
Exploring the Reactivity of Na[W2(μ-Cl)3Cl4(THF)2]∙(THF)3 towards the Polymerization of Selected Cycloolefins


 and
and
Abstract
:
1. Introduction
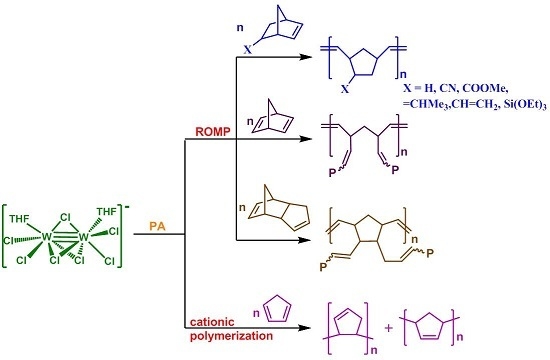
2. Results and Discussion
2.1. Catalyst and Polymerization Reactions

{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | Solvent | 1/PA/Monomer Molar Ratio | t (h) | Yield (%) | Mw × 10−3 g | Mw/Mn g |
|---|---|---|---|---|---|---|---|
| 1 | NBE [14] | THF | 1/0/500 a | 19 | 12 | 86.2 | 1.2 |
| 2 | CH2Cl2 | 1 | 96 | 529 | 1.2 | ||
| 3 | dme | 48 | - f | - | - | ||
| 4 | CH3CN | 48 | - f | - | - | ||
| 5 | toluene | 24 | 37 | 296 | 2.9 | ||
| 6 | Et2O | 20 | 94 | 422 | 1.4 | ||
| 7 | NBE/PA | THF | 1/20/500 b | 20 | >99 | 184 | 1.5 |
| 8 | CH2Cl2 | 0.1 | >99 | 413 | 1.3 | ||
| 9 | dme | 20 | 18 | 3.2 | 1.5 | ||
| 10 | CH3CN | 48 | - f | - | - | ||
| 11 | toluene | 20 | 34 | 11 | 1.5 | ||
| 12 | Et2O | 20 | 72 | 392 | 1.4 | ||
| 13 | NBE/PA | THF | 1/20/1000 c | 1 | 95 | 22.3 | 2.5 |
| 14 | CH2Cl2 | 0.1 | 97 | 300 | 1.2 | ||
| 15 | toluene | 0.3 | 95 | 62 | 4.5 | ||
| 16 | VNBE [14] | CH2Cl2 | 1/0/500 a | 8 | >99 | 974 | 2.6 |
| 17 | VNBE/PA | CH2Cl2 | 1/20/500 b | 1 | >99 | 97 | 2.7 |
| 18 | NBE-COOMe [14] | CH2Cl2 | 1/0/500 a | 12 | >99 | 685 | 1.15 |
| 19 | NBE-COOMe/PA | CH2Cl2 | 1/20/500 b | 6 | 90 | - h | - |
| 20 | - | 1/20/1000 c | 1 | 40 | 810 | 1.2 | |
| 21 | NBE-CN [14] | CH2Cl2 | 1/0/500 a | 48 | - g | - | - |
