
Development and Validation of 697 Novel Polymorphic Genomic and EST-SSR Markers in the American Cranberry (Vaccinium macrocarpon Ait.)

Abstract
:1. Introduction
2. Results and Discussion
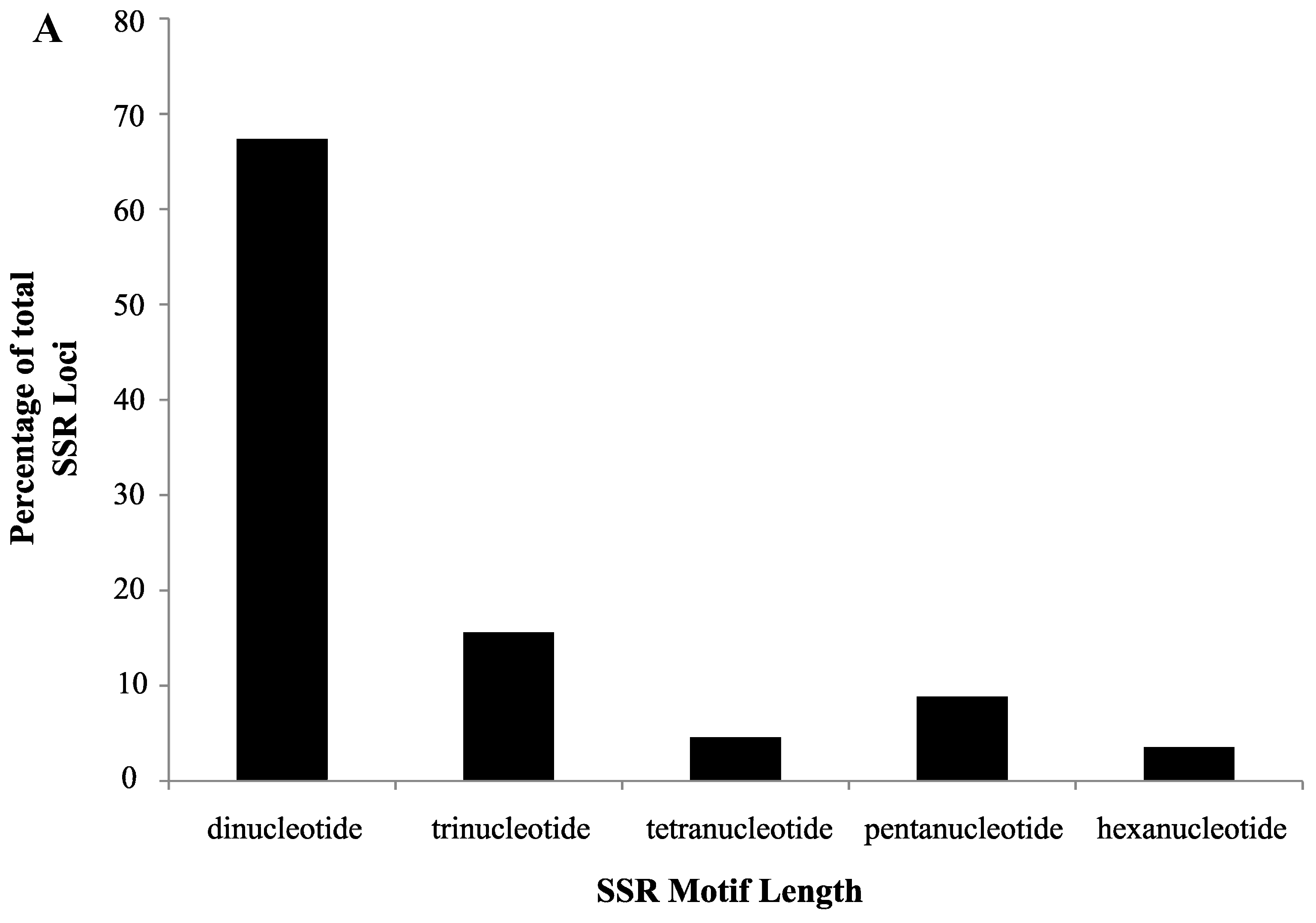
2.1. SSR Search and Primer Design
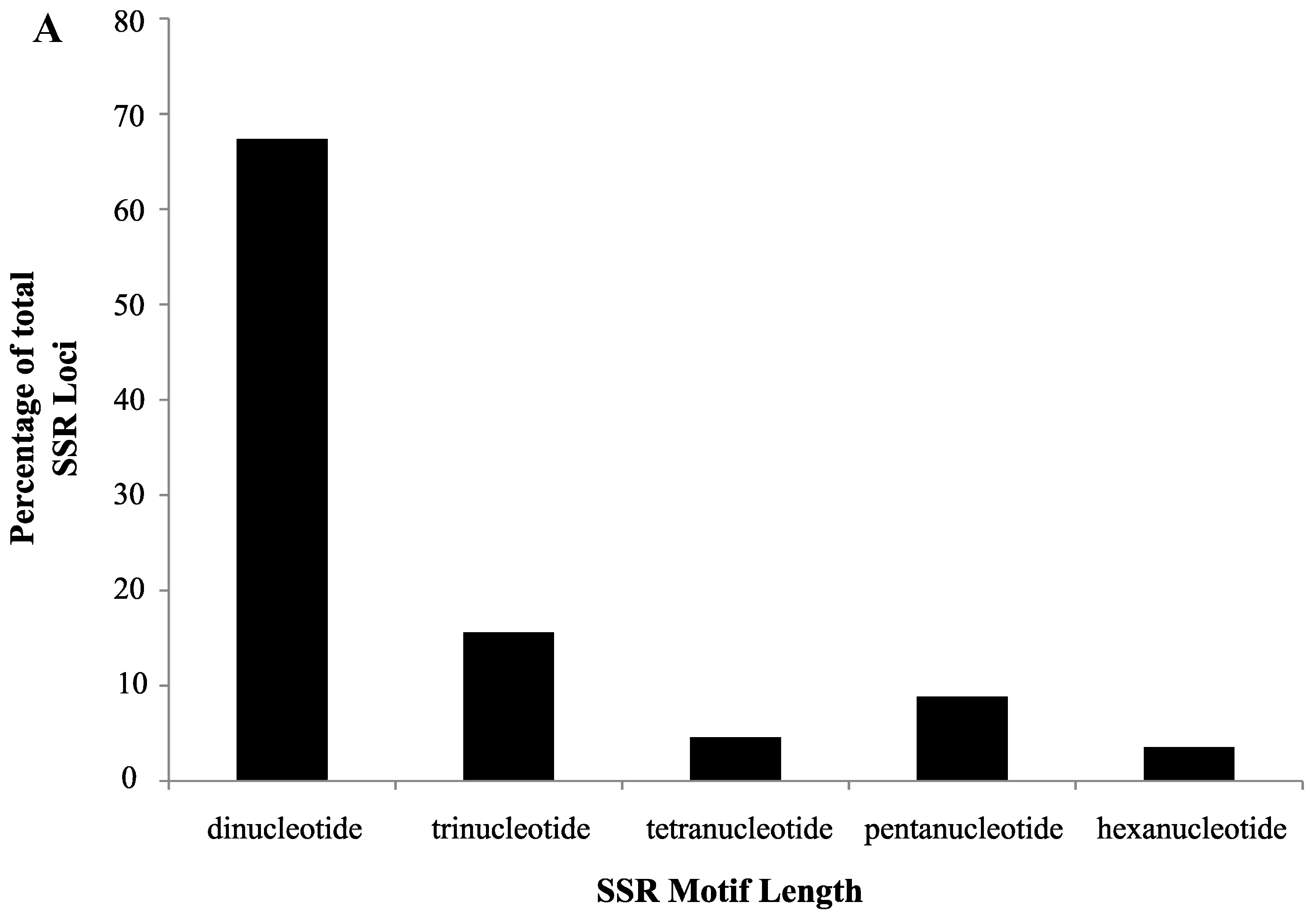
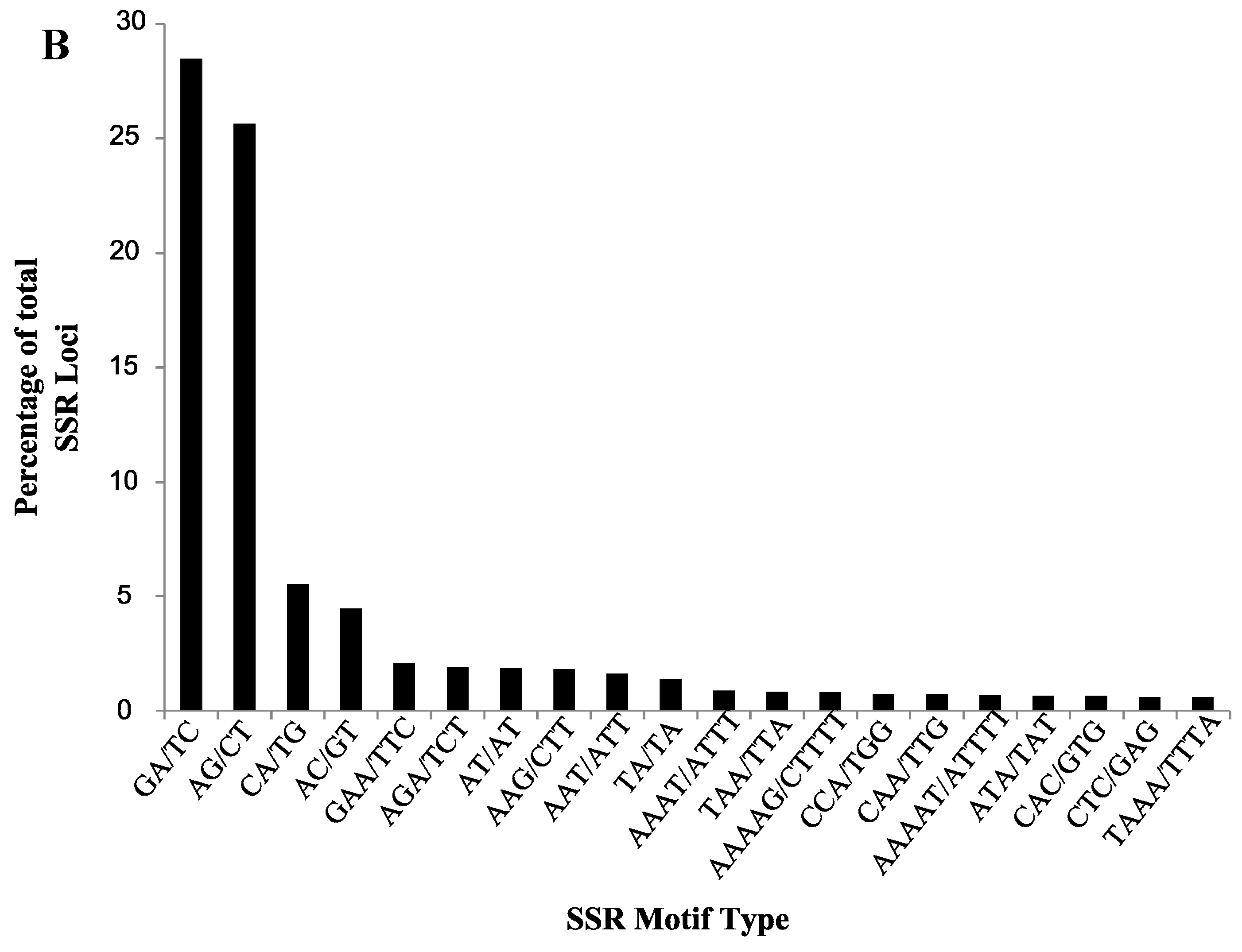
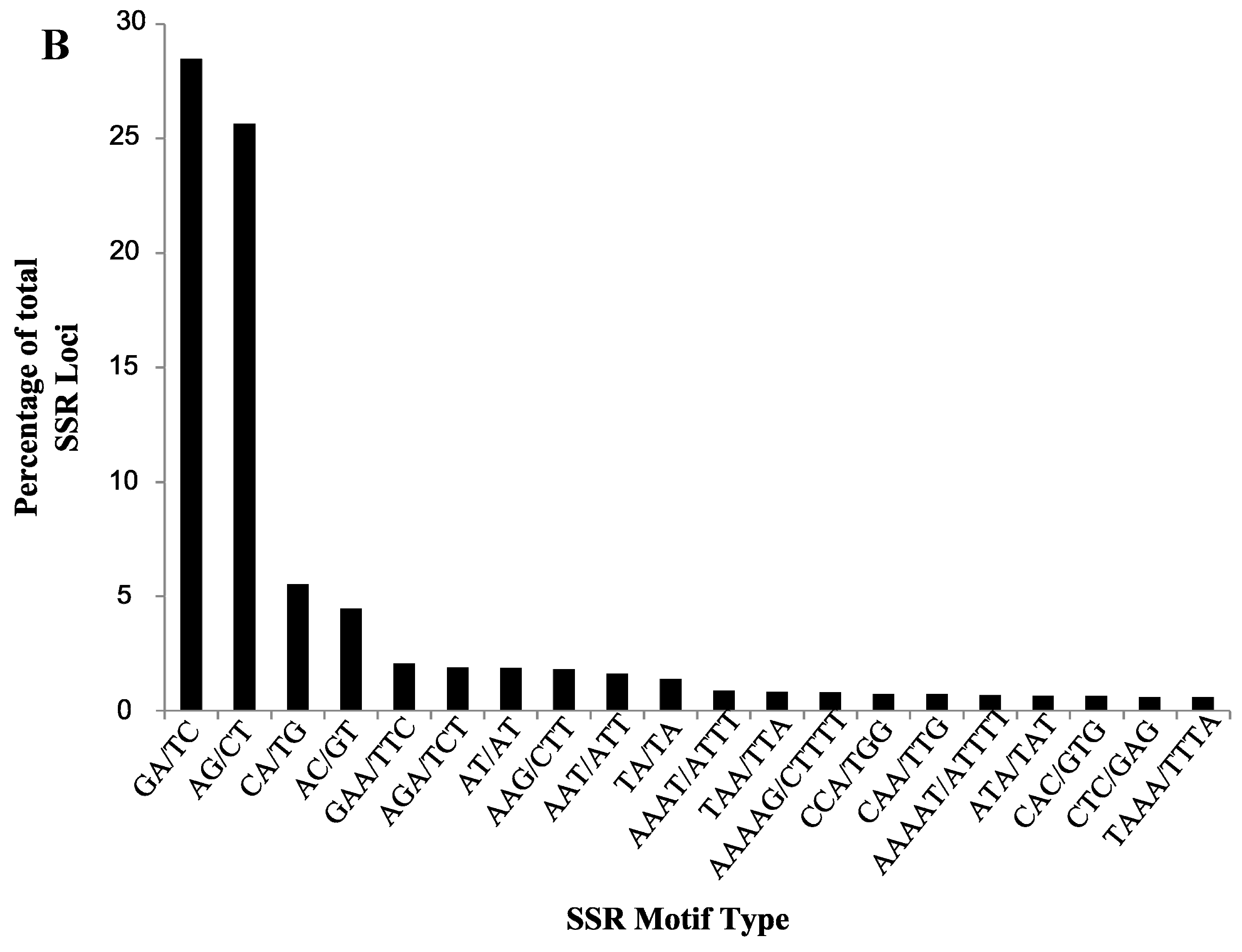
2.2. Validation of Cranberry SSR Loci

{kind=link}
{kind=link}
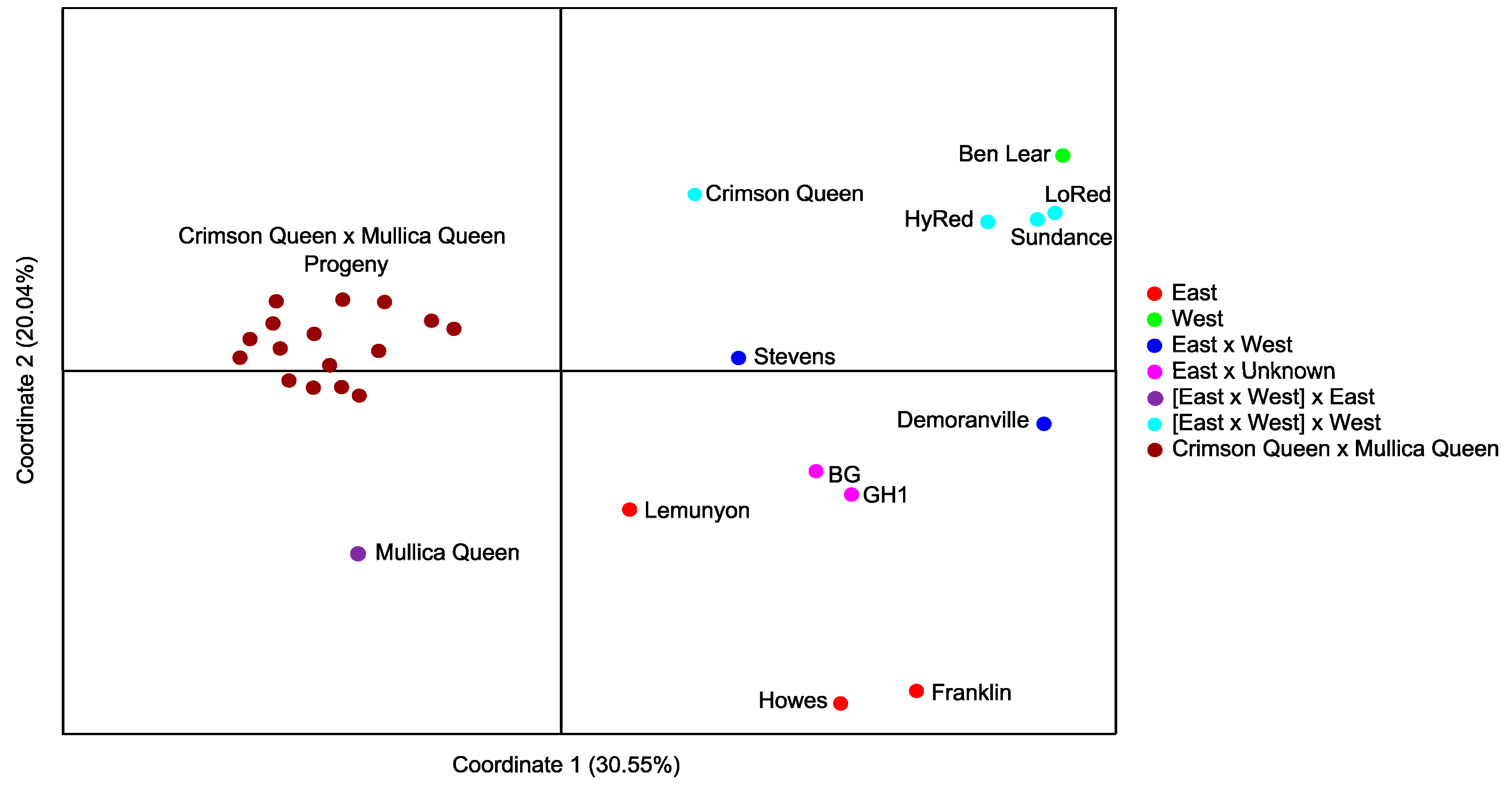{kind=link}
| Status | Name | Source | Pedigree | Release Date |
|---|---|---|---|---|
| Cultivar | Stevens | NCGR (PI 614078) | McFarlin * × Potters Favorite * | 1950 |
| Cultivar | GH1 | E. Grygleski, Wisconsin | McFarlin * × Searles * | 2004 |
| Cultivar | Ben Lear * | NCGR (PI 554983) | Native Selection, WI, USA | 1901 |
| Cultivar | Crimson Queen | Rutgers University | Stevens × Ben Lear * | 2006 |
| Cultivar | Demoranville | Rutgers University | Franklin × Ben Lear * | 2006 |
| Cultivar | Franklin | NCGR (PI 554998) | Early Black * × Howes * | 1950 |
| Cultivar | Howes * | NCGR (PI 614076) | Native Selection, MA, USA | 1843 |
| Cultivar | Lemunyon * | NCGR (PI 554985) | Native Selection, NJ, USA | 1960 |
| Cultivar | Sundance | UW-Madison | Stevens × Ben Lear * | 2011 |
| Cultivar | HyRed | UW-Madison | Stevens × Ben Lear * | 2003 |
| Cultivar | LoRed | UW-Madison | Stevens × Ben Lear * | Unreleased |
| Cultivar | Mullica Queen | Rutgers University | (Howes * × Searles *) × LeMunyon * | 2007 |
| Cultivar | BG | E. Grygleski, Wisconsin | Beckwith × GH1 | 2012 |
