
Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology


Abstract
:1. Introduction
2. Results and Discussion
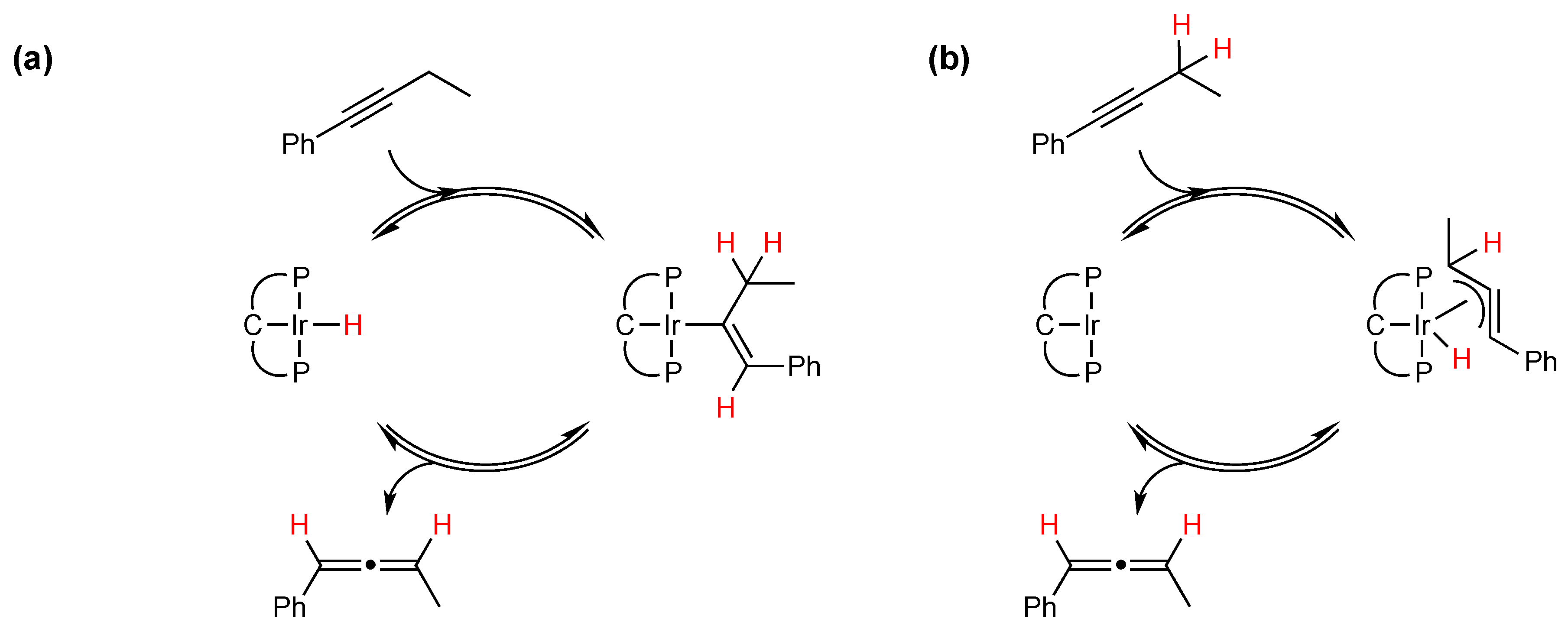