| 22 | NBE-CN/PA | CH2Cl2 | 1/20/500 b | 6 | 17 | - i | - |
| 23 | - | 1/20/1000 c | 20 | 30 | - i | - | |
| 24 | NBE-EN | CH2Cl2 | 1/0/500 a | 0.3 | 97 | 452 | 1.3 |
| 25 | NBE-EN/PA | CH2Cl2 | 1/20/500 b | 4 | >99 | 583 | 1.2 |
| 26 | - | 1/20/1000 c | 20 | >99 | 974 | 2.6 | |
| 27 | NBE-SiM | CH2Cl2 | 1/0/500 a | 20 | 60 | - i | - |
| 28 | NBE-SiE | CH2Cl2 | 1/0/500 a | 20 | 60 | - i | - |
| 29 | NBE-SiE/PA | CH2Cl2 | 1/20/500 b | 8 | 75 | - i | - |
| 30 | NBD [14] | THF | 1/0/500 a | 4 | >99 | - j | - |
| 31 | NBD/PA | THF | 1/20/500 b | 0.5 | 38 | - j | - |
| 32 | 1/20/1000 c | 21 | 61 | - j | |||
| 33 | DCPD | CH2Cl2 | 1/0/500 a | 17 | 10 | - j | - |
| 34 | toluene | 17 | 20 | - j | - | ||
| 35 | - | 17 | 20 | - j | - | ||
| 36 | DCPD/PA | CH2Cl2 | 1/20/500 b | 17 | >99 | - j | - |
| 37 | toluene | 17 | >99 | - j | - | ||
| 38 | - | 1/20/1000 c | 20 | traces | - | - | |
| 39 | CPD | CH2Cl2 | 1/0/250 d | 20 | 90 | 29.7 | 2.1 |
| 40 | THF | 20 | 5 | - | - | ||
| 41 | toluene | 20 | 30 | 3.3 | 1.5 | ||
| 42 | - | 20 | 30 | 6.3 | 2.7 | ||
| 43 | CPD/PA | CH2Cl2 | 1/10/250 e | 8 | 50 | 11.1 | 1.3 |
| 44 | toluene | 8 | 25 | 1.2 | 1.6 |
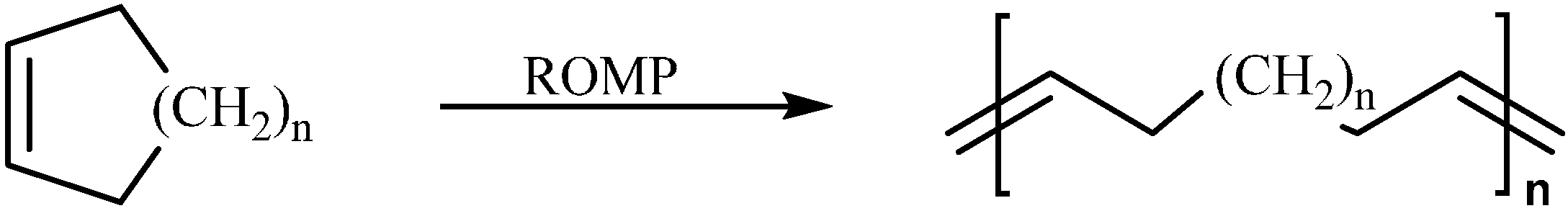
2.2. Ring Opening Metathesis Polymerization Reactions
2.3. Cationic Polymerization of CPD
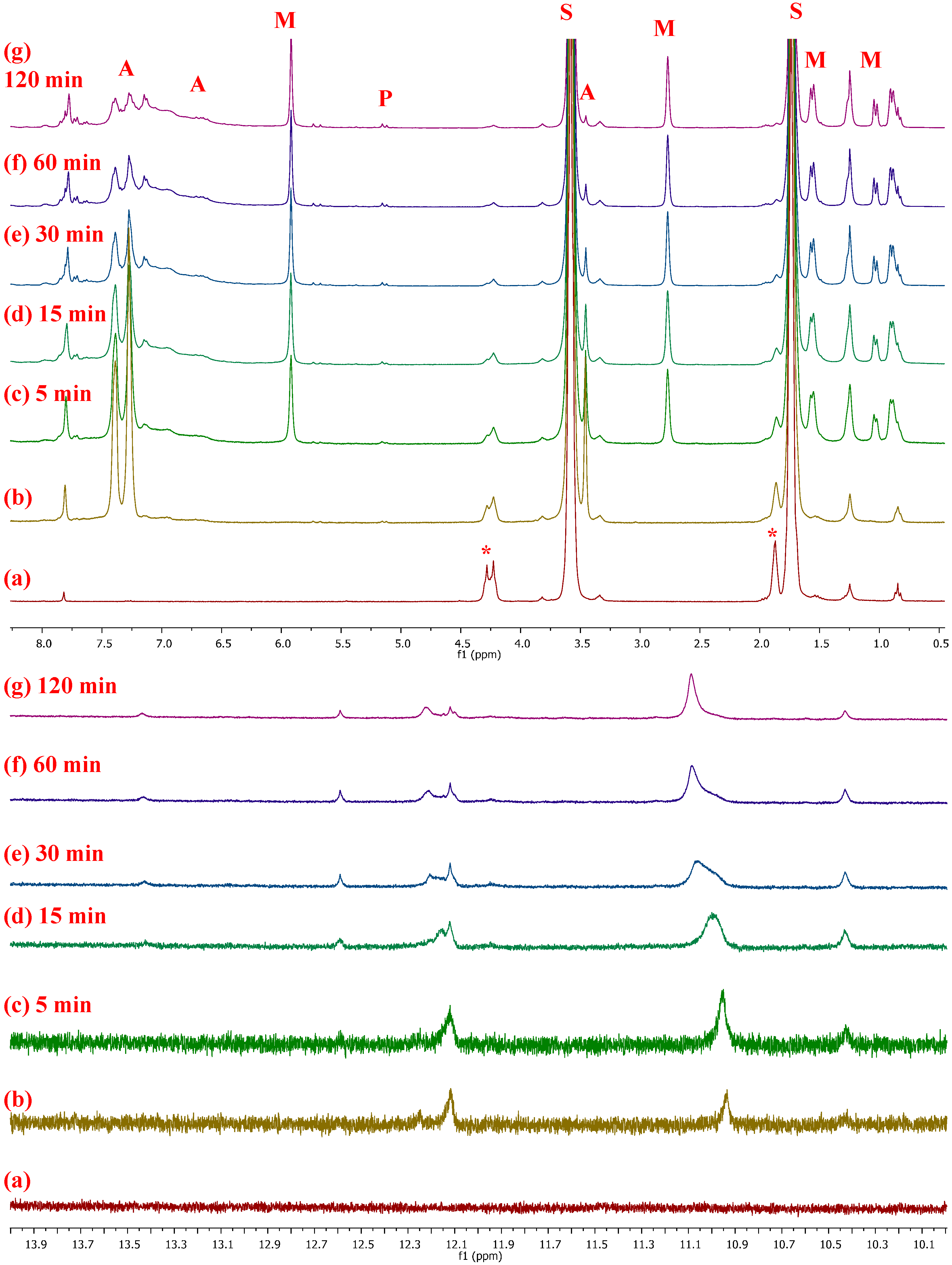
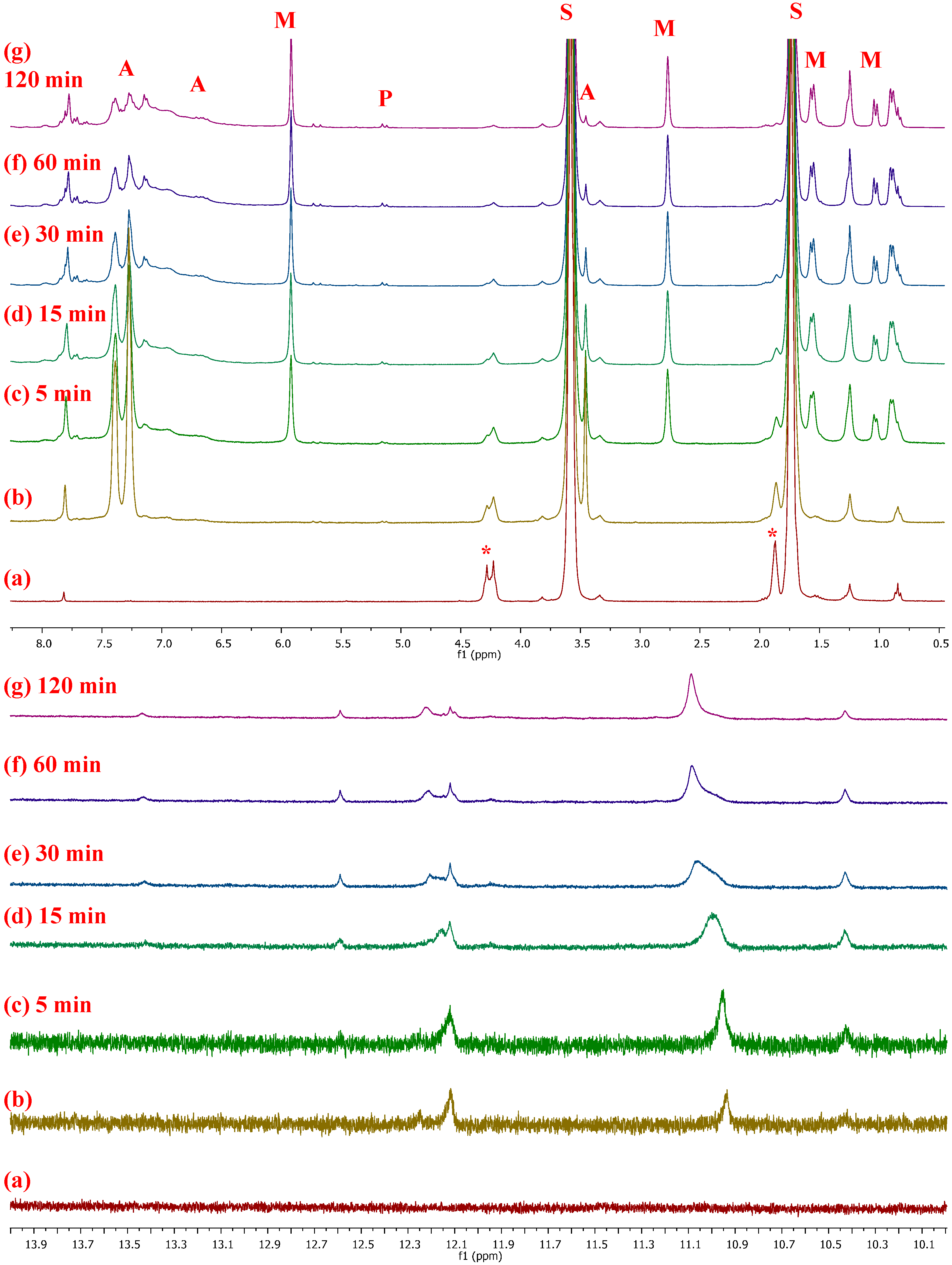