| Progeny | CNJ02-1-159 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-160 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-161 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-162 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-163 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-164 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-165 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-166 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-168 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-169 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-170 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-173 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-174 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-175 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-176 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
| Progeny | CNJ02-1-178 | Rutgers University | Mullica Queen × Crimson Queen | Unreleased |
2.3. Genetic Diversity and Segregation Analyses
| Motif Length | Number of Loci | Total Na | Average NA | Average NE | Average PIC | Average HO | Average HE |
|---|---|---|---|---|---|---|---|
| Dinucleotide | 427 | 1983 | 4.64 | 3.24 | 0.59 | 0.72 | 0.63 |
| Trinucleotide | 72 | 272 | 3.78 | 2.87 | 0.51 | 0.67 | 0.58 |
| Tetranucleotide | 1 | 5 | 5.00 | 2.27 | 0.51 | 0.62 | 0.56 |
| Pentanucleotide | 7 | 18 | 2.57 | 1.85 | 0.37 | 0.66 | 0.45 |
| Total | 507 | 2278 | 4.49 | 3.17 | 0.57 | 0.72 | 0.62 |
| Motif Type | Number of Loci | Total NA | Total NE | Average NA | Average NE | Average PIC | Average HO | Average HE |
|---|---|---|---|---|---|---|---|---|
| AC/GT | 30 | 130 | 92.76 | 4.33 | 3.09 | 0.57 | 0.71 | 0.60 |
| AG/CT | 141 | 650 | 456.31 | 4.61 | 3.24 | 0.59 | 0.74 | 0.64 |
| AT/AT | 10 | 53 | 38.54 | 5.30 | 3.85 | 0.67 | 0.61 | 0.71 |
| CA/TG | 38 | 142 | 93.89 | 3.74 | 2.47 | 0.46 | 0.60 | 0.52 |
