
Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
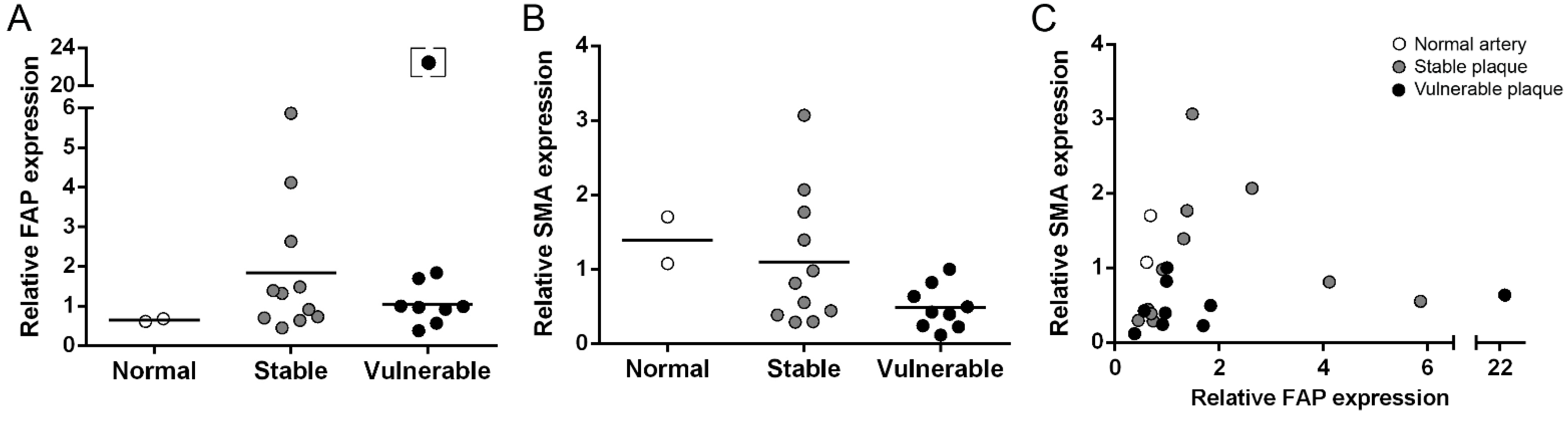
2.1. Gene Expression Analysis of FAP and SMA in Human Carotid Plaques
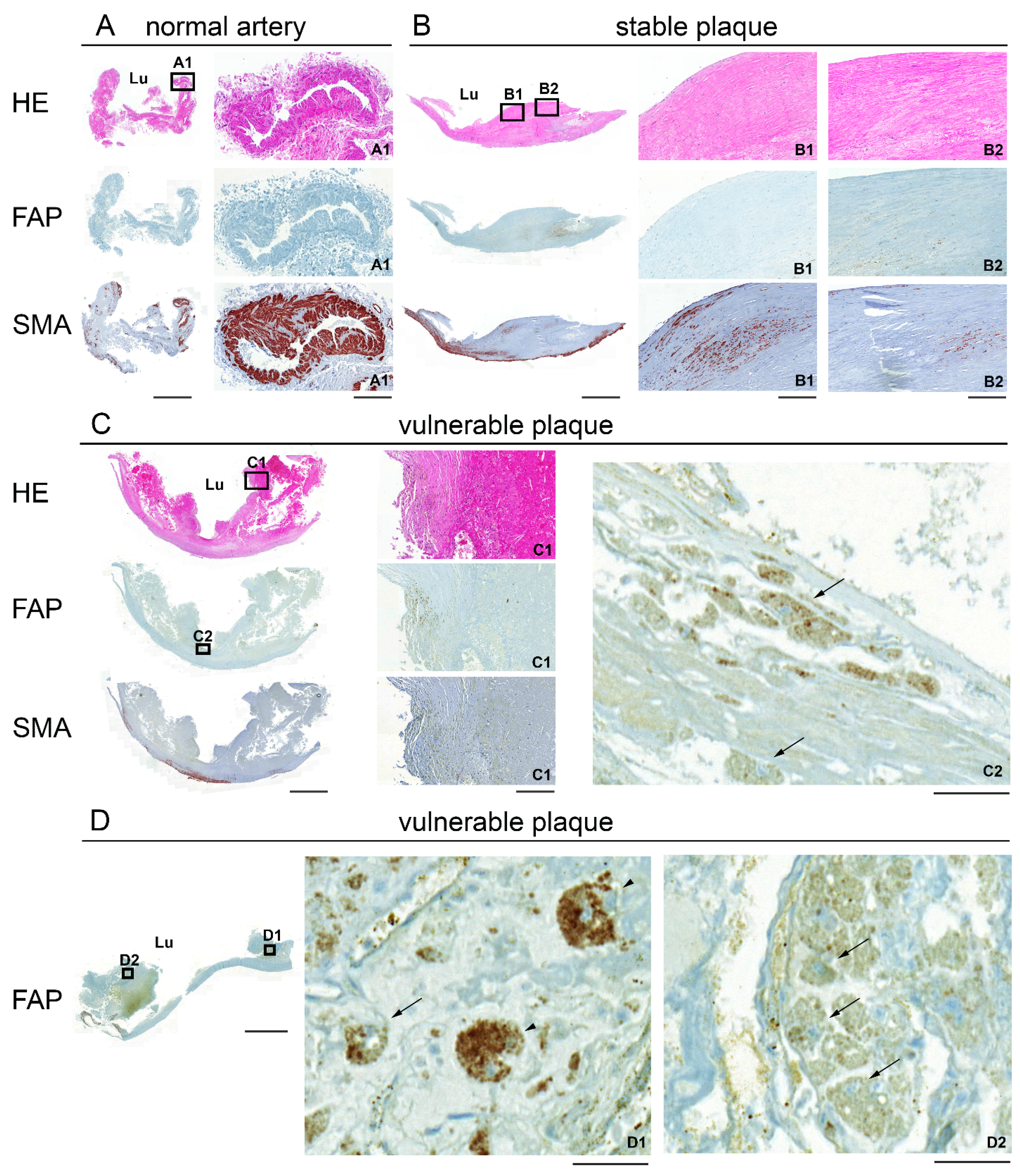
2.2. Immunohistochemical Staining of Human Carotid Plaques for FAP and SMA
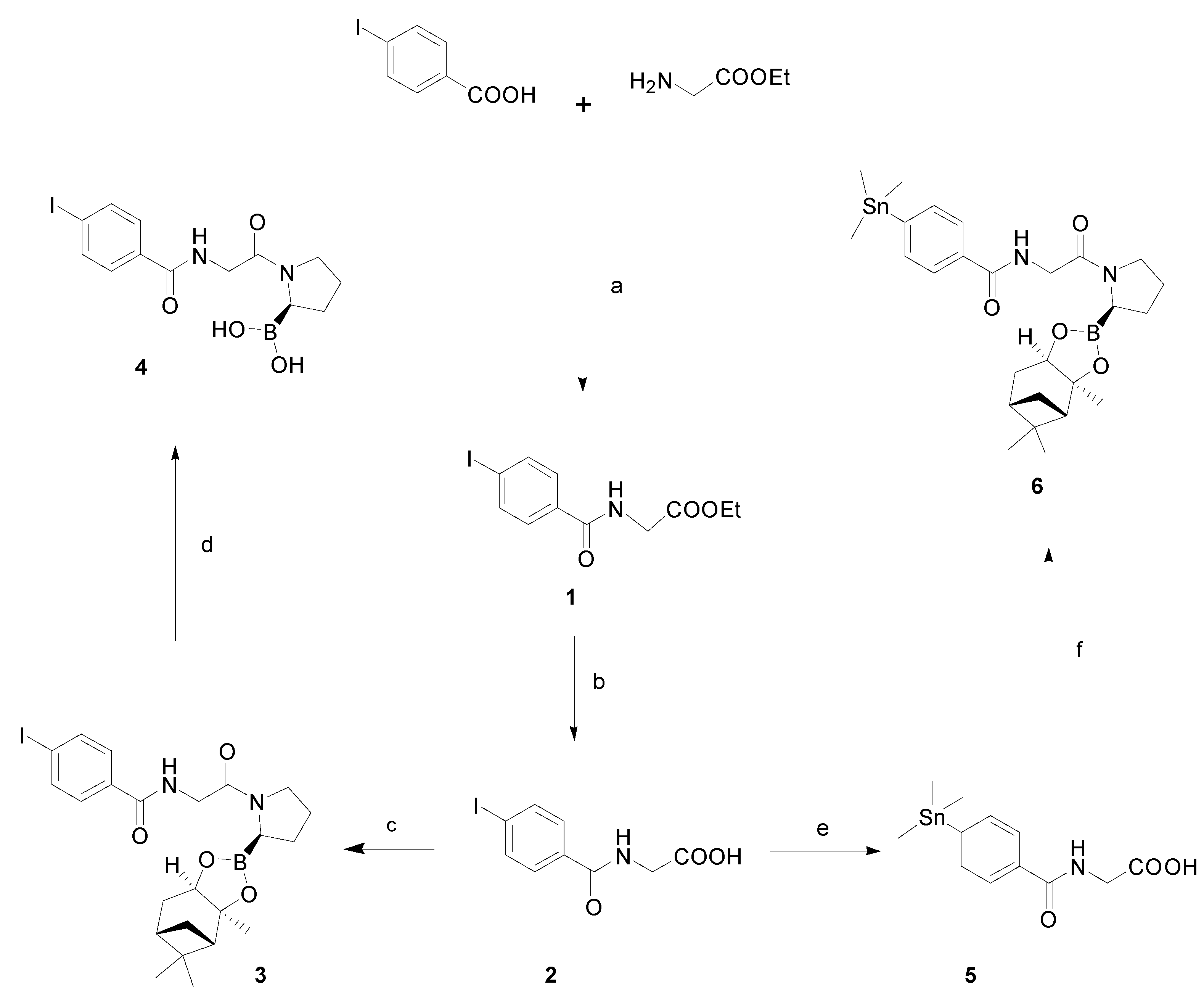
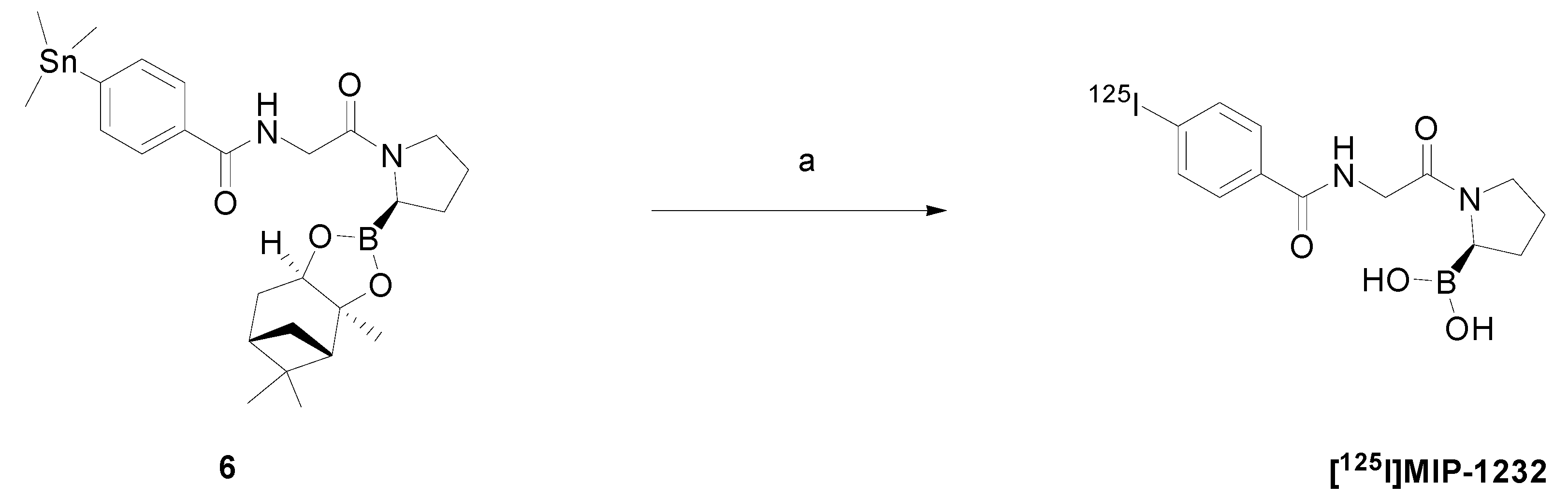
2.3. Chemistry and Radiochemistry
2.4. In Vitro Autoradiography
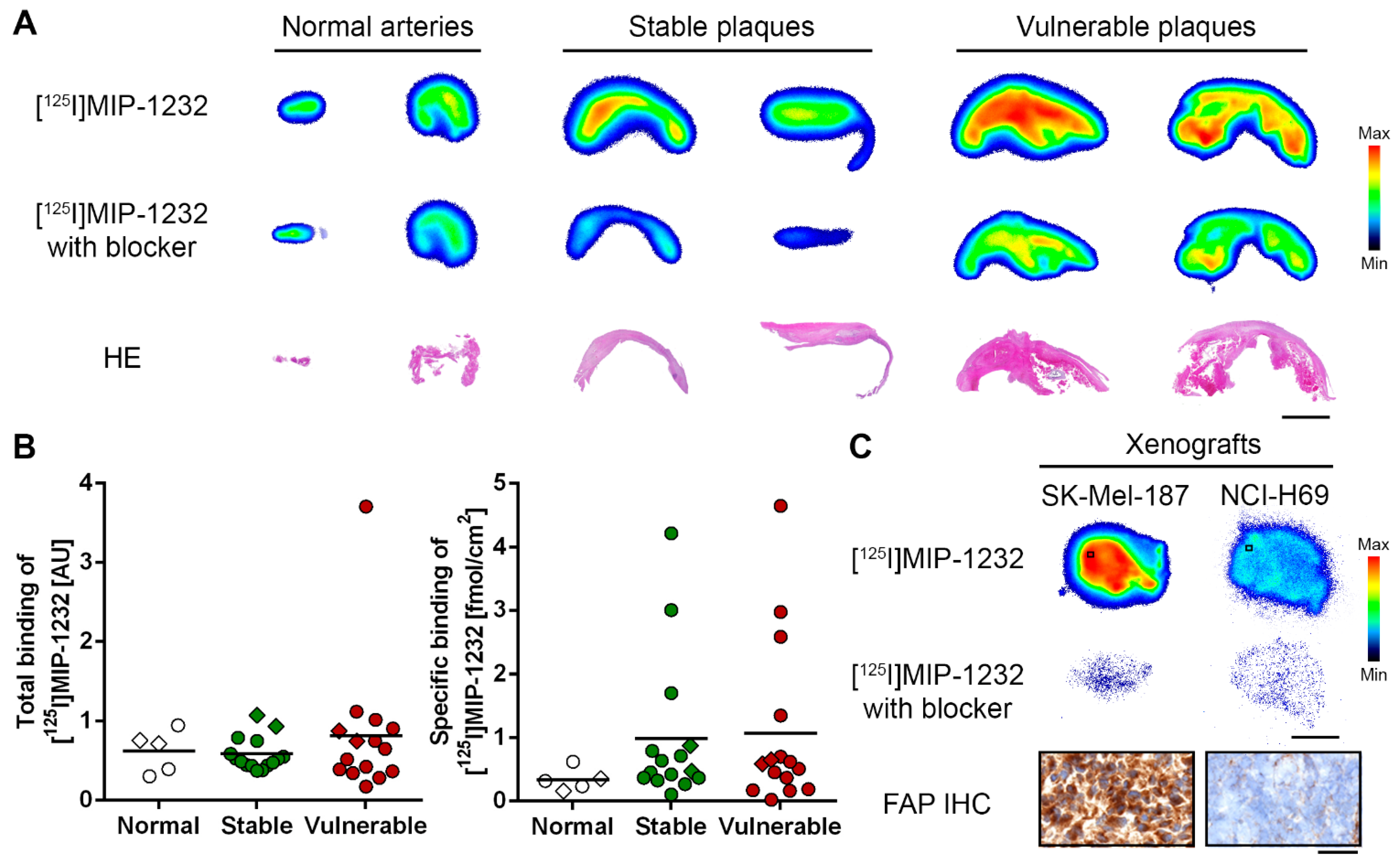
2.5. Discussion
3. Experimental Section
3.1. Patient Characteristics and Human Carotid Tissue Banking
3.2. RNA Isolation, Reverse-Transcription and Real-Time Polymerase Chain Reaction
3.3. Histology and Immunohistochemistry
3.4. Chemicals and Reagents
3.5. Chemistry
3.6. Radiochemistry
3.7. In Vitro Autoradiography
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ylä-Herttuala, S.; Bentzon, J.F.; Daemen, M.; Falk, E.; Garcia-Garcia, H.M.; Herrmann, J.; Hoefer, I.; Jukema, J.W.; Krams, R.; Kwak, B.R.; et al. Stabilisation of atherosclerotic plaques. Position paper of the european society of cardiology (ESC) working group on atherosclerosis and vascular biology. Thromb. Haemost. 2011, 106, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Rimoldi, O.E.; Gaemperli, O.; Libby, P. Non-invasive anatomic and functional imaging of vascular inflammation and unstable plaque. Eur. Heart J. 2012, 33, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.; Millon, A.; Fayad, Z.A. Molecular imaging in atherosclerosis: FDG PET. Curr. Atheroscler. Rep. 2012, 14, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, A.C.; Becker, A.E.; van der Loos, C.M.; Das, P.K. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Lutgens, S.P.; Cleutjens, K.B.; Daemen, M.J.; Heeneman, S. Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 2007, 21, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C.; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; et al. Fibroblast activation protein is induced by inflammation and degrades type i collagen in thin-cap fibroatheromata. Eur Heart J. 2011, 32, 2713–2722. [Google Scholar] [CrossRef]
- Müller, A.; Krämer, S.D.; Meletta, R.; Beck, K.; Selivanova, S.V.; Rancic, Z.; Kaufmann, P.A.; Vos, B.; Meding, J.; Stellfeld, T.; et al. Gene expression levels of matrix metalloproteinases in human atherosclerotic plaques and evaluation of radiolabeled inhibitors as imaging agents for plaque vulnerability. Nucl. Med. Biol. 2014, 41, 562–569. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Raj, B.K.; Calvo, B.; Garin-Chesa, P.; Sanz-Moncasi, M.P.; Healey, J.H.; Old, L.J.; Rettig, W.J. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. USA 1994, 91, 5657–5661. [Google Scholar] [CrossRef] [PubMed]
- Pineiro-Sanchez, M.L.; Goldstein, L.A.; Dodt, J.; Howard, L.; Yeh, Y.; Tran, H.; Argraves, W.S.; Chen, W.T. Identification of the 170-kda melanoma membrane-bound gelatinase (seprase) as a serine integral membrane protease. J. Biol. Chem. 1997, 272, 7595–7601. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.A.; Ghersi, G.; Pineiro-Sanchez, M.L.; Salamone, M.; Yeh, Y.; Flessate, D.; Chen, W.T. Molecular cloning of seprase: A serine integral membrane protease from human melanoma. Biochim. Biophys. Acta 1997, 1361, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Edosada, C.Y.; Quan, C.; Tran, T.; Pham, V.; Wiesmann, C.; Fairbrother, W.; Wolf, B.B. Peptide substrate profiling defines fibroblast activation protein as an endopeptidase of strict gly(2)-pro(1)-cleaving specificity. FEBS Lett. 2006, 580, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.T.; McCaughan, G.W.; Abbott, C.A.; Park, J.E.; Cunningham, A.M.; Müller, E.; Rettig, W.J.; Gorrell, M.D. Fibroblast