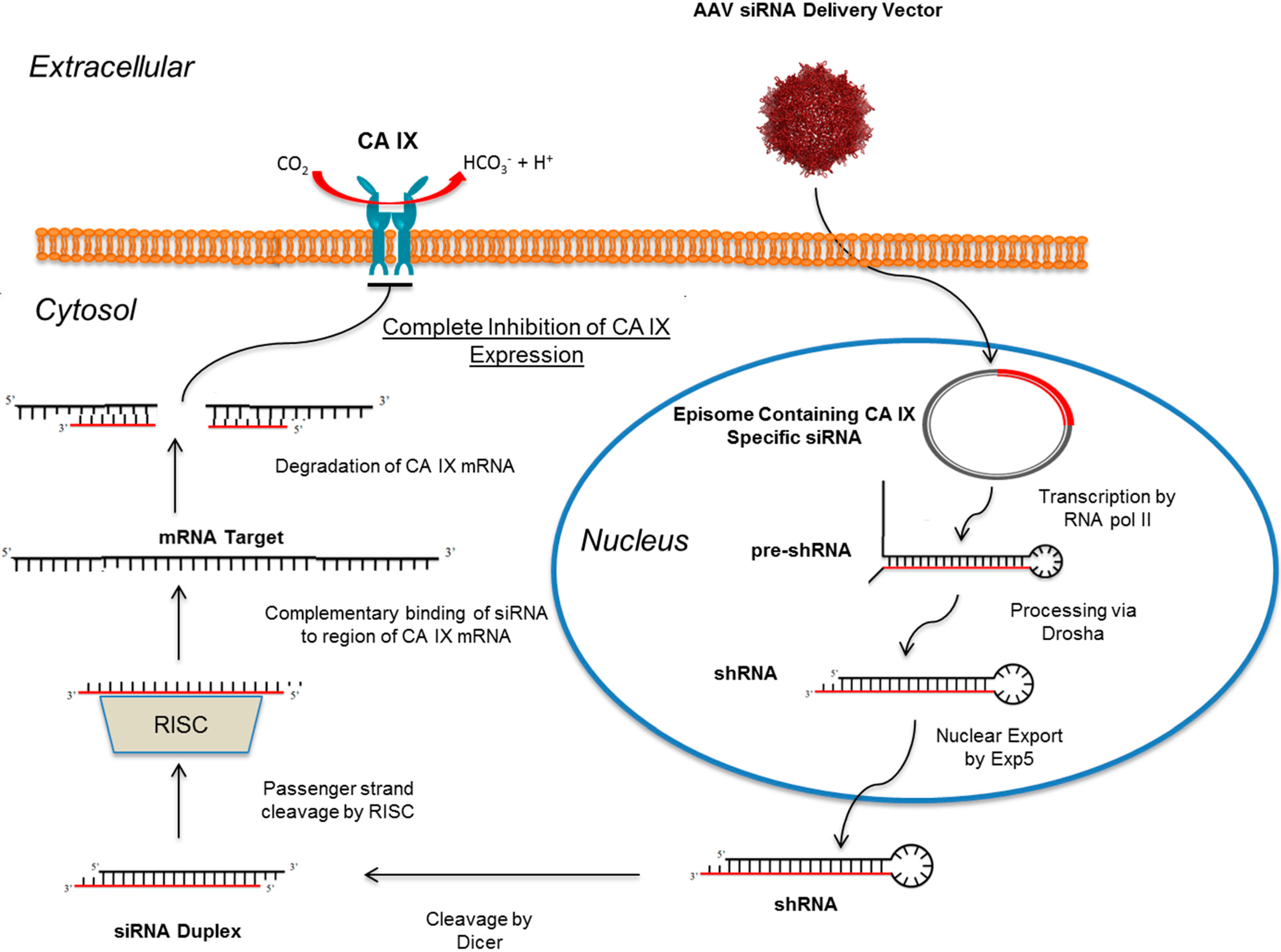
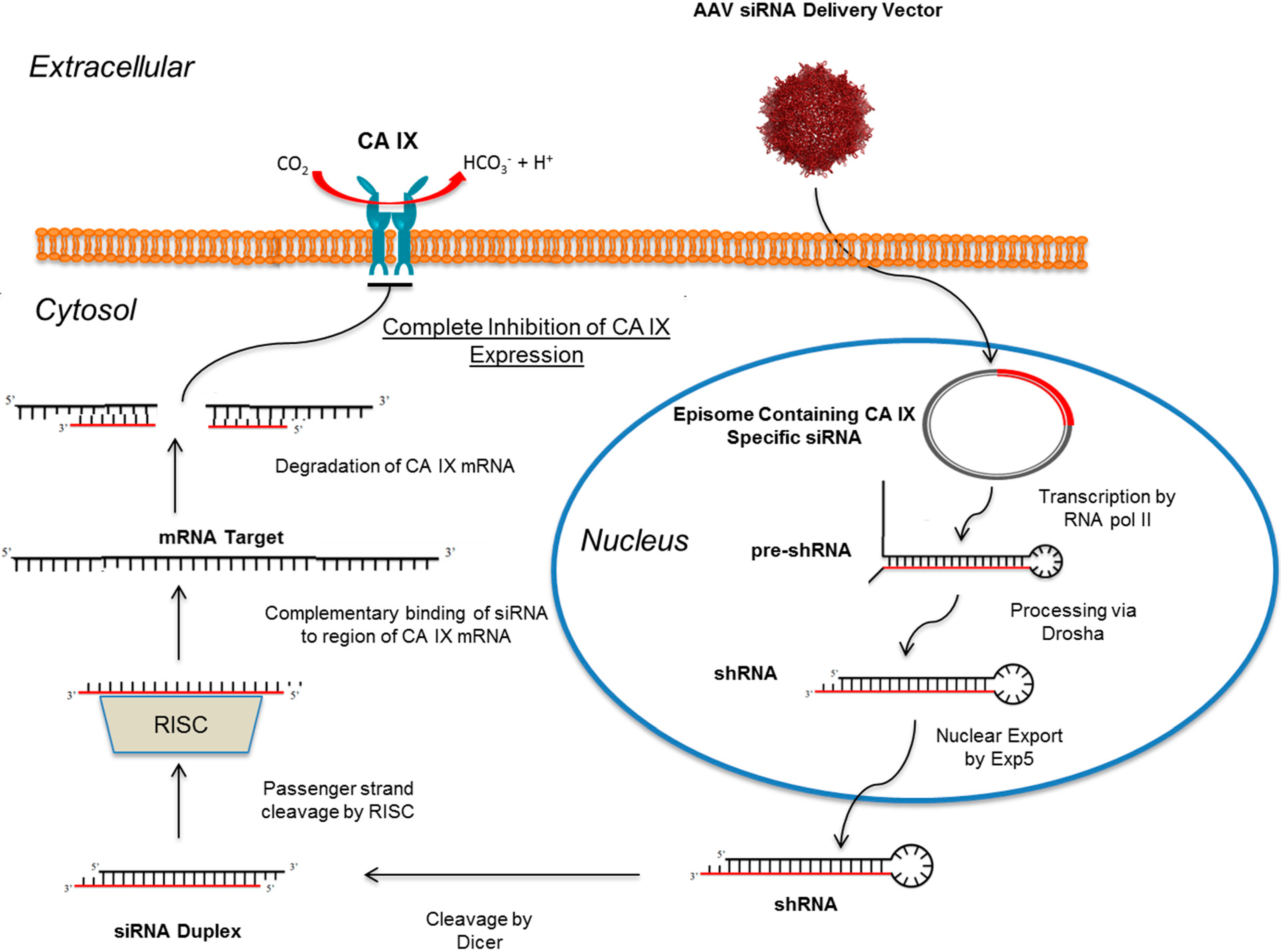
10. RNAi Mediated Knockdown of CA IX as a Means to Abolish the Hypoxic Tumor Milieu
11. Targeting CA IX as a Combinatorial Treatment for Cancer; Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moulder, J.E.; Rockwell, S. Tumor hypoxia: Its impact on cancer therapy. Cancer Metastasis Rev. 1987, 5, 313–341. [Google Scholar] [PubMed]
- Peskin, B.; Carter, M.J. Chronic cellular hypoxia as the prime cause of cancer: What is the de-oxygenating role of adulterated and improper ratios of polyunsaturated fatty acids when incorporated into cell membranes? Med. Hypotheses 2008, 70, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
- Racker, E. Warburg effect revisited. Science 1981, 213, 1313. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Chia, S.K.; Wykoff, C.C.; Watson, P.H.; Han, C.; Leek, R.D.; Pastorek, J.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J. Clin. Oncol. 2001, 19, 3660–3668. [Google Scholar] [PubMed]
- McDonald, P.C.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [PubMed]
- Luo, D.; Wang, Z.; Wu, J.; Jiang, C.; Wu, J. The Role of Hypoxia Inducible Factor-1 in Hepatocellular Carcinoma. BioMed Res. Int. 2014, 2014, e409272. [Google Scholar]