| GA/TC | 202 | 979 | 680.26 | 4.85 | 3.37 | 0.60 | 0.75 | 0.65 |
| TA/TA | 6 | 29 | 23.13 | 4.83 | 3.86 | 0.66 | 0.73 | 0.71 |
| AAC/TTG | 2 | 4 | 3.73 | 2.00 | 1.87 | 0.35 | 0.54 | 0.46 |
| AAG/CTT | 8 | 39 | 27.03 | 4.88 | 3.38 | 0.62 | 0.71 | 0.66 |
| AAT/ATT | 6 | 24 | 18.05 | 4.00 | 3.01 | 0.54 | 0.66 | 0.58 |
| ACA/TGT | 1 | 2 | 1.90 | 2.00 | 1.90 | 0.36 | 0.46 | 0.47 |
| AGA/TCT | 12 | 50 | 42.58 | 4.17 | 3.55 | 0.58 | 0.80 | 0.65 |
| AGC/GCT | 2 | 6 | 3.99 | 3.00 | 2.00 | 0.41 | 0.64 | 0.47 |
| AGG/CCT | 1 | 2 | 1.99 | 2.00 | 1.99 | 0.37 | 0.77 | 0.50 |
| CAA/TTG | 5 | 14 | 10.46 | 2.80 | 2.09 | 0.40 | 0.66 | 0.48 |
| CAG/CTG | 1 | 4 | 2.25 | 4.00 | 2.25 | 0.51 | 0.62 | 0.56 |
| ATG/CAT | 2 | 4 | 3.95 | 2.00 | 1.98 | 0.37 | 0.85 | 0.49 |
| CCA/TGG | 3 | 8 | 5.52 | 2.67 | 1.84 | 0.36 | 0.43 | 0.42 |
| CTA/TAG | 1 | 2 | 2.00 | 2.00 | 2.00 | 0.38 | 1.00 | 0.50 |
| GAA/TTC | 9 | 41 | 30.30 | 4.56 | 3.37 | 0.55 | 0.57 | 0.60 |
| CTC/GAG | 1 | 3 | 2.77 | 3.00 | 2.77 | 0.57 | 0.92 | 0.64 |
| ATC/GAT | 2 | 6 | 5.00 | 3.00 | 2.50 | 0.51 | 0.85 | 0.60 |
| GCA/TGC | 3 | 9 | 6.24 | 3.00 | 2.08 | 0.43 | 0.54 | 0.51 |
| GTA/TAC | 2 | 10 | 8.20 | 5.00 | 4.10 | 0.72 | 0.85 | 0.76 |
| CAC/GTG | 1 | 2 | 1.74 | 2.00 | 1.74 | 0.34 | 0.31 | 0.43 |
| TCA/TGA | 2 | 6 | 4.35 | 3.00 | 2.17 | 0.46 | 0.65 | 0.54 |
| GGA/TCC | 3 | 6 | 5.54 | 2.00 | 1.85 | 0.35 | 0.53 | 0.45 |
| TCG/CGA | 1 | 3 | 1.89 | 3.00 | 1.89 | 0.42 | 0.62 | 0.47 |
| TAA/TTA | 4 | 27 | 17.41 | 6.75 | 4.35 | 0.65 | 0.65 | 0.74 |
| TATG/CATA | 1 | 5 | 2.27 | 5.00 | 2.27 | 0.51 | 0.62 | 0.56 |
| AAAAT/ATTTTT | 1 | 3 | 1.75 | 3.00 | 1.75 | 0.39 | 0.54 | 0.43 |
| AAACA/TGTTT | 1 | 3 | 1.48 | 3.00 | 1.48 | 0.29 | 0.38 | 0.32 |