activation protein: A cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology 1999, 29, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, V.J.; Jackson, K.W.; Lee, K.N.; McKee, P.A. Effect of fibroblast activation protein and alpha2-antiplasmin cleaving enzyme on collagen types i, iii, and iv. Arch. Biochem. Biophys. 2007, 457, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, G.; Dong, H.; Goldstein, L.A.; Yeh, Y.; Hakkinen, L.; Larjava, H.S.; Chen, W.T. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J. Biol. Chem. 2002, 277, 29231–29241. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Albright, C.F.; Fish, B.H.; George, H.J.; Selling, B.H.; Hollis, G.F.; Wynn, R. Expression, purification, and kinetic characterization of full-length human fibroblast activation protein. Protein Expr. Purif. 2002, 24, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.D.; Dunbrack, R.L., Jr.; Valianou, M.; Rogatko, A.; Alpaugh, R.K.; Weiner, L.M. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002, 62, 4767–4772. [Google Scholar] [PubMed]
- Huber, M.A.; Kraut, N.; Park, J.E.; Schubert, R.D.; Rettig, W.J.; Peter, R.U.; Garin-Chesa, P. Fibroblast activation protein: Differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Investig. Dermatol. 2003, 120, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Eager, R.M.; Cunningham, C.C.; Senzer, N.; Richards, D.A.; Raju, R.N.; Jones, B.; Uprichard, M.; Nemunaitis, J. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin. Oncol. (R. Coll. Radiol.) 2009, 21, 464–472. [Google Scholar] [CrossRef]