2.4. Mechanistic Considerations
3. Experimental Section
3.1. General
3.2. Catalytic Reactions
3.3. Catalytic Reactions in NMR Tubes
3.4. Polymer Microstructure
 | PNBE. 1H-NMR (CDCl3, 300 MHz): 5.25 (s, 2H, H2,3 t), 5.10 (s, 2H, H2,3 c), 2.73 (br, s, 2H, H1,4 c), 2.37 (br, s, 2H, H1,4 t), 1.95–1.60 (br, m, 3H, H5a,6a,7a), 1.50–1.20 (br, m, 2H, H5b,6b), 1.20–0.85 ppm (br, m, 1H, H7b); 13C-NMR (CDCl3, 75.4 MHz): 133.99 (s, C2,3 ccc), 133.21 (m, C2,3 ctt/ttt/ctc), 133.08 (s, C2,3 ttc), 43.67 (s, C1,4 tc), 43.44 (s, C1,4 tt), 42.88 (s, C7 cc), 42.25 (s, C7 ct/tc), 41.52 (s, C7 tt), 38.88 (s, C1,4 cc), 38.67 (s, C1,4 ct), 33.39 (s, C5,6 cc), 33.19 (s, C5,6 ct), 32.61 (s, C5,6 tc), 32.43 ppm (s, C5,6 tt). |
 | PVNBE. 1H-NMR (CDCl3, 300 MHz): 5.76 (br, 1H, H8), 5.30 (br, 2H, H2,3), 4.89–4.96 (br, 2H, H9), 2.19–3.10 (br, 3H, H1,4,5), 1.10–2.10 ppm (br, m, 2H, H6,7); 13C-NMR (CDCl3, 75.4 MHz): 141.7, 140.6 (s, C8), 135.6–130.3 (m, C2,3), 113.6, 113.1 (s, C9), 50.1, 47.9 (s, C5),45.6, 37.5 (s, C1), 45.6, 41.3 (s, C4), 42.8, 41.3, 39.5 (s, C7), 41.3 ppm (s, C6). |
 | PNBE-COOME. 1H-NMR (CDCl3, 300 MHz): 5.26–5.37 (br, 2H, H2,3), 2.76–3.10 (br, s, 1H, H4), 2.26 (br, s, 1H, H5), 1.80–2.10 (br, m, 3H, H9), 1.10–1.60 ppm (br, m, 3H, H1,6,7); 13C-NMR (CDCl3, 75.4 MHz): 170.7 (s, C8), 140.9–127.4 (m, C2,3), 51.1 (s, C9), 48.6–34.8 ppm (s, C1,4,5,6,7). |