| AAACT/AGTTT | 1 | 2 | 1.95 | 2.00 | 1.95 | 0.37 | 0.69 | 0.49 |
| CACCT/AGGTG | 1 | 3 | 1.70 | 3.00 | 1.70 | 0.35 | 0.38 | 0.41 |
| GTTTG/CAAAC | 1 | 2 | 2.00 | 2.00 | 2.00 | 0.38 | 1.00 | 0.50 |
| TTGGT/ACCAA | 1 | 3 | 2.05 | 3.00 | 2.05 | 0.46 | 0.62 | 0.51 |
| TTTTA/TAAAA | 1 | 2 | 2.00 | 2.00 | 2.00 | 0.38 | 1.00 | 0.50 |
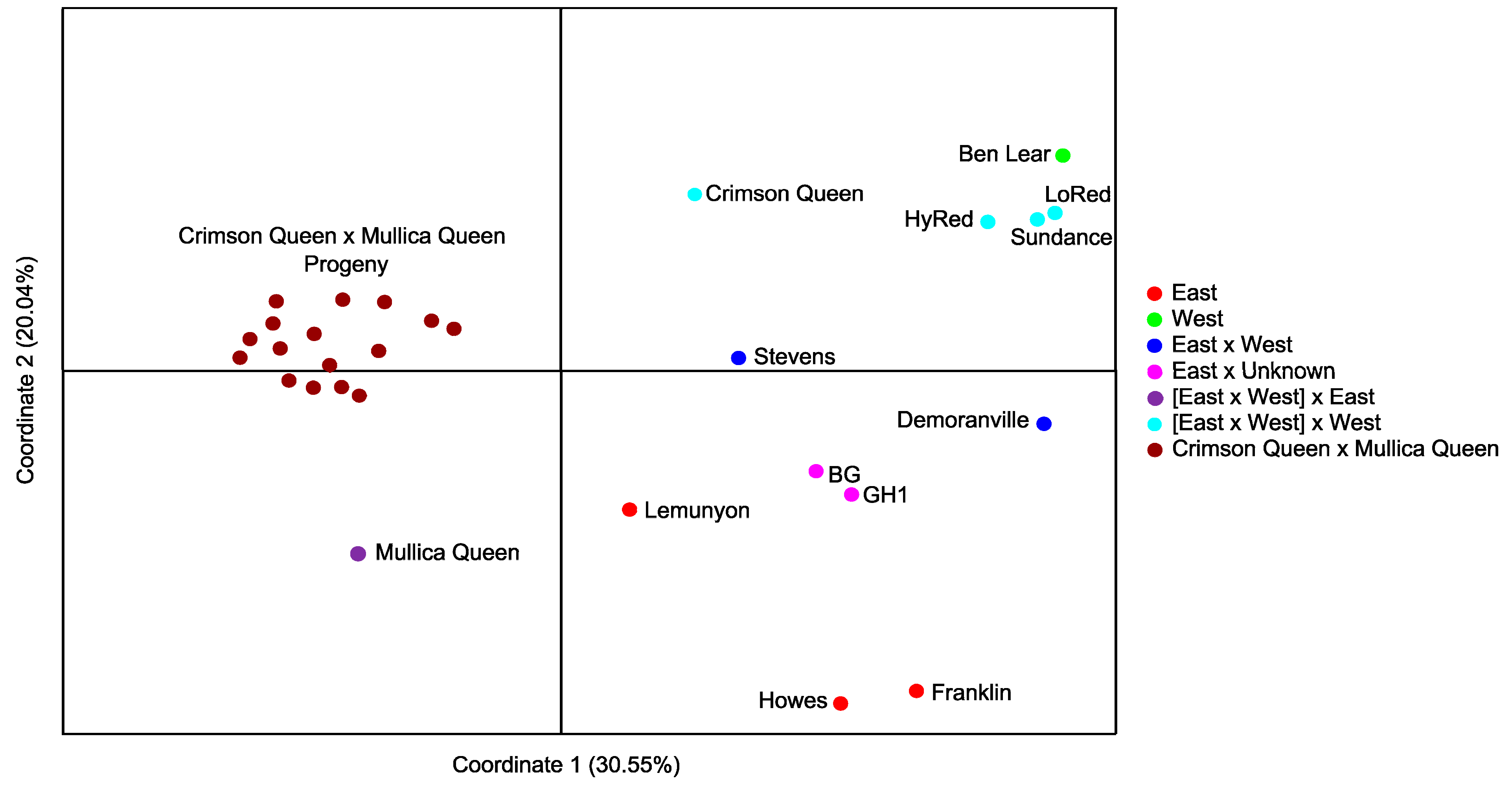
3. Experimental Section
3.1. Plant Material
3.2. Detection of Genomic and EST-SSR Markers
3.3. PCR Amplification and Fragment Analysis
3.4. Validation of SSR Polymorphism and Genetic Diversity Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kron, K.A.; Judd, W.S.; Stevens, P.F.; Crayn, D.M.; Anderberg, A.A.; Gadek, P.A.; Quinn, C.J.; Luteyn, J.L. Phylogenetic Classification of Ericaceae: Molecular and Morphological Evidence. Bot. Rev. 2002, 68, 335–423. [Google Scholar] [CrossRef]
- Fajardo, D.; Senalik, D.; Ames, M.; Zhu, H.; Steffan, S.A.; Harbut, R.; Polashock, J.; Vorsa, N.; Gillespie, E.; Kron, K.; et al. Complete plastid genome sequence of Vaccinium macrocarpon: Structure, gene content, and rearrangements revealed by next generation sequencing. Tree Genet. Genomes 2012, 9, 489–498. [Google Scholar] [CrossRef]
- Seeram, N.P. Berry fruits: Compositional elements, biochemical activities, and the impact of their intake on human health, performance, and disease. J. Agric. Food Chem. 2008, 56, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Feghali, K.; Feldman, M.; La, V.D.; Santos, J.; Grenier, D. Cranberry Proanthocyanidins: Natural Weapons against Periodontal Diseases. J. Agric. Food Chem. 2012, 60, 5728–5735. [Google Scholar] [CrossRef] [PubMed]