- Eager, R.M.; Cunningham, C.C.; Senzer, N.N.; Stephenson, J., Jr.; Anthony, S.P.; O’Day, S.J.; Frenette, G.; Pavlick, A.C.; Jones, B.; Uprichard, M.; et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer 2009, 9. [Google Scholar] [CrossRef]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase ii trial of single agent val-boropro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.M.; de Meester, I.; Augustyns, K.; et al. Selective inhibitors of fibroblast activation protein (fap) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Hu, Y.; Ma, L.; Wu, M.; Wong, M.S.; Li, B.; Corral, S.; Yu, Z.; Nomanbhoy, T.; Alemayehu, S.; Fuller, S.R.; et al. Synthesis and structure-activity relationship of N-alkyl gly-boro-pro inhibitors of dpp4, fap, and dpp7. Bioorganic Med. Chem. Lett. 2005, 15, 4239–4242. [Google Scholar] [CrossRef]
- Tran, T.; Quan, C.; Edosada, C.Y.; Mayeda, M.; Wiesmann, C.; Sutherlin, D.; Wolf, B.B. Synthesis and structure-activity relationship of N -acyl-gly-, N-acyl-sar- and N-blocked-boropro inhibitors of FAP, DPP4, and POP. Bioorganic Med. Chem. Lett. 2007, 17, 1438–1442. [Google Scholar] [CrossRef]
- Edosada, C.Y.; Quan, C.; Wiesmann, C.; Tran, T.; Sutherlin, D.; Reynolds, M.; Elliott, J.M.; Raab, H.; Fairbrother, W.; Wolf, B.B. Selective inhibition of fibroblast activation protein protease based on dipeptide substrate specificity. J. Biol. Chem. 2006, 281, 7437–7444. [Google Scholar] [CrossRef] [PubMed]
- Poplawski, S.E.; Lai, J.H.; Li, Y.; Jin, Z.; Liu, Y.; Wu, W.; Wu, Y.; Zhou, Y.; Sudmeier, J.L.; Sanford, D.G.; et al. Identification of selective and potent inhibitors of fibroblast activation protein and prolyl oligopeptidase. J. Med. Chem. 2013, 56, 3467–3477. [Google Scholar] [CrossRef]
- Marquis, J.; Wang, J.; Maresca, K.; Hillier, S.; Zimmermann, C.; Joyal, J.; Babich, J. Targeting tumor microenvironment with radiolabeled inhibitors of seprase (FAPa). In Proceedings of the 100th Annual Meeting of the American Association for Cancer Research, Denver, CO, USA, 18–22 April 2009; AACR: Philadelphia, PA; USA, 2009. Abstract number: 4467. [Google Scholar]