 | PNBE-CN. 1H-NMR (CDCl3, 300 MHz): 5.15–5.75 (2H, H2,3), 3.52 (1H, H4), 2.80–3.35 (1H, H1), 2.00–2.68 (br 1H, H5), 1.40–1.95 (br, 2H, H6), 1.10–1.40 and 2.50–2.56 ppm (2H, H7); 13C-NMR (CDCl3, 75.4 MHz): 125.0–140.0 (C2,3), 120.0–125.0 (C8), 32.5–48.5 ppm (C1,4,5,6,7). |
 | PNBE-EN. 1H-NMR (CDCl3, 300 MHz): 5.06–5.50 (br, m, 3H, H2,3,8), 2.86 (br, s, 2H, H1,4 c), 2.50 (br, s, 2H, H1,4 t), 1.80–2.20 (br, m, 2H, H6), 1.57 (s, 3H, H9), 1.10–1.30 ppm (br, s, 2H, H7); 13C-NMR (CDCl3, 75.4 MHz): 146.0 (C5 t), 145.6 (C5 c), 134.6 (C2 t, TH), 134.5 (C2 c, TH), 134.0 (C2 c, TT), 133.7 (C2 t, TT), 132.9 (C3 c,t, HH), 132.4 (C3 c, HT), 132.0 (C3 t, HT), 117.0 (C8 t), 116.2 (C8 c), 48.9 (C4 t), 44.2 (C4 c), 42.9–42.7 (C7), 41.8 (C6), 38.1–36.4 (C1), 14.9–14.0 ppm (C9). |
 | PNBE-SiE (ROMP). 1H-NMR (CDCl3, 300 MHz): 5.10–5.80 (br, m, 2H, H2,3), 3.85 (br, s, 6H, H8), 1.40–2.20 (br, m, 7H, H1,4,5,6,7), 1.22 ppm (br, s, 9H, H9); 13C-NMR (CDCl3, 75.4 MHz): 130.0–140.0 (C2,3), 58.4 (C8), 40.0–50.0 (C1,4,6,7), 26.0–27.0 (C5), 18.2 ppm (C9). |
 | PNBE-SiE (cationic). 1H-NMR (CDCl3, 300 MHz): 3.60–3.90 (br, m, 6H, H8), 1,30–2,00 (br, 9H, H1,2,3,4,5,6,7), 1.10–1.30 ppm (m, 9H, H9); 13C-NMR (CDCl3, 75.4 MHz): 58.8 (C8), 18.8 ppm (C9). |
  | PCPD 1,2. 1H-NMR (CDCl3, 300 MHz): 5.50–5.75 (Η3,4), 2.62 (Η5), 2.42 ppm (Η1,2); 13C-NMR (CDCl3, 75.4 MHz): 133.04 (C3), 130.03 (C4), 55.09 (C2), 44.70 (C1), 36.40 ppm (C5). PCPD 1,4. 1H-NMR (CDCl3, 300 MHz): 5.50–5.75 (H2,3), 2.02 (H1,4), 1.62 ppm (H5); 13C-NMR (CDCl3, 75.4 MHz): 134.17 (C2,3), 50.53 (C1,4), 32.03 ppm (C5). |
4. Conclusions
- (a)
- The 1/PA catalytic system catalyzes the ROMP of norbornene (NBE) and a number of substituted norbornenes efficiently, providing high molecular weight polymers in high yields and high stereoselectivity (86% cis for PNBE). Monomers bearing strongly-coordinating pendant groups (–COOH, –OH, –CN) were either not activated by 1/PA (the first two) or provided polymers in low yields (case of –CN), while those bearing weaker ones (–COOMe, –CH=CH2, =CHCH3) showed high reactivity. It should be noted that less strained double bonds remained unaffected (–CH=CH2, =CHCH3). Norbornadiene (NBD) and dicyclopentadiene (DCPD) were also activated quickly and quantitatively, giving insoluble, highly crosslinked polymers.
- (b)
- 1 activated cyclopentadiene (CPD), but in a different fashion, providing oligomers or low molecular weight polymers, not via metathesis, but via the cationic polymerization pathway.
- (c)
- Compared to 1, the catalytic system 1/PA was in general more active towards the ROMP of all monomers studied, in all solvents, as well as in bulk. The molecular weights of the polymers obtained were higher (with very few exceptions), while the molecular weight distributions were either retained or improved. The cis specificity of PNBE was the same (86% cis) with either system.