- Sadri, N.; Zhang, P.J. Hypoxia-inducible factors: Mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers 2013, 5, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Boone, C.D.; Kondeti, B.; McKenna, R. Structural annotation of human carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2013, 28, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C. Physiological functions of the alpha class of carbonic anhydrases. Subcell. Biochem. 2014, 75, 9–30. [Google Scholar] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Oosterwijk, E.; Selman, Y.; Mira, J.C.; Medrano, T.; Shiverick, K.T.; Frost, S.C. Antibody-specific detection of CAIX in breast and prostate cancers. Biochem. Biophys. Res. Commun. 2009, 386, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Türeci, O.; Sahin, U.; Vollmar, E.; Siemer, S.; Göttert, E.; Seitz, G.; Parkkila, A.K.; Shah, G.N.; Grubb, J.H.; Pfreundschuh, M.; et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 7608–7613. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.I.; Hofman, V.; Ortholan, C.; Ammadi, R.E.; Bonnetaud, C.; Havet, K.; Venissac, N.; Mouroux, J.; Mazure, N.M.; Pouysségur, J.; et al. Overexpression of carbonic anhydrase XII in tissues from resectable non-small cell lung cancers is a biomarker of good prognosis. Int. J. Cancer 2011, 128, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.Y.; Lerman, M.I.; Stanbridge, E.J. Expression of transmembrane carbonic anhydrases, CAIX and CAXII, in human development. BMC Dev. Biol. 2009, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Opavský, R.; Pastoreková, S.; Zelník, V.; Gibadulinová, A.; Stanbridge, E.J.; Závada, J.; Kettmann, R.; Pastorek, J. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics 1996, 33, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Supuran, C.T. Carbonic anhydrase IX: Biochemical and crystallographic characterization of a novel antitumor target. Biochim. Biophys. Acta 2010, 1804, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Baranauskiene, L.; Salzano, A.M.; Scaloni, A.; Matulis, D.; Innocenti, A.; Scozzafava, A.; Monti, S.M.; Di Fiore, A.; De Simone, G.; et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J. Biol. Chem. 2008, 283, 27799–27809. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Mahon, B.P.; McKenna, R. Probing the Surface of Human Carbonic Anhydrase for Clues towards the Design of Isoform Specific Inhibitors. BioMed Res. Int. 2014, in press. [Google Scholar]
- Barathova, M.; Takacova, M.; Holotnakova, T.; Gibadulinova, A.; Ohradanova, A.; Zatovicova, M.; Hulikova, A.; Kopacek, J.; Parkkila, S.; Supuran, C.T.; et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumour phenotype. Br. J. Cancer 2008, 98, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–531. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; McKenna, R. Regulation and role of carbonic anhydrase IX and use as a biomarker and therapeutic target in cancer. Res. Trends Curr. Top. Biochem. Res. 2013, 15, 1–21. [Google Scholar]
- The PyMOL Molecular Graphics System; Version 1.5.0.4; Schrödinger, LLC: New York, NY, USA, 2013.