- Milo, L.J., Jr.; Lai, J.H.; Wu, W.; Liu, Y.; Maw, H.; Li, Y.; Jin, Z.; Shu, Y.; Poplawski, S.E.; Wu, Y.; et al. Chemical and biological evaluation of dipeptidyl boronic acid proteasome inhibitors for use in prodrugs and pro-soft drugs targeting solid tumors. J. Med. Chem. 2011, 54, 4365–4377. [Google Scholar] [CrossRef]
- Zimmerman, C.; Babich, J.W.; Joyal, J.; Marquis, J.; Wang, J. Selective Seprase Inhibitors. Patent US 2010/0098633 A1, 22 April 2010. [Google Scholar]
- Wilbur, D.S.; Chyan, M.K.; Hamlin, D.K.; Kegley, B.B.; Risler, R.; Pathare, P.M.; Quinn, J.; Vessella, R.L.; Foulon, C.; Zalutsky, M.; et al. Reagents for astatination of biomolecules: Comparison of the in vivo distribution and stability of some radioiodinated/astatinated benzamidyl and nido-carboranyl compounds. Bioconjugate Chem. 2004, 15, 203–223. [Google Scholar] [CrossRef]
- Coutts, S.J.; Adams, J.; Krolikowski, D.; Snow, R.J. Two efficient methods for the cleavage of pinanediol boronate esters yielding the free boronic acids. Tetrahedron Lett. 1994, 35, 5109–5112. [Google Scholar] [CrossRef]
- Fischer, E.; Chaitanya, K.; Wuest, T.; Wadle, A.; Scott, A.M.; van den Broek, M.; Schibli, R.; Bauer, S.; Renner, C. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin. Cancer Res. 2012, 18, 6208–6218. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Eisenblatter, M.; Voller, T.; Zenker, S.; Hermann, S.; van Lent, P.; Faust, A.; Geyer, C.; Petersen, B.; Roebrock, K.; et al. Alarmin s100a8/s100a9 as a biomarker for molecular imaging of local inflammatory activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Dimastromatteo, J.; Broisat, A.; Perret, P.; Ahmadi, M.; Boturyn, D.; Dumy, P.; Fagret, D.; Riou, L.M.; Ghezzi, C. In vivo