- (d)
- In situ monitoring of the reaction (1/PA/NBE) by 1H-NMR spectroscopy revealed the formation of active alkylidenes of the propagating chains, in agreement with our previous studies, but a detailed mechanistic study of the catalytic system is underway.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ivin, K.J.; Mol, J.C. Olefin Metathesis and Metathesis Polymerization; Academic Press, Inc.: San Diego, CA, USA, 1997. [Google Scholar]
- Dragutan, V.; Streck, R. Catalytic Polymerization of Cycloolefins. Ionic, Ziegler-Natta and Ring-Opening Metathesis Polymerization; Elsevier Science B.V.: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Grubbs, R.H.; Wenzel, A.G.; O’Leary, D.J.; Khosravi, E. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Frenzel, U.; Müller, B.K.M.; Nuyken, O. Handbook of Polymer Synthesis, 2nd ed.; Kricheldorf, H.R., Nuyken, O., Swift, G., Eds.; Marcel Dekker: New York, NY, USA, 2005. [Google Scholar]
- Buchmeiser, M.R. Homogeneous metathesis polymerization by well-defined group VI and group VIII transition-metal alkylidenes: Fundamentals and applications in the preparation of advanced materials. Chem. Rev. 2000, 100, 1565–1604. [Google Scholar] [CrossRef] [PubMed]
- Buchmeiser, M.R. Ring-Opening Metathesis Polymerization, in Synthesis of Polymers: New Structures and Methods, 1st ed.; Schlüter, A.D., Hawker, C.J., Sakamoto, J., Eds.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Szymańska-Buzar, T. Structure and reactivity of tungsten(II) and molybdenum(II) compounds containing an M–M bond. Coord. Chem. Rev. 2005, 249, 2195–2202. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Fujita, A.; Suzuki, N.; Ito, T. Metathesis polymerization of norbornene and terminal acetylenes catalyzed by bis(acetonitrile) complexes of molybdenum and tungsten. J. Mol. Catal. A Chem. 2005, 240, 226–232. [Google Scholar] [CrossRef]
- Kozmer, V.A.; Poletayeva, I.A.; Yufa, T.L. Cycloolefin polymerization initiated by transition metal-π-allylic complexes. J. Polym. Sci. A 1972, 10, 251–258. [Google Scholar]
- Mutch, A.; Leconte, A.; Lefebvre, F.; Basset, J.-M. Effect of alcohols and epoxides on the rate of ROMP of norbornene by a ruthenium trichloride catalyst. J. Mol. Catal. A Chem. 1998, 133, 191–199. [Google Scholar] [CrossRef]
- Grubbs, R.H. Olefin Metathesis Catalysts for the Preparation of Molecules and Materials. Angew. Chem. Int. Ed. 2006, 45, 3760–3765. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Multiple Metal–Carbon Bonds for Catalytic Metathesis Reactions. Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Raptopoulos, G.; Grigoropoulos, A.; Mertis, K.; Paraskevopoulou, P.; Pitsikalis, M. Multinuclear transition metal catalysts for metathesis polymerization. Current developments and future perspectives. In Recent Research Developments in Polymer Science; Transworld Research Network: Trivandrum, India, 2014; Volume 12, pp. 83–106. [Google Scholar]
- Saragas, N.; Floros, G.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Hadjichristidis, N.; Mertis, K. Ring opening metathesis polymerization of norbornene and norbornadiene by bimetallic multiply bonded tungsten complexes. Polymers 2012, 4, 1657–1673. [Google Scholar]
- Chisholm, M.H.; Eichhorn, B.W.; Folting, K.; Huffman, J.C.; Ontiveros, C.D.; Streib, W.E.; van der Sluys, W.G. Preparation and characterization of NaW2Cl7(THF)5. A synthetically useful precursor for X3W≡WX3 compounds where X = CH2-t-Bu, NMe2, and O-t-Bu. Inorg. Chem. 1987, 26, 3182–3186. [Google Scholar] [CrossRef]
- Saragas, N.; Floros, G.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Mertis, K. Polymerization of terminal alkynes with a triply bonded ditungsten halo-complex. J. Mol. Catal. A Chem. 2009, 303, 124–131. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer: New York, NY, USA, 2005. [Google Scholar]