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; de Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors: an editorial. Expert Opin. Ther. Pat. 2013, 23, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic Anhydrase IX: Regulation and Role in Cancer. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S.C., McKenna, R., Eds.; Subcellular Biochemistry: Springer, The Netherlands, 2014; pp. 199–219. [Google Scholar]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [PubMed]
- Block, K.; Gorin, Y.; Hoover, P.; Williams, P.; Chelmicki, T.; Clark, R.A.; Yoneda, T.; Abboud, H.E. NAD(P)H Oxidases Regulate HIF-2 Protein Expression. J. Biol. Chem. 2007, 282, 8019–8026. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, E.; Brattain, M.G.; Chowdhury, S. Cell survival and metastasis regulation by AKT signaling in colorectal cancer. Cell. Signal. 2013, 25, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Willam, C.; Masson, N.; Tian, Y.M.; Mahmood, S.A.; Wilson, M.I.; Bicknell, R.; Eckardt, K.U.; Maxwell, P.H.; Ratcliffe, P.J.; Pugh, C.W. Peptide blockade of HIFα degradation modulates cellular metabolism and angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 10423–10428. [Google Scholar] [CrossRef] [PubMed]
- Gallou, C.; Joly, D.; Méjean, A.; Staroz, F.; Martin, N.; Tarlet, G.; Orfanelli, M.T.; Bouvier, R.; Droz, D.; Chrétien, Y.; et al. Mutations of the VHL gene in sporadic renal cell carcinoma: Definition of a risk factor for VHL patients to develop an RCC. Hum. Mutat. 1999, 13, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Bono, F.; Maxwell, P.; Dor, Y.; Dewerchin, M.; Collen, D.; Herbert, J.M.; Carmeliet, P. Hypoxia-inducible factor-2alpha (HIF-2alpha) is involved in the apoptotic response to hypoglycemia but not to hypoxia. J. Biol. Chem. 2001, 276, 39192–39196. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Kohler, I.; Fellmann, C.; Lowe, S.W.; Hengartner, M.O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010, 465, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W. Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: A novel calcium-insensitive mechanism. FASEB J. 2002, 16, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Saarnio, J.; Parkkila, S.; Parkkila, A.K.; Waheed, A.; Casey, M.C.; Zhou, X.Y.; Pastoreková, S.; Pastorek, J.; Karttunen, T.; Haukipuro, K.; et al. Immunohistochemistry of carbonic anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J. Histochem. Cytochem. 1998, 46, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Saarnio, J.; Parkkila, S.; Parkkila, A.K.; Haukipuro, K.; Pastoreková, S.; Pastorek, J.; Kairaluoma, M.I.; Karttunen, T.J. Immunohistochemical study of colorectal tumors for expression of a novel transmembrane carbonic anhydrase, MN/CA IX, with potential value as a marker of cell proliferation. Am. J. Pathol. 1998, 153, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Leibovich, B.C.; Sheinin, Y.; Lohse, C.M.; Thompson, R.H.; Cheville, J.C.; Zavada, J.; Kwon, E.D. Carbonic anhydrase IX is not an independent predictor of outcome for patients with clear cell renal cell carcinoma. J. Clin. Oncol. 2007, 25, 4757–4764. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Liao, S.Y.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle Georget. Tex 2013, 12, 1791–1801. [Google Scholar] [CrossRef]
- Eichhorn, M. Mode of action, clinical profile and relevance of carbonic anhydrase inhibitors in glaucoma therapy. Klinische Monatsblätter für Augenheilkunde 2013, 230, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.D.; Pinard, M.; McKenna, R.; Silverman, D. Catalytic Mechanism of α-Class Carbonic Anhydrases: CO2 Hydration and Proton Transfer. Subcell. Biochem. 2014, 75, 31–52. [Google Scholar] [PubMed]
- Pinard, M.A.; Boone, C.D.; Rife, B.D.; Supuran, C.T.; McKenna, R. Structural study of interaction between brinzolamide and dorzolamide inhibition of human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 7210–7215. [Google Scholar] [CrossRef] [PubMed]
- McKenna, R.; Supuran, C.T. Carbonic anhydrase inhibitors drug design. Subcell. Biochem. 2014, 75, 291–323. [Google Scholar] [PubMed]
- Kolayli, S.; Karahalil, F.; Sahin, H.; Dincer, B.; Supuran, C.T. Characterization and inhibition studies of an α-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J. Enzym. Inhib. Med. Chem. 2011, 26, 895–900. [Google Scholar] [CrossRef]