molecular imaging of atherosclerotic lesions in apoe-/- mice using vcam-1-specific, 99mtc-labeled peptidic sequences. J. Nucl. Med. 2013, 54, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Gaemperli, O.; Shalhoub, J.; Owen, D.R.; Lamare, F.; Johansson, S.; Fouladi, N.; Davies, A.H.; Rimoldi, O.E.; Camici, P.G. Imaging intraplaque inflammation in carotid atherosclerosis with 11C-PK11195 positron emission tomography/computed tomography. Eur. Heart J. 2012, 33, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Mu, L.; Meletta, R.; Beck, K.; Rancic, Z.; Drandarov, K.; Kaufmann, P.A.; Ametamey, S.M.; Schibli, R.; Borel, N.; et al. Towards non-invasive imaging of vulnerable atherosclerotic plaques by targeting co-stimulatory molecules. Int. J. Cardiol. 2014, 174, 503–515. [Google Scholar] [CrossRef]
- Müller, A.; Beck, K.; Rancic, Z.; Müller, C.; Fischer, C.R.; Betzel, T.; Kaufmann, P.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Imaging atherosclerotic plaque inflammation via folate receptor targeting using a novel 18F-folate radiotracer. Mol. Imaging 2014, 13, 1–11. [Google Scholar] [PubMed]
- Katsuda, S.; Kaji, T. Atherosclerosis and extracellular matrix. J. Atheroscler. Thromb. 2003, 10, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Herrera, A.; Dominguez, G.; Silva, J.; Garcia, V.; Garcia, J.M.; Gomez, I.; Soldevilla, B.; Munoz, C.; Provencio, M.; et al. Cancer-associated fibroblast and m2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013, 104, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-alpha and heme oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Levin, I.; Shi, L.; Snell, G.P.; Jennings, A.; Prasad, G.S.; Zhang, Y.; Kraus, M.L.; Salakian, S.; Sridhar, V.; et al. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J. Biol. Chem. 2005, 280, 19441–19444. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Extended structure-activity relationship and pharmacokinetic investigation of (4-quinolinoyl)glycyl-2-cyanopyrrolidine inhibitors of fibroblast activation protein (fap). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.R.; Lee, H.O.; Lee, J.S.; Klein-Szanto, A.; Watts, P.; Ross, E.A.; Chen, W.T.; Cheng, J.D. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 2007, 13, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, H.; Liu, L.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Nett, P.C.; Zund, G.; Pretre, R.; Niederhouser, U.; Vogt, P.R.; Turina, M. A 20-year follow-up of internal carotid artery endarterectomy with bifurcation advancement. Thorac. Cardiovasc. Surg. 2000, 48, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, J.N.; Gallagher, P.; Lovett, J.K.; Rothwell, P.M. Critical cap thickness and rupture in symptomatic carotid plaques: The oxford plaque study. Stroke 2008, 39, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meletta, R.; Müller Herde, A.; Chiotellis, A.; Isa, M.; Rancic, Z.; Borel, N.; Ametamey, S.M.; Krämer, S.D.; Schibli, R. Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging. Molecules 2015, 20, 2081-2099. https://doi.org/10.3390/molecules20022081
Meletta R, Müller Herde A, Chiotellis A, Isa M, Rancic Z, Borel N, Ametamey SM, Krämer SD, Schibli R. Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging. Molecules. 2015; 20(2):2081-2099. https://doi.org/10.3390/molecules20022081
Chicago/Turabian StyleMeletta, Romana, Adrienne Müller Herde, Aristeidis Chiotellis, Malsor Isa, Zoran Rancic, Nicole Borel, Simon M. Ametamey, Stefanie D. Krämer, and Roger Schibli. 2015. "Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging" Molecules 20, no. 2: 2081-2099. https://doi.org/10.3390/molecules20022081