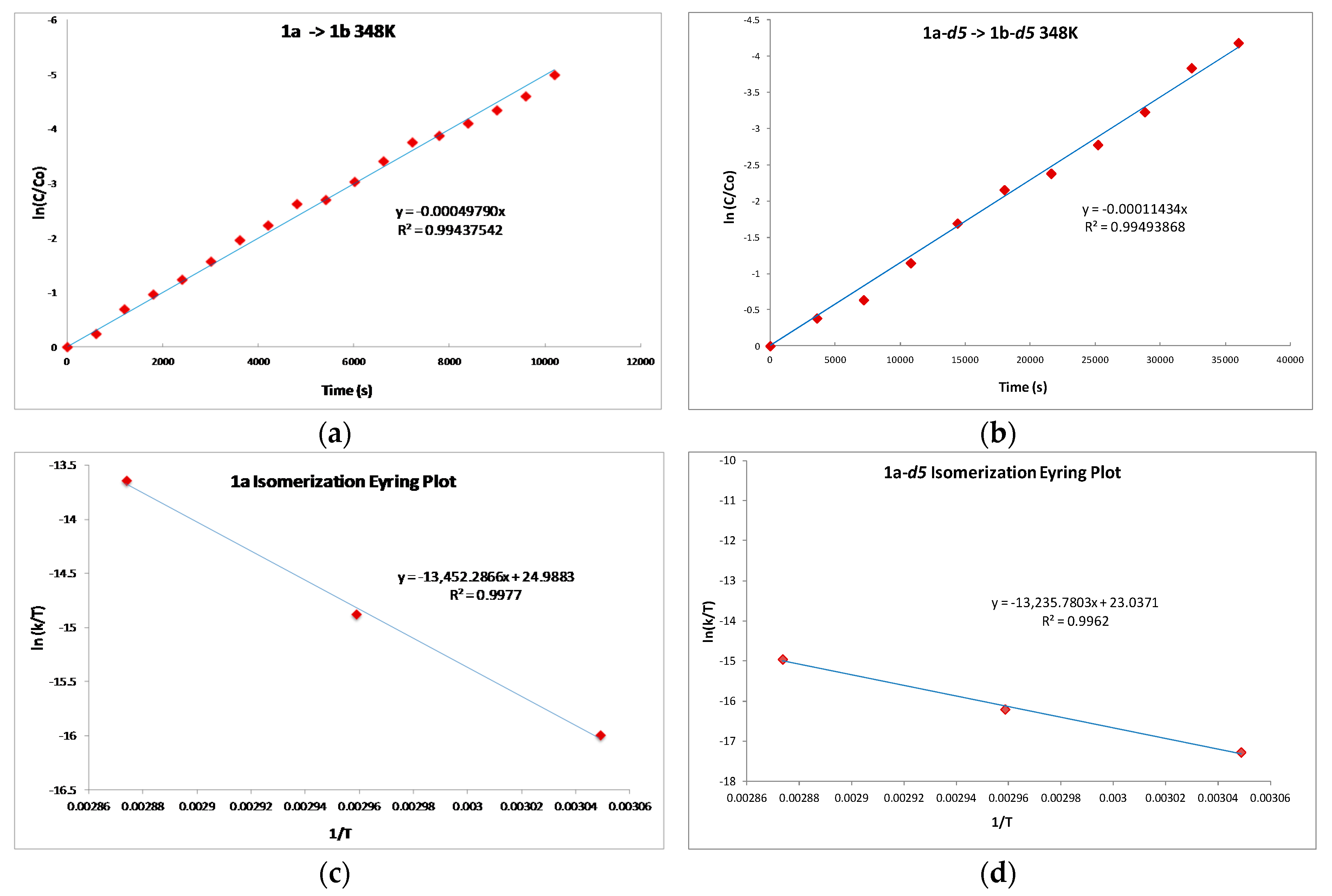
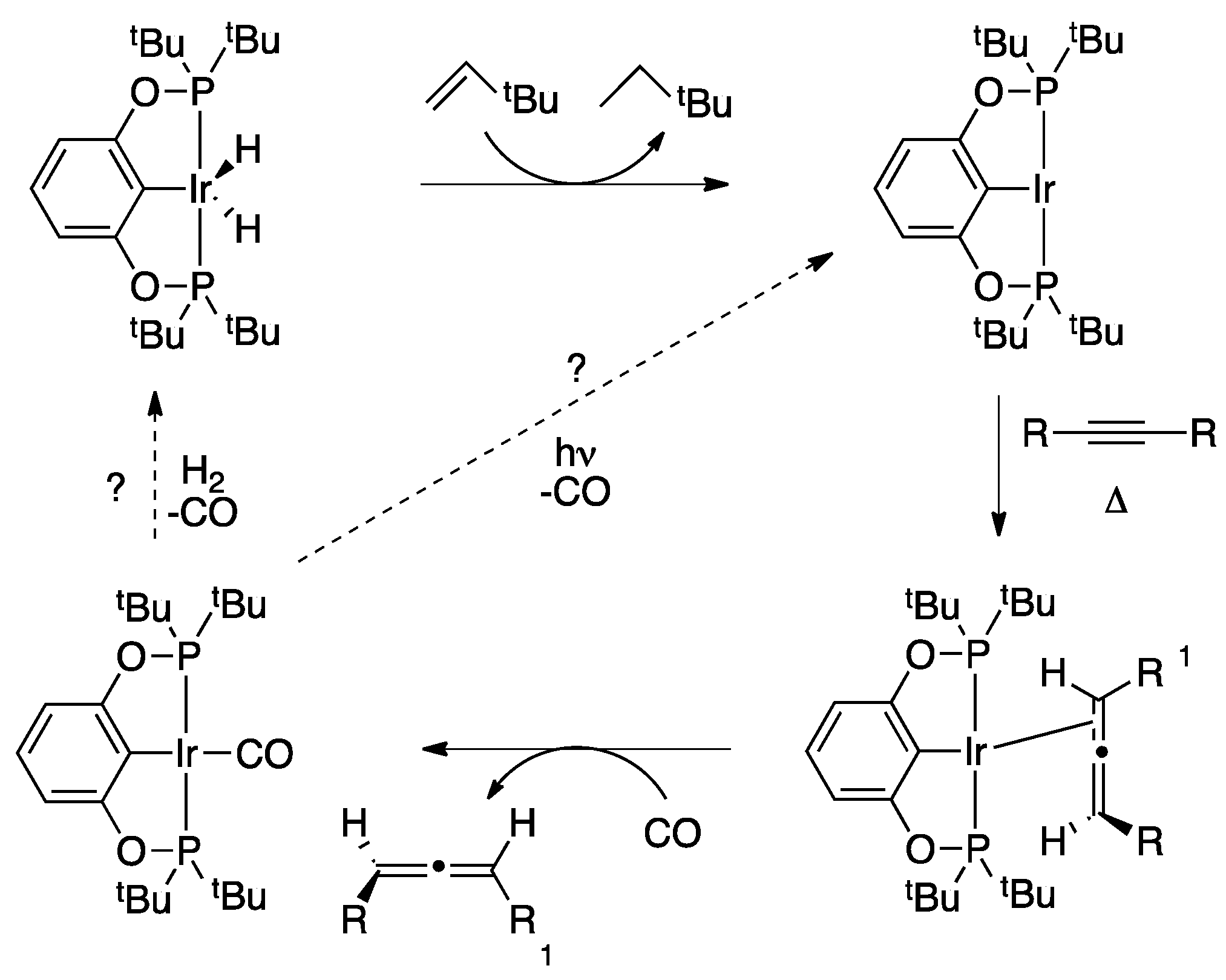
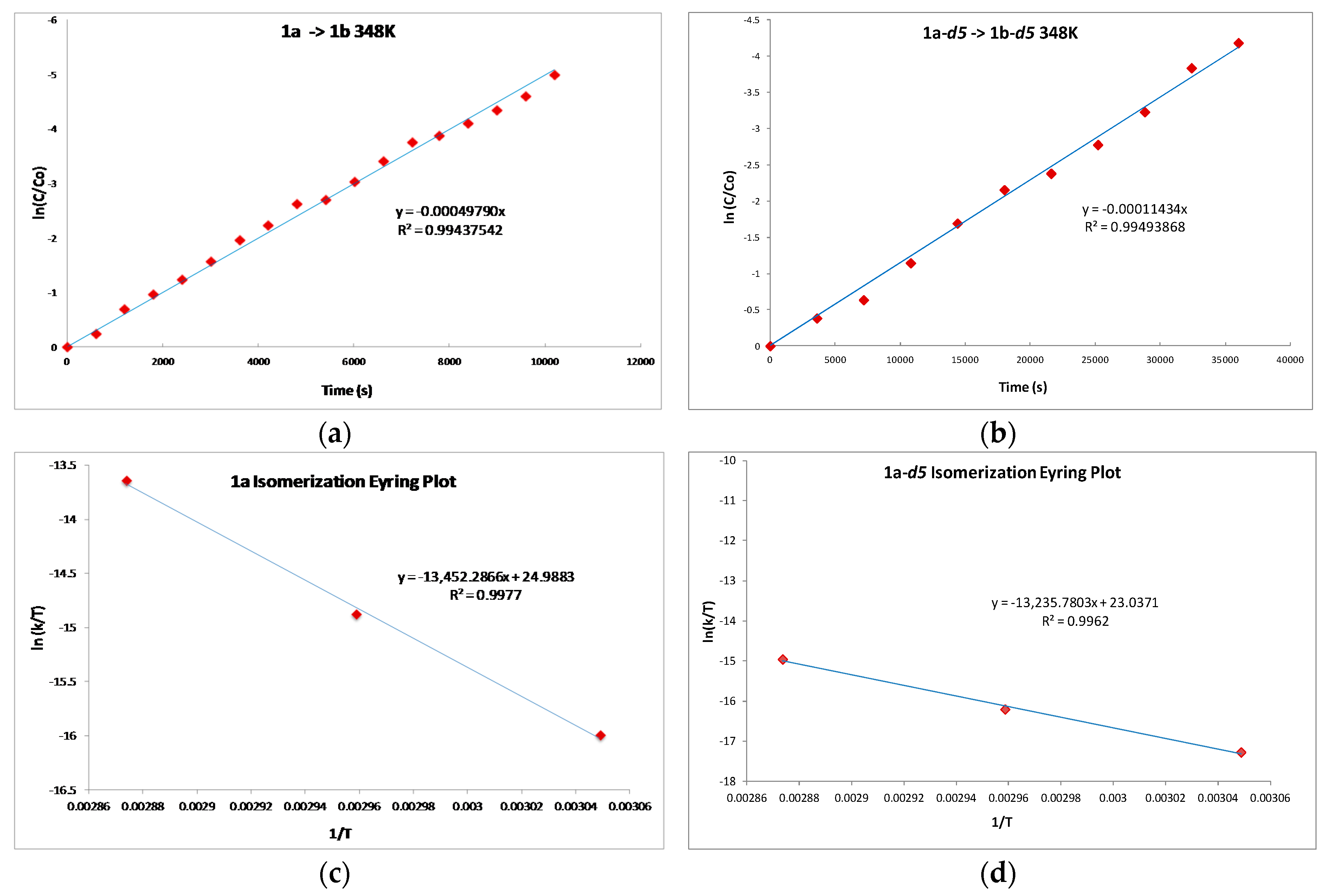