- Avvaru, B.S.; Kim, C.U.; Sippel, K.H.; Gruner, S.M.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. A short, strong hydrogen bond in the active site of human carbonic anhydrase II. Biochemistry 2010, 49, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Moeker, J.; Mahon, B.P.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; McKenna, R.; Poulsen, S.A. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J. Med. Chem. 2014, 57, 8635–8645. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Hendon, A.M.; Driscoll, J.M.; Rankin, G.M.; Poulsen, S.A.; Supuran, C.T.; McKenna, R. Saccharin: A Lead Compound for Structure-Based Drug Design of Carbonic Anhydrase IX Inhibitors. Bioorg. Med. Chem. 2014, in press. [Google Scholar]
- Siebels, M.; Rohrmann, K.; Oberneder, R.; Stahler, M.; Haseke, N.; Beck, J.; Hofmann, R.; Kindler, M.; Kloepfer, P.; Stief, C. A clinical phase I/II trial with the monoclonal antibody cG250 (RENCAREX®) and interferon-alpha-2a in metastatic renal cell carcinoma patients. World J. Urol. 2011, 29, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Macis, G.; Di Giovanni, S.; Di Franco, D.; Bonomo, L. Future perspectives for diagnostic imaging in urology: from anatomic and functional to molecular imaging. Urologia 2013, 80, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Závada, J.; Závadová, Z.; Pastorek, J.; Biesová, Z.; Jezek, J.; Velek, J. Human tumour-associated cell adhesion protein MN/CA IX: identification of M75 epitope and of the region mediating cell adhesion. Br. J. Cancer 2000, 82, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Tomura, H.; Wang, J.Q.; Liu, J.P.; Komachi, M.; Damirin, A.; Mogi, C.; Tobo, M.; Nochi, H.; Tamoto, K.; Im, D.S.; et al. Cyclooxygenase-2 expression and prostaglandin E2 production in response to acidic pH through OGR1 in a human osteoblastic cell line. J. Bone Miner. Res. 2008, 23, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Morgan, P.E.; Vullo, D.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J. Med. Chem. 2004, 47, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Groves, K.; Bao, B.; Zhang, J.; Handy, E.; Kennedy, P.; Cuneo, G.; Supuran, C.T.; Yared, W.; Peterson, J.D.; Rajopadhye, M. Synthesis and evaluation of near-infrared fluorescent sulfonamide derivatives for imaging of hypoxia-induced carbonic anhydrase IX expression in tumors. Bioorg. Med. Chem. Lett. 2012, 22, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Tinker, J.P.; Coulson, R.; Weiner, I.M. Dextran-bound inhibitors of carbonic anhydrase. J. Pharmacol. Exp. Ther. 1981, 218, 600–607. [Google Scholar] [PubMed]
- Maren, T.H. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol. Rev. 1967, 47, 595–781. [Google Scholar]
- Pastorekova, S.; Casini, A.; Scozzafava, A.; Vullo, D.; Pastorek, J.; Supuran, C.T. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg. Med. Chem. Lett. 2004, 14, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J. Enzym. Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef]
- Winum, J.Y.; Poulsen, S.A.; Supuran, C.T. Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med. Res. Rev. 2009, 29, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Cousins, K.R. Computer review of ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef] [PubMed]
- Moeker, J.; Peat, T.S.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Cyclic secondary sulfonamides: Unusually good inhibitors of cancer-related carbonic anhydrase enzymes. J. Med. Chem. 2014, 57, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Bornaghi, L.F.; Innocenti, A.; Vullo, D.; Charman, S.A.; Supuran, C.T.; Poulsen, S.A. Sulfonamide Linked Neoglycoconjugates−A New Class of Inhibitors for Cancer-Associated Carbonic Anhydrases. J. Med. Chem. 2010, 53, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Vitavska, O.; Wieczorek, H. Identification of an animal sucrose transporter. J. Cell Sci. 2011, 124, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Solberg, R.; Jacobsen, L.L.; Voreland, A.L.; Rustan, A.C.; Thoresen, G.H.; Johansen, H.T. Simvastatin inhibits glucose metabolism and legumain activity in human myotubes. PLoS One 2014, 9, e85721. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Eck, P.; Chen, S.; Corpe, C.P.; Lee, J.H.; Kruhlak, M.; Levine, M. Inhibition of the intestinal glucose transporter GLUT2 by flavonoids. FASEB J. 2007, 21, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Carroux, C.J.; Rankin, G.M.; Moeker, J.; Bornaghi, L.F.; Katneni, K.; Morizzi, J.; Charman, S.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. A prodrug approach toward cancer-related carbonic anhydrase inhibition. J. Med. Chem. 2013, 56, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Giang, I.; Boland, E.L.; Poon, G.M.K. Prodrug Applications for Targeted Cancer Therapy. AAPS J. 2014, 16, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Tiefenthaler, G.; Georges, G. Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genomics Proteomics 2014, 11, 67–79. [Google Scholar] [PubMed]
- Moody, C.L.; Wheelhouse, R.T. The medicinal chemistry of imidazotetrazine prodrugs. Pharm. Basel Switz. 2014, 7, 797–838. [Google Scholar]
- Kehayova, P.D.; Woodrell, C.D.; Dostal, P.J.; Chandra, P.P.; Jain, A. Phototriggered delivery of hydrophobic carbonic anhydrase inhibitors. Photochem. Photobiol. Sci. 2002, 1, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Grandane, A.; Tanc, M.; Zalubovskis, R.; Supuran, C.T. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. Lett. 2014, 24, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Woltersdorf, O.W.; Schwam, H.; Bicking, J.B.; Brown, S.L.; deSolms, S.J.; Fishman, D.R.; Graham, S.L.; Gautheron, P.D.; Hoffman, J.M.; Larson, R.D. Topically active carbonic anhydrase inhibitors. 1. O-acyl derivatives of 6-hydroxybenzothiazole-2-sulfonamide. J. Med. Chem. 1989, 32, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, L.M.; Kaufman, P.L.; Robin, A.L.; Weinreb, R.N.; Crawford, K.; Shaw, B. New developments in the drug treatment of glaucoma. Drugs 1991, 41, 514–532. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Gautheron, P.; Plazonnet, B.; Sugrue, M.F. Ocular distribution studies of the topical carbonic anhydrase inhibitors L-643,799 and L-650,719 and related alkyl prodrugs. J. Ocul. Pharmacol. 1988, 4, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Barrese, A.A.; Genis, C.; Fisher, S.Z.; Orwenyo, J.N.; Kumara, M.T.; Dutta, S.K.; Phillips, E.; Kiddle, J.J.; Tu, C.; Silverman, D.N.; et al. Inhibition of carbonic anhydrase II by thioxolone: A mechanistic and structural study. Biochemistry 2008, 47, 3174–3184. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Vitale, R.M.; Di Fiore, A.; Pedone, C.; Scozzafava, A.; Montero, J.L.; Winum, J.Y.; Supuran, C.T. Carbonic anhydrase inhibitors: Hypoxia-activatable sulfonamides incorporating disulfide bonds that target the tumor-associated isoform IX. J. Med. Chem. 2006, 49, 5544–5551. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. 5- and 6-membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Reich, R.; Hoffman, A.; Veerendhar, A.; Maresca, A.; Innocenti, A.; Supuran, C.T.; Breuer, E. Carbamoylphosphonates control tumor cell proliferation and dissemination by simultaneously inhibiting carbonic anhydrase IX and matrix metalloproteinase-2. Toward nontoxic chemotherapy targeting tumor microenvironment. J. Med. Chem. 2012, 55, 7875–7882. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Lindskog, S. Effects of high concentrations of salt on the esterase activity of human carbonic anhydrase. FEBS Lett. 1972, 24, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C. K.; Zhang, H.; Qin, L.; Chen, H.; Fang, C.; Lu, A.; Yang, Z. Carbonic anhydrase IX-directed immunoliposomes for targeted drug delivery to human lung cancer cells in vitro. Drug Des. Dev. Ther. 2014, 8, 993–1001. [Google Scholar]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J. L.; Supuran, C.T.; Neri, D. A Small-Molecule Drug Conjugate for the Treatment of Carbonic Anhydrase IX Expressing Tumors. Angew. Chem. Int. Ed. 2014, 53, 4231–4235. [Google Scholar] [CrossRef]
- Shinkai, M.; Le, B.; Honda, H.; Yoshikawa, K.; Shimizu, K.; Saga, S.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Targeting hyperthermia for renal cell carcinoma using human MN antigen-specific magnetoliposomes. Jpn. J. Cancer Res. Gann 2001, 92, 1138–1145. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Neri, D. A bivalent small molecule-drug conjugate directed against carbonic anhydrase IX can elicit complete tumour regression in mice. Chem. Sci. 2014, 5, 3640–3644. [Google Scholar] [CrossRef]