- Foo, L.Y.; Lu, Y.; Howell, A.B.; Vorsa, N. A-type proanthocyanidin trimers from cranberry that inhibit adherence of uropathogenic P-fimbriated Escherichia coli. J. Nat. Prod. 2000, 63, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.B.; Vorsa, N.; Der Marderosian, A.; Foo, L.Y. Inhibition of the adherence of P-fimbriated Escherichia coli to uroepithelial-cell surfaces by proanthocyanidin extracts from cranberries. N. Engl. J. Med. 1998, 339, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, D.; Morales, J.; Zhu, H.; Steffan, S.A.; Harbut, R.; Bassil, N.; Zalapa, J. Discrimination of American Cranberry Cultivars and Assessment of Clonal Heterogeneity Using Microsatellite Markers. Plant Mol. Biol. Report. 2013, 31, 264–271. [Google Scholar] [CrossRef]
- Rodriguez-Saona, C.; Vorsa, N.; Singh, A.P.; Johnson-Cicalese, J.; Szendrei, Z.; Mescher, M.C.; Frost, C.J. Tracing the history of plant traits under domestication in cranberries: Potential consequences on anti-herbivore defences. J. Exp. Bot. 2011, 62, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Georgi, L.; Herai, R.H.; Vidal, R.; Carazzolle, M.F.; Pereira, G.G.; Polashock, J.; Vorsa, N. Cranberry microsatellite marker development from assembled next-generation genomic sequence. Mol. Breed. 2011, 30, 227–237. [Google Scholar] [CrossRef]
- Georgi, L.; Johnson-Cicalese, J.; Honig, J.; Das, S.P.; Rajah, V.D.; Bhattacharya, D.; Bassil, N.; Rowland, L.J.; Polashock, J.; Vorsa, N. The first genetic map of the American cranberry: Exploration of synteny conservation and quantitative trait loci. Theor. Appl. Genet. 2013, 126, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Senalik, D.; McCown, B.H.; Zeldin, E.L.; Speers, J.; Hyman, J.; Bassil, N.; Hummer, K.; Simon, P.W.; Zalapa, J.E. Mining and validation of pyrosequenced simple sequence repeats (SSRs) from American cranberry (Vaccinium macrocarpon Ait.). Theor. Appl. Genet. 