- Düz, B.; Elbistan, C.K.; Ece, A.; Sevin, F. Application of carbon arc-generated Mo- and W-based catalyst systems to the ROMP of norbornene. Appl. Organometal. Chem. 2009, 23, 359–364. [Google Scholar] [CrossRef]
- Mauldin, T.C.; Kessler, M.R. Enhanced bulk catalyst dissolution for self-healing materials. J. Mater. Chem. 2010, 20, 4198–4206. [Google Scholar] [CrossRef]
- Gorski, M.; Szymańska-Buzar, T. Tungsten(II)-initiated ring-opening metathesis polymerization and other C–C bond forming reactions of 5-vinyl-2-norbornene. J. Mol. Cat. A Chem. 2006, 257, 41–47. [Google Scholar] [CrossRef]
- Hakala, J.; Hänninen, M.M.; Lehtonen, A. Imidotungsten(VI) complexes with chelating phenols as ROMP catalysts. Inorg. Chem. Commun. 2011, 14, 1362–1364. [Google Scholar] [CrossRef]
- Nomura, K.; Suzuki, K.; Katao, S.; Matsumoto, Y. Ring-opening polymerization of THF by aryloxo-modified (imido)vanadium(V)-alkyl complexes and ring-opening metathesis polymerization by highly active V(CHSiMe3)(NAd)(OC6F5)(PMe3)2. Organometallics 2012, 31, 5114–5120. [Google Scholar] [CrossRef]
- Alb, A.M.; Enohnyaket, P.; Craymer, J.F.; Eren, T.; Coughlin, E.B.; Reed, W.F. Online monitoring of ring-opening metathesis polymerization of cyclooctadiene and a functionalized norbornene. Macromolecules 2007, 40, 444–451. [Google Scholar] [CrossRef]
- Vygodskii, Y.S.; Shaplov, A.S.; Lozinskaya, E.I.; Filippov, O.A.; Shubina, E.S.; Bandari, R.; Buchmeiser, M.R. Ring-opening metathesis polymerization (ROMP) in ionic liquids: Scope and limitations. Macromolecules 2006, 39, 7821–7830. [Google Scholar] [CrossRef]
- Balcar, H.; Bek, D.; Sedláček, J.; Dědeček, J.; Bastl, Z.; Lamač, M. RuCl2(p-cymene)(PCy3) immobilized on mesoporous molecular sieves as catalyst for ROMP of norbornene and its derivatives. J. Mol. Catal. A Chem. 2010, 332, 19–24. [Google Scholar] [CrossRef]
- Greene, R.M.E.; Ivin, K.J.; Rooney, J.J.; Kress, J.; Osborn, J.A. Preparation of block copolymers by ring-opening polymerization of cycloalkenes initiated by a tungsten-cyclopentylidene complex. Makromol. Chem. 1988, 189, 2797–2805. [Google Scholar] [CrossRef]
- Aïssa, B.; Nechache, R.; Haddad, E.; Jamroz, W.; Merle, P.G.; Rosei, F. Ruthenium Grubbs’ catalyst nanostructures grown by UV-excimer-laser ablation for self-healing applications. Appl. Surf. Sci. 2012, 258, 9800–9804. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, X.; Yoon, S.H.; Kessler, M.R. Thermal analysis of ring-opening metathesis polymerized healing agents. J. Polym. Sci. B Polym. Phys. 2007, 45, 1771–1780. [Google Scholar] [CrossRef]
- Trzaska, S.T.; Lee, L.-B.W.; Register, R.A. Synthesis of narrow-distribution “perfect” polyethylene and its block copolymers by polymerization of cyclopentene. Macromolecules 2000, 33, 9215–9221. [Google Scholar] [CrossRef]
- Gringolts, M.L.; Bermeshev, M.V.; Rogan, Y.V.; Moskvicheva, M.V.; Filatova, M.P.; Finkelshtein, E.S.; Bondarenko, G.N. Comparative reactivity of Me3Si-substituted norbornene derivatives in ring-opening metathesis polymerization. Silicon 2015, 7, 107–115. [Google Scholar] [CrossRef]
- Makovetsky, K.L.; Finkelshtein, E.S.; Ostrovskaya, I.A.; Portnykh, E.B.; Gorbacheva, L.I.; Goldberg, A.I.; Ushakov, N.V.; Yampolsky, Y.P. Ring-opening metathesis polymerization of substituted norbornenes. J. Mol. Catal. 1992, 76, 107–121. [Google Scholar] [CrossRef]
- Bell, B.; Hamilton, J.G.; Law, E.E.; Rooney, J.J. A one-pot synthesis of comb polymers and hydrogels by the ring-opening metathesis polymerization reaction. Macromol. Rapid Commun. 1994, 15, 543–550. [Google Scholar] [CrossRef]