- Borel, F.; Kay, M.A.; Mueller, C. Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 692–701. [Google Scholar] [CrossRef]
- Banerjee, D.; Slack, F. Control of developmental timing by small temporal RNAs: a paradigm for RNA-mediated regulation of gene expression. BioEssays News Rev. Mol. Cell. Dev. Biol. 2002, 24, 119–129. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Tiram, G.; Scomparin, A.; Ofek, P.; Satchi-Fainaro, R. Interfering cancer with polymeric siRNA nanomedicines. J. Biomed. Nanotechnol. 2014, 10, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Hyodo, M.; Akita, H.; Harashima, H. Gene silencing via RNAi and siRNA quantification in tumor tissue using MEND, a liposomal siRNA delivery system. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1195–1203. [Google Scholar] [CrossRef]
- Cheng, B.; Ling, C.; Dai, Y.; Lu, Y.; Glushakova, L.G.; Gee, S.W. Y.; McGoogan, K.E.; Aslanidi, G.V.; Park, M.; Stacpoole, P.W.; et al. Development of optimized AAV3 serotype vectors: Mechanism of high-efficiency transduction of human liver cancer cells. Gene Ther. 2012, 19, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Yue, Y.; Engelhardt, J.F. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: A quantitative comparison. Mol. Ther. J. Am. Soc. Gene Ther. 2001, 4, 383–391. [Google Scholar] [CrossRef]
- Ling, C.; Lu, Y.; Cheng, B.; McGoogan, K.E.; Gee, S.W.Y.; Ma, W.; Li, B.; Aslanidi, G.V.; Srivastava, A. High-efficiency transduction of liver cancer cells by recombinant adeno-associated virus serotype 3 vectors. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Weng, Y.; Fei, B.; Chi, A.L.; Cai, M. Inhibition of gastric cancer cell growth in vivo by overexpression of adeno-associated virus-mediated survivin mutant C84A. Oncol. Res. 2013, 20, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.G.; Luo, R.Q.; Zhou, X.; Han, R.F.; Zeng, G.W. Potent antitumor activity of the combination of HSV-TK and endostatin by adeno-associated virus vector for bladder cancer in vivo. Clin. Lab. 2013, 59, 1147–1158. [Google Scholar] [PubMed]
- Pandya, J.; Ortiz, L.; Ling, C.; Rivers, A.E.; Aslanidi, G. Rationally designed capsid and transgene cassette of AAV6 vectors for dendritic cell-based cancer immunotherapy. Immunol. Cell Biol. 2014, 92, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.G.; Luo, R.Q.; Zhou, X.; Han, R.F.; Zeng, G.W. Suppression of bladder cancer growth by adeno-associated virus vector-mediated combination of HSV-TK and endostatin in vitro. Clin. Lab. 2013, 59, 1077–1089. [Google Scholar] [PubMed]
- Rajendran, S.; Collins, S.; van Pijkeren, J.P.; O’Hanlon, D.; O’Sullivan, G.C.; Tangney, M. Targeting of breast metastases using a viral gene vector with tumour-selective transcription. Anticancer Res. 2011, 31, 1627–1635. [Google Scholar] [PubMed]
- Kozłowska, A.; Mackiewicz, J.; Mackiewicz, A. Therapeutic gene modified cell based cancer vaccines. Gene 2013, 525, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lebron, E.; Denovan-Wright, E.M.; Nash, K.; Lewin, A.S.; Mandel, R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 618–633. [Google Scholar] [CrossRef]
- Ojano-Dirain, C.; Glushakova, L.G.; Zhong, L.; Zolotukhin, S.; Muzyczka, N.; Srivastava, A.; Stacpoole, P.W. An animal model of PDH deficiency using AAV8-siRNA vector-mediated knockdown of pyruvate dehydrogenase E1α. Mol. Genet. Metab. 2010, 101, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.; Justilien, V.; Liu, J.; Hauswirth, W.W.; Lewin, A.S. Suppression of mouse rhodopsin expression in vivo by AAV mediated siRNA delivery. Vis. Res. 2007, 47, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Hagemann, C.; Carta, F.; Katzer, A.; Polat, B.; Staab, A.; Scozzafava, A.; Anacker, J.; Vince, G.H.; Flentje, M.; et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg. Med. Chem. 2013, 21, 3949–3957. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; McDonald, P.C.; Lou, Y.; Serrano, I.; Chafe, S.C.; Ostlund, C.; Aparicio, S.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013, 32, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Radvak, P.; Repic, M.; Svastova, E.; Takacova, M.; Csaderova, L.; Strnad, H.; Pastorek, J.; Pastorekova, S.; Kopacek, J. Suppression of carbonic anhydrase IX leads to aberrant focal adhesion and decreased invasion of tumor cells. Oncol. Rep. 2013, 29, 1147–1153. [Google Scholar] [PubMed]