2012, 124, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fugate, K.K.; Fajardo, D.; Schlautman, B.; Ferrareze, J.P.; Bolton, M.D.; Campbell, L.G.; Wiesman, E.; Zalapa, J. Generation and Characterization of a Sugarbeet Transcriptome and Transcript-Based SSR Markers. Plant Genome 2014, 7. [Google Scholar] [CrossRef]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Polashock, J.; Zelzion, E.; Fajardo, D.; Zalapa, J.; Georgi, L.; Bhattacharya, D. The American cranberry: First insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014, 14, 165. [Google Scholar] [PubMed]
- Fajardo, D.; Schlautman, B.; Steffan, S.; Polashock, J.; Vorsa, N.; Zalapa, J. The American cranberry mitochondrial genome reveals the presence of selenocysteine (tRNA-Sec and SECIS) insertion machinery in land plants. Gene 2014, 536, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Da Maia, L.C.; Palmieri, D.A.; de Souza, V.Q.; Kopp, M.M.; de Carvalho, F.I.F.; Costa de Oliveira, A. SSR Locator: Tool for Simple Sequence Repeat Discovery Integrated with Primer Design and PCR Simulation. Int. J. Plant Genomics 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.S.; Lucas, D.C.S.; de Souza Neves, K.F.; Bertioli, D.J. WebSat—A web software for microsatellite marker development. Bioinformation 2009, 3, 282–283. [Google Scholar] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.G.; Fitch, A.J.; Bertozzi, T.; Lowe, A.J. Rise of the machines—Recommendations for ecologists when using next generation sequencing for microsatellite development. Mol. Ecol. Resour. 