2.1. Mechanistic Studies
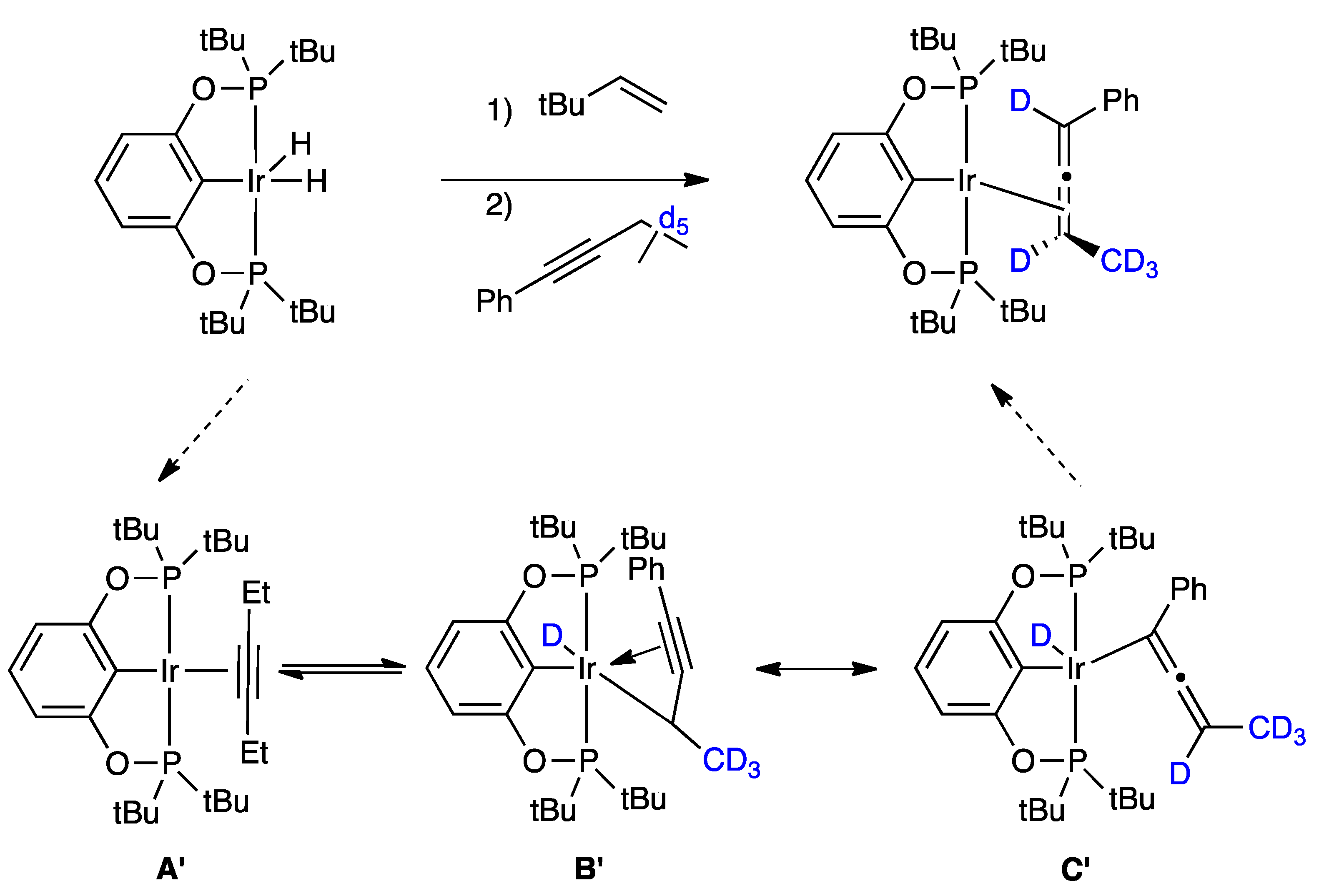

{kind=link}
{kind=link}
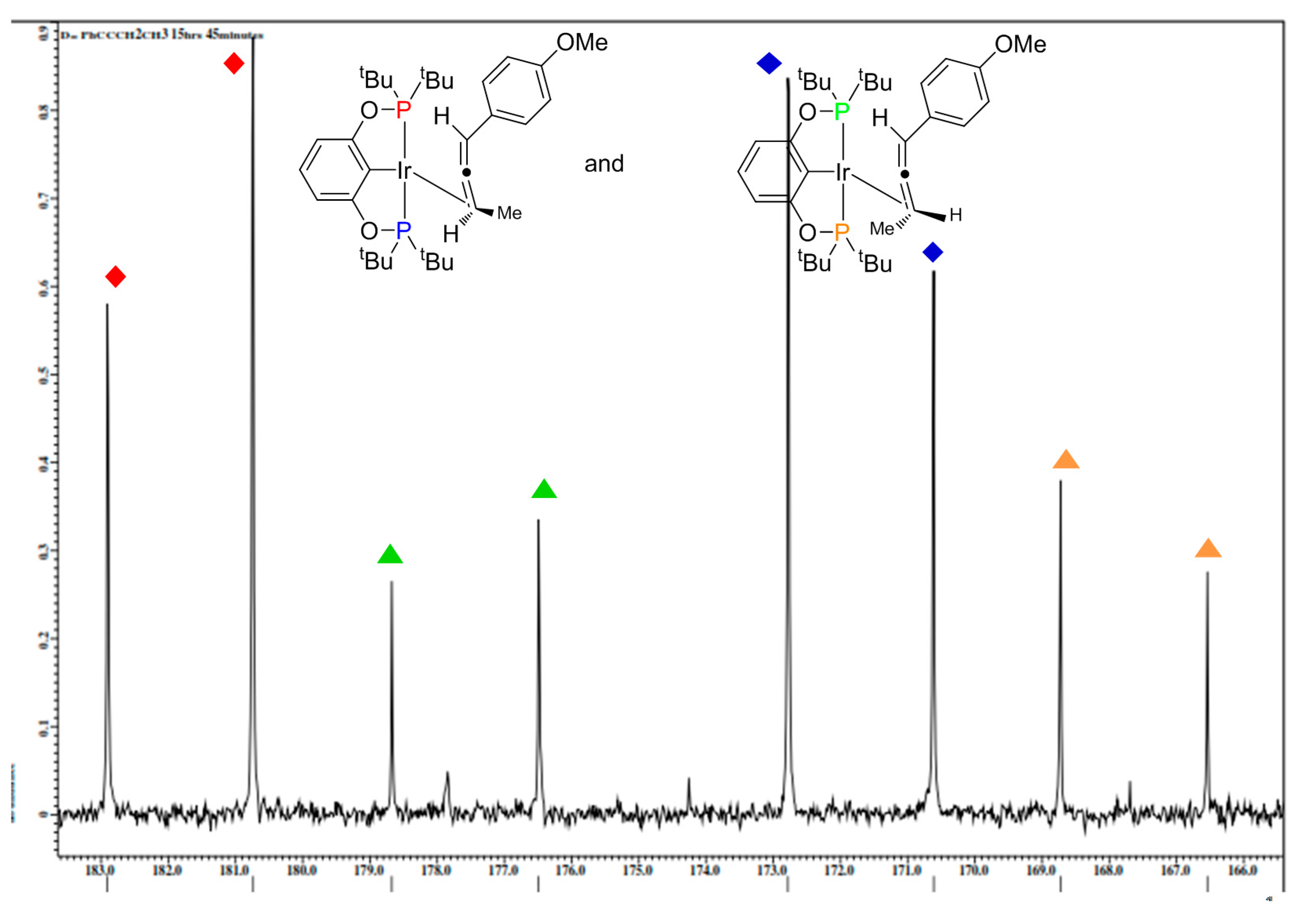{kind=link}