- Yu, S.; Yoon, J.; Lee, J.; Myung, S.; Jang, E.; Kwak, M.; Cho, E.; Jang, J.; Kim, Y.; Lee, H. Inhibition of hypoxia-inducible carbonic anhydrase-IX enhances hexokinase II inhibitor-induced hepatocellular carcinoma cell apoptosis. Acta Pharmacol. Sin. 2011, 32, 912–920. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, E.P. siRNAs and shRNAs: Tools for Protein Knockdown by Gene Silencing. Mater. Methods 2013, 3. [Google Scholar] [CrossRef]
- Lerch, T.F.; Xie, Q.; Chapman, M.S. The structure of adeno-associated virus serotype 3B (AAV-3B): Insights into receptor binding and immune evasion. Virology 2010, 403, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.S.; Sharma, A.; Kumari, R.; Mohammad, N.; Singh, S.V.; Bhat, M.K. Inherent and acquired resistance to paclitaxel in hepatocellular carcinoma: Molecular events involved. PLoS One 2013, 8, e61524. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int. J. Mol. Sci. 2013, 14, 24706–24725. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, T.; Lowndes, N.F.; Ceredig, R. Hypoxia enhances the radioresistance of mouse mesenchymal stromal cells. Stem Cells Dayt. Ohio 2014, 32, 2188–2200. [Google Scholar] [CrossRef]
- Wang, M.; Li, X.; Qu, Y.; Xu, O.; Sun, Q. Hypoxia promotes radioresistance of CD133-positive Hep-2 human laryngeal squamous carcinoma cells in vitro. Int. J. Oncol. 2013, 43, 131–140. [Google Scholar] [PubMed]
- Marampon, F.; Gravina, G.L.; Zani, B.M.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Di Genova, D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1α functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [PubMed]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noël, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef] [PubMed]
- Crowder, S.W.; Balikov, D.A.; Hwang, Y.S.; Sung, H.J. Cancer Stem Cells under Hypoxia as a Chemoresistance Factor in Breast and Brain. Curr. Pathobiol. Rep. 2014, 2, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [PubMed]
- Vordermark, D.; Kaffer, A.; Riedl, S.; Katzer, A.; Flentje, M. Characterization of carbonic anhydrase IX (CA IX) as an endogenous marker of chronic hypoxia in live human tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Ostheimer, C.; Bache, M.; Güttler, A.; Kotzsch, M.; Vordermark, D. A pilot study on potential plasma hypoxia markers in the radiotherapy of non-small cell lung cancer. Osteopontin, carbonic anhydrase IX and vascular endothelial growth factor. Strahlenther. Onkol. 2014, 190, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.J.; Jirstrom, K.; Kronblad, A.; Millikan, R.C.; Landberg, G.; Duffy, M.J.; Rydén, L.; Gallagher, W.M.; O’Brien, S.L. CA IX is an independent prognostic marker in premenopausal breast cancer patients with one to three positive lymph nodes and a putative marker of radiation resistance. Clin. Cancer Res. 2006, 12, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Anai, S.; Shiverick, K.; Medrano, T.; Nakamura, K.; Goodison, S.; Brown, B.D.; Rosser, C.J. Downregulation of BCL-2 induces downregulation of carbonic anhydrase IX, vascular endothelial growth factor, and pAkt and induces radiation sensitization. Urology 2007, 70, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.; Peeters, S.G.J.A.; van Kuijk, S.J.A.; Yaromina, A.; Lieuwes, N.G.; Saraya, R.; Biemans, R.; Rami, M.; Parvathaneni, N.K.; Vullo, D.; et al. Targeting carbonic anhydrase IX by nitroimidazole based sulfamides enhances the therapeutic effect of tumor irradiation: A new concept of dual targeting drugs. Radiother. Oncol. 2013, 108, 523–528. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahon, B.P.; Pinard, M.A.; McKenna, R. Targeting Carbonic Anhydrase IX Activity and Expression. Molecules 2015, 20, 2323-2348. https://doi.org/10.3390/molecules20022323
Mahon BP, Pinard MA, McKenna R. Targeting Carbonic Anhydrase IX Activity and Expression. Molecules. 2015; 20(2):2323-2348. https://doi.org/10.3390/molecules20022323
Chicago/Turabian StyleMahon, Brian P., Melissa A. Pinard, and Robert McKenna. 2015. "Targeting Carbonic Anhydrase IX Activity and Expression" Molecules 20, no. 2: 2323-2348. https://doi.org/10.3390/molecules20022323