2011, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Kimmel, M.; Stivers, D.N.; Davison, L.J.; Deka, R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc. Natl. Acad. Sci. USA 1997, 94, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Kumar Yadav, H.; Ranjan, A.; Asif, M.H.; Mantri, S.; Sawant, S.V.; Tuli, R. EST-derived SSR markers in Jatropha curcas L.: Development, characterization, polymorphism, and transferability across the species/genera. Tree Genet. Genomes 2010, 7, 207–219. [Google Scholar] [CrossRef]
- Hamblin, M.T.; Warburton, M.L.; Buckler, E.S. Empirical comparison of Simple Sequence Repeats and single nucleotide polymorphisms in assessment of maize diversity and relatedness. PLoS One 2007, 2, e1367. [Google Scholar] [CrossRef] [PubMed]
- Merdinoglu, D.; Butterlin, G.; Bevilacqua, L.; Chiquet, V.; Adam-Blondon, A.-F.; Decroocq, S. Development and characterization of a large set of microsatellite markers in grapevine (Vitis vinifera L.) suitable for multiplex PCR. Mol. Breed. 2005, 15, 349–366. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Boches, P.S.; Bassil, N.V.; Rowland, L.J. Microsatellite markers for Vaccinium from EST and genomic libraries. Mol. Ecol. Notes 2005, 5, 657–660. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Liu, D.; Wei, Y.; Liu, C.; Yang, Y.; Tao, C.; Liu, W. Exploiting EST databases for the development and characterization of EST-SSR markers in blueberry (Vaccinium) and their cross-species transferability in Vaccinium spp. Sci. Hortic. (Amsterdam) 2014, 176, 319–329. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments A poor man’s approach to genotyping for research and high-throughput diagnostics. Nat. Biotechnol. 2000, 18, 1–2. [Google Scholar] [PubMed]