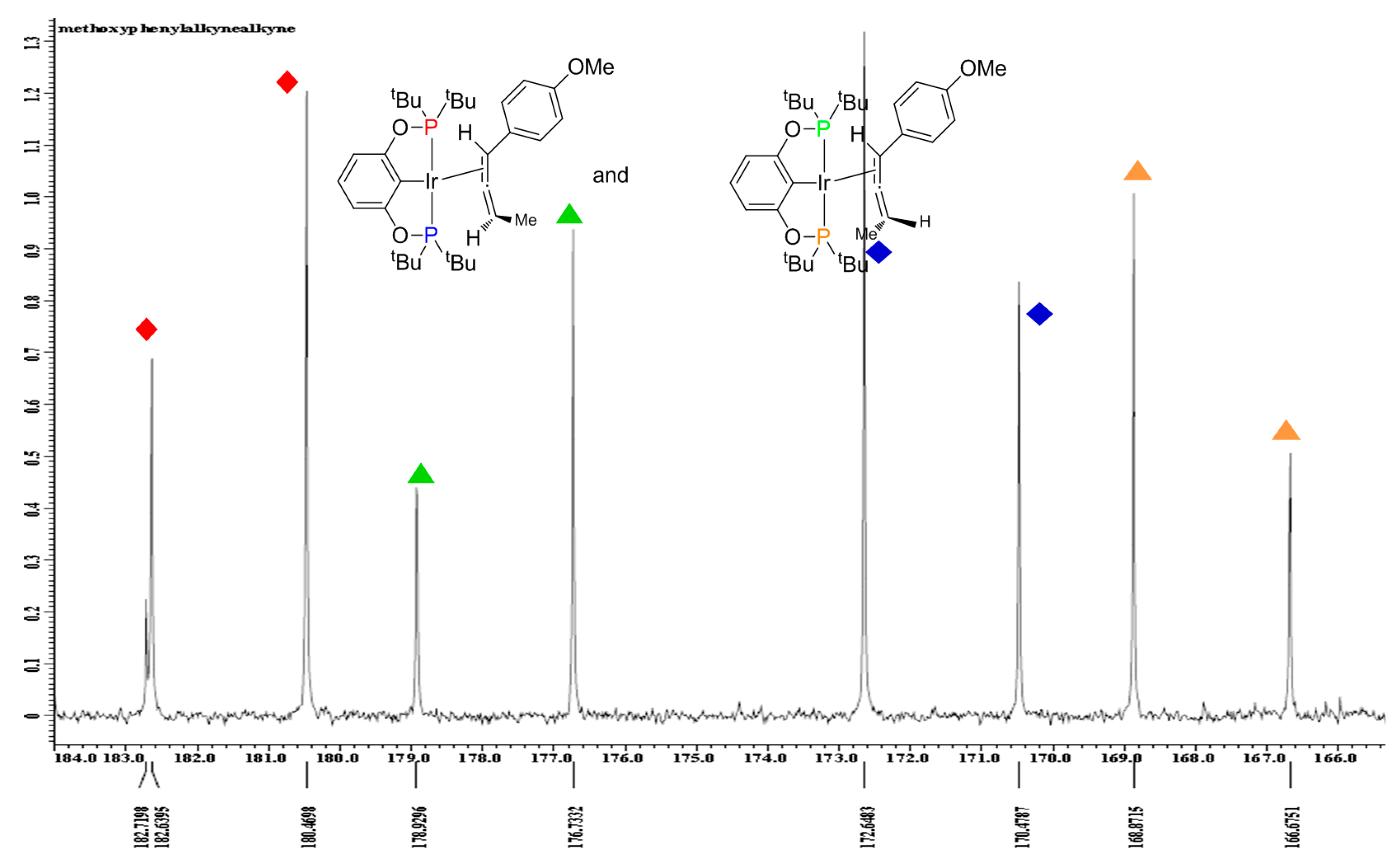{kind=link}
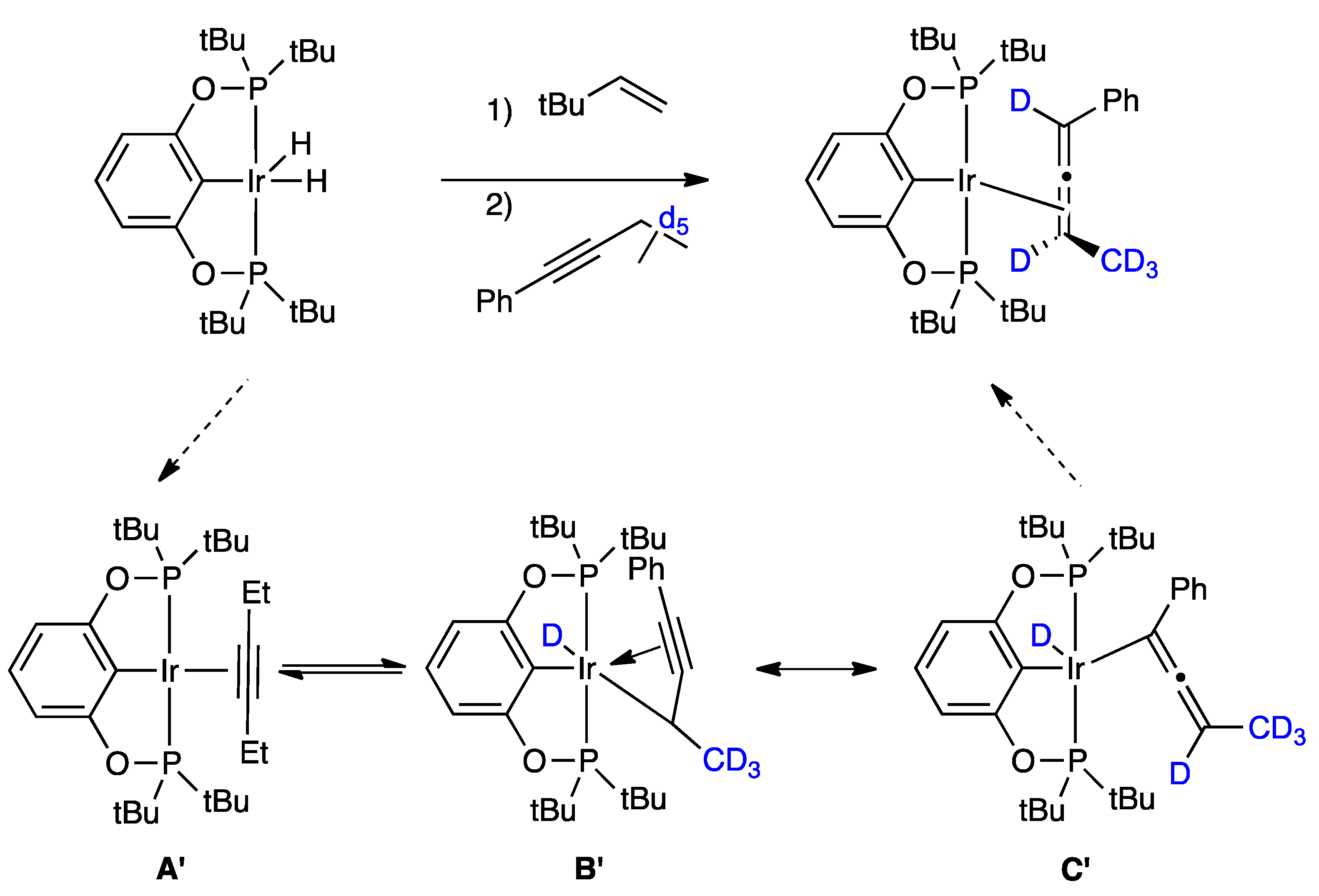{kind=link}
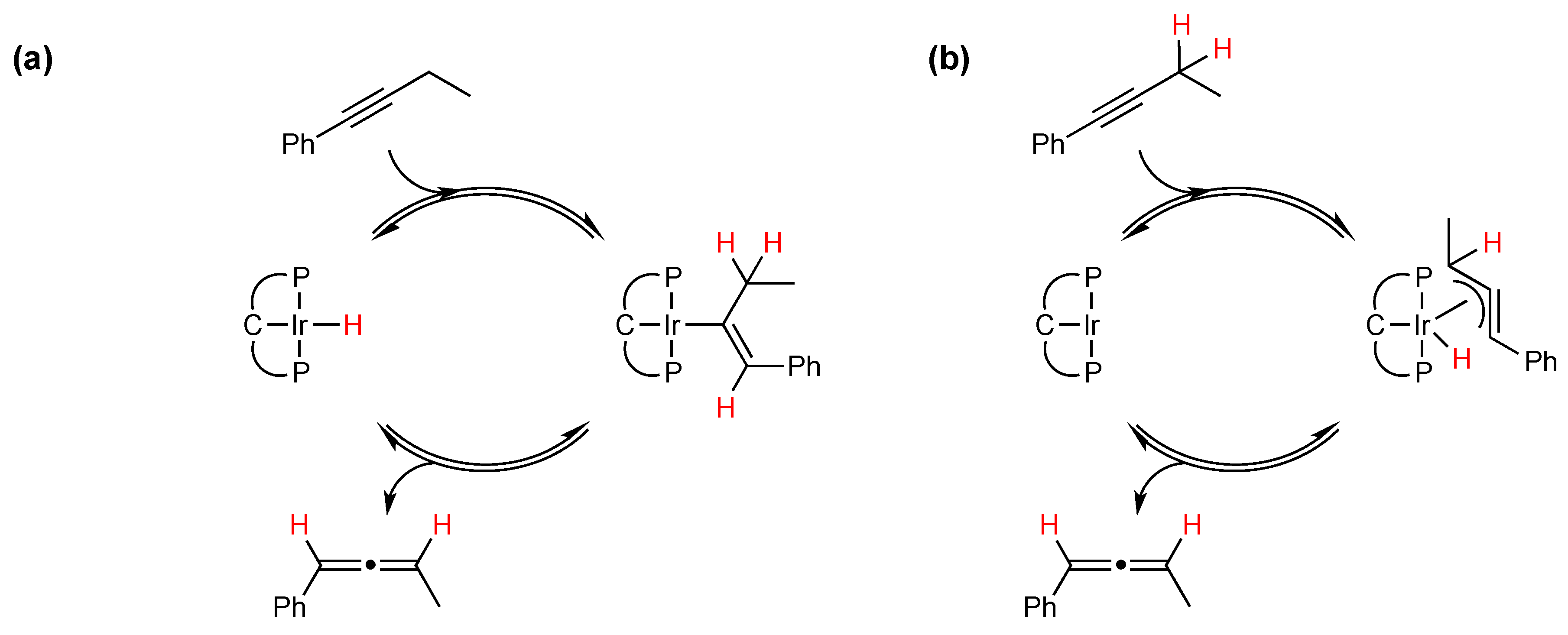{kind=link}
{kind=link}
| kH/kD | T (K) |
|---|---|
| 3.61 | 328.0 |
| 3.77 | 338.0 |
| 3.75 | 348.0 |
2.2. Substrate Scope
| Entry | Alkyne (a) | Complex (b) |
|---|---|---|
| 1 | 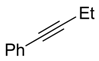 | 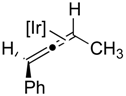 |
| 1-d5 |  | 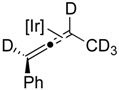 |
| 2 |  | 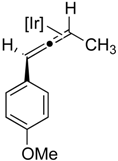 |
| 3 |  |  |
| 4 | 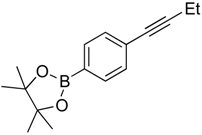 |  |
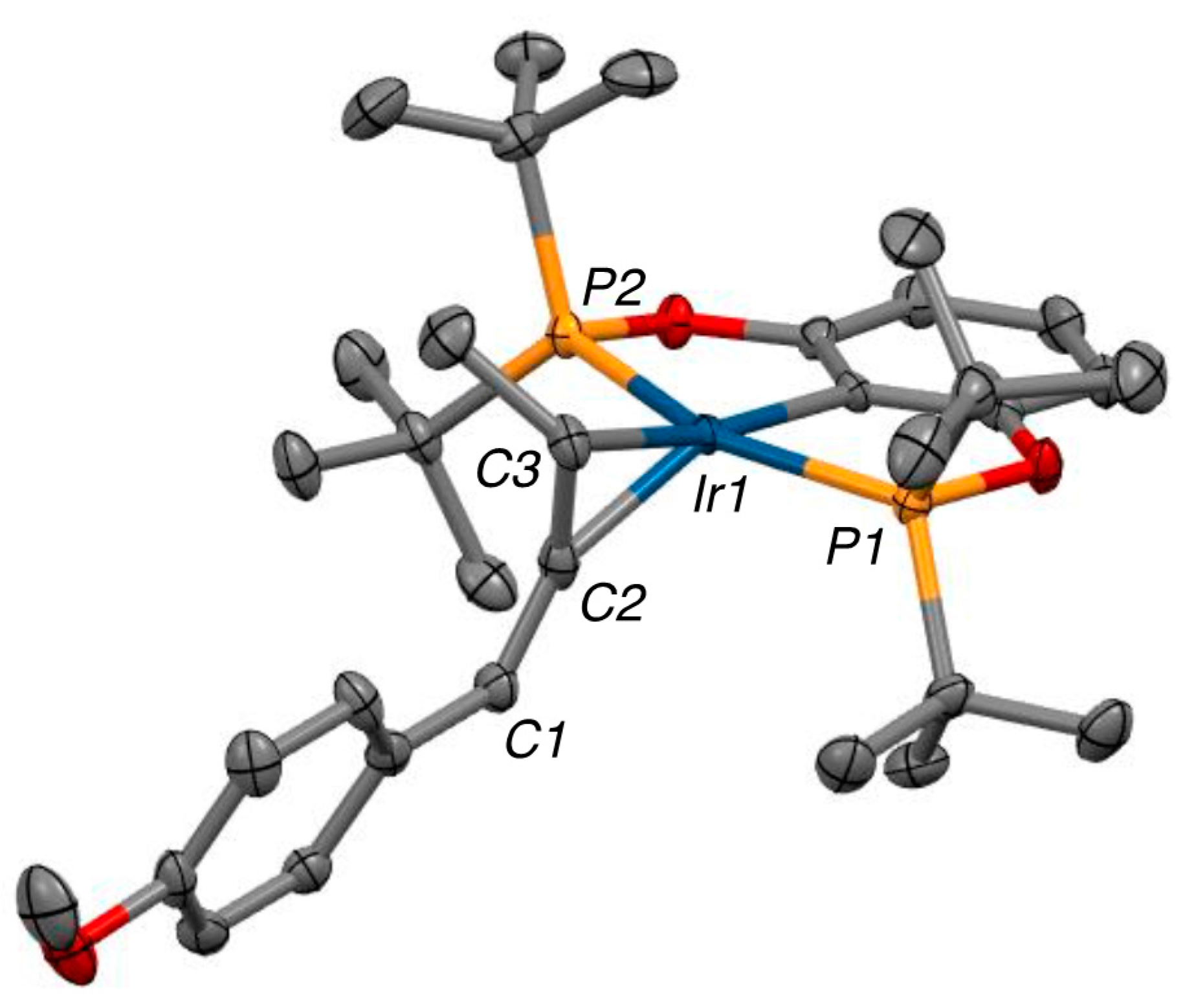
2.3. X-ray Structure
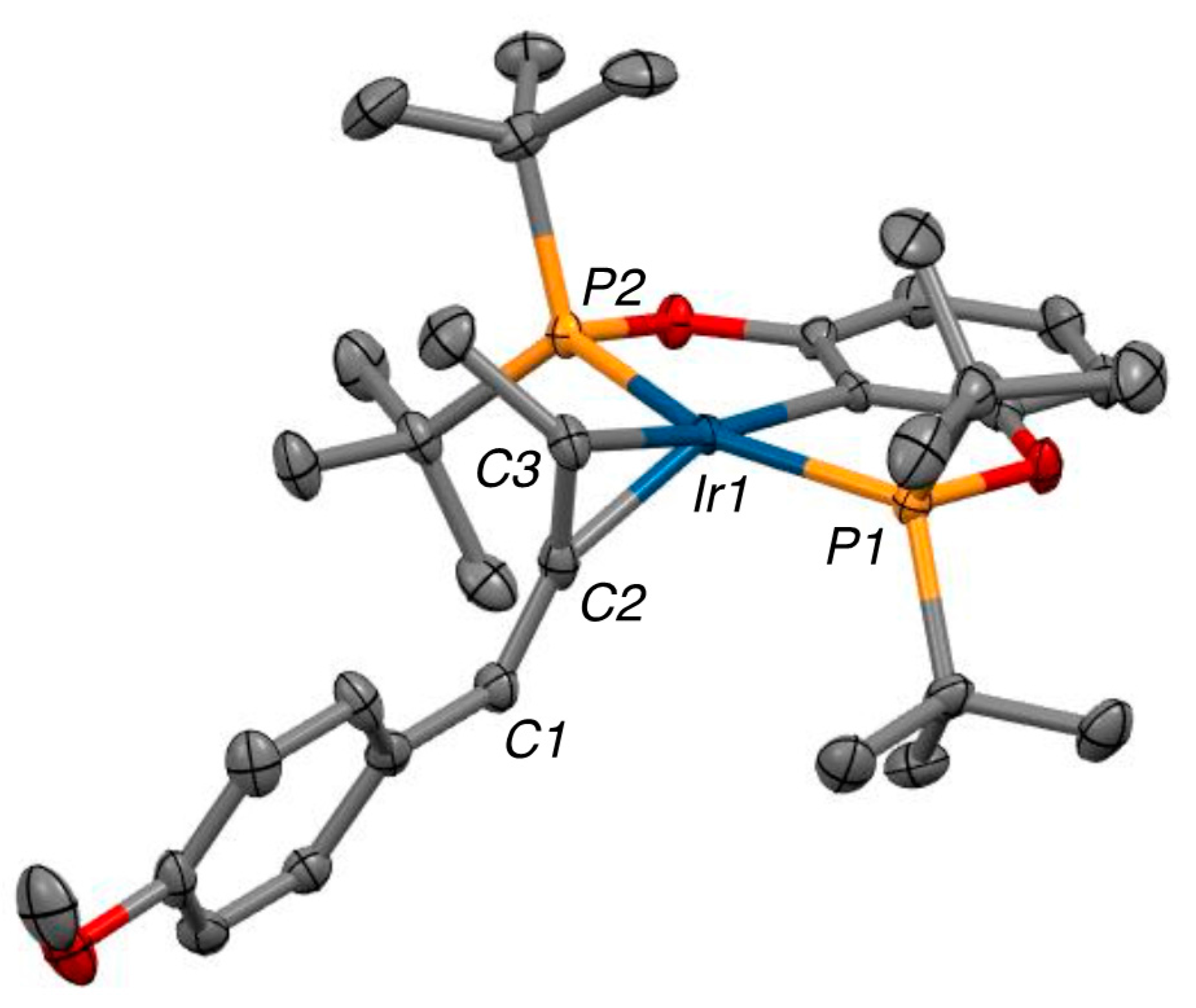