- Nishihara, Y.; Inoue, Y.; Nakayama, Y.; Shiono, T.; Takagi, K. Comparative reactivity of exo- and endo-isomers in the Ru-initiated ring-opening metathesis polymerization of doubly functionalized norbornenes with both cyano and ester groups. Macromolecules 2006, 39, 7458–7460. [Google Scholar] [CrossRef]
- Muhlebach, A.; van der Schaaf, P.A.; Hafner, A.; Kolly, R.; Rime, F.; Kimer, H.-J. Ruthenium Catalysts for Ring-Opening Metathesis Polymerization (ROMP) and Related Chemistry. In Ring Opening Metathesis Polymerisation and Related Chemistry; Khosravi, E., Szymanska-Buzar, T., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; pp. 23–44. [Google Scholar]
- Davidson, T.A.; Wagener, K.B.; Priddy, D.B. Polymerization of dicyclopentadiene: A tale of two mechanisms. Macromolecules 1996, 29, 786–788. [Google Scholar] [CrossRef]
- McCann, M.; MacGiolla Coda, E. [Mo2(MeCN)8][BF4]4 and [Mo2(μ-O2CCH3)2(MeCN)6][BF4]2 as catalysts for the cationic polymerization of cyclopentadiene and dicyclopentadiene. J. Mol. Catal. A Chem. 1996, 109, 99–111. [Google Scholar] [CrossRef]
- Bencze, L.; Kraut-Vass, A.; Prókai, L. Mechanism of initiation of the metathesis of norbornene using W(CO)3Cl2(AsPh3)2 as catalyst. J. Chem. Soc. Chem. Commun. 1985, 13, 911–912. [Google Scholar] [CrossRef]
- Czeluśniak, I.; Szymańska-Buzar, T. Ring-opening metathesis polymerization of norbornene and norbornadiene by tungsten(II) and molybdenum(II) complexes. J. Mol. Catal. A Chem. 2002, 190, 131–143. [Google Scholar] [CrossRef]
- Bencze, L.; Szalai, G.; Overton, T.L. Investigation of the catalytic properties of the thermally activated dichloro-tetracarbonyl-tungsten in olefin metathesis reaction. Inorg. Chim. Acta 1997, 254, 5–7. [Google Scholar] [CrossRef]
- Bencze, L.; Biró, N.; Szabó-Ravasz, B.; Mihichuk, L. Chemical transformations of cis-W(CO)4(C5H5N)2 in the ring-opening metathesis polymerization of norbornene. Can. J. Chem. 2004, 82, 499–503. [Google Scholar] [CrossRef]
- Farona, M.F.; Tucker, R.L. Studies on the mechanism of olefin metathesis promoted by a heterogeneous catalyst. J. Mol. Catal. 1980, 8, 85–90. [Google Scholar] [CrossRef]
- Szymańska-Buzar, T.; Głowiak, T.; Czeluśniak, I. The initiation of ring-opening metathesis polymerisation of norbornadiene by seven-coordinate molybdenum(II) compounds. X-ray crystal structure of [Mo(μ-Cl)(SnCl3)(CO)3(η4-NBD)]. J. Organomet. Chem. 2001, 640, 72–78. [Google Scholar] [CrossRef]
- Szymańska-Buzar, T.; Głowiak, T.; Czeluśniak, I. Reactivity of [Mo(μ-Cl)(SnCl3)(CO)3(NCMe)2] towards norbornadiene. X-ray crystal structure of [Mo(μ-Cl)(SnCl3)(CO)2(η4-C7H8)(NCMe)]. Polyhedron 2002, 21, 2505–2513. [Google Scholar] [CrossRef]
- Veige, A.S.; Wolczanski, P.T.; Lobkovsky, E.B. Dehydrogenation of [{(silox)3Nb}2(η-1,2;η-5,6-C8H8)] (silox = tBu3SiO) to [{(silox)3Nb}2(η-1,2;η-5,6-C8H6)] and its subsequent alkene-to-alkylidene rearrangement. Angew. Chem. Int. Ed. 2001, 40, 3629–3632. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saragas, N.; Floros, G.; Raptopoulos, G.; Pitsikalis, M.; Paraskevopoulou, P.; Mertis, K. Exploring the Reactivity of Na[W2(μ-Cl)3Cl4(THF)2]∙(THF)3 towards the Polymerization of Selected Cycloolefins. Molecules 2015, 20, 21896-21908. https://doi.org/10.3390/molecules201219810
Saragas N, Floros G, Raptopoulos G, Pitsikalis M, Paraskevopoulou P, Mertis K. Exploring the Reactivity of Na[W2(μ-Cl)3Cl4(THF)2]∙(THF)3 towards the Polymerization of Selected Cycloolefins. Molecules. 2015; 20(12):21896-21908. https://doi.org/10.3390/molecules201219810
Chicago/Turabian StyleSaragas, Nikolaos, Georgios Floros, Grigorios Raptopoulos, Marinos Pitsikalis, Patrina Paraskevopoulou, and Konstantinos Mertis. 2015. "Exploring the Reactivity of Na[W2(μ-Cl)3Cl4(THF)2]∙(THF)3 towards the Polymerization of Selected Cycloolefins" Molecules 20, no. 12: 21896-21908. https://doi.org/10.3390/molecules201219810