- Brownstein, M.J.; Carpten, J.D.; Smith, J.R. Modulation of non-templated nucleotide addition by taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques 1996, 20, 1004–1006, 1008–1010. [Google Scholar] [PubMed]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Slate, J.; Marshall, T.; Pemberton, J. A retrospective assessment of the accuracy of the paternity inference program CERVUS. Mol. Ecol. 2000, 9, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlautman, B.; Fajardo, D.; Bougie, T.; Wiesman, E.; Polashock, J.; Vorsa, N.; Steffan, S.; Zalapa, J. Development and Validation of 697 Novel Polymorphic Genomic and EST-SSR Markers in the American Cranberry (Vaccinium macrocarpon Ait.). Molecules 2015, 20, 2001-2013. https://doi.org/10.3390/molecules20022001
Schlautman B, Fajardo D, Bougie T, Wiesman E, Polashock J, Vorsa N, Steffan S, Zalapa J. Development and Validation of 697 Novel Polymorphic Genomic and EST-SSR Markers in the American Cranberry (Vaccinium macrocarpon Ait.). Molecules. 2015; 20(2):2001-2013. https://doi.org/10.3390/molecules20022001
Chicago/Turabian StyleSchlautman, Brandon, Diego Fajardo, Tierney Bougie, Eric Wiesman, James Polashock, Nicholi Vorsa, Shawn Steffan, and Juan Zalapa. 2015. "Development and Validation of 697 Novel Polymorphic Genomic and EST-SSR Markers in the American Cranberry (Vaccinium macrocarpon Ait.)" Molecules 20, no. 2: 2001-2013. https://doi.org/10.3390/molecules20022001
APA StyleSchlautman, B., Fajardo, D., Bougie, T., Wiesman, E., Polashock, J., Vorsa, N., Steffan, S., & Zalapa, J. (2015). Development and Validation of 697 Novel Polymorphic Genomic and EST-SSR Markers in the American Cranberry (Vaccinium macrocarpon Ait.). Molecules, 20(2), 2001-2013. https://doi.org/10.3390/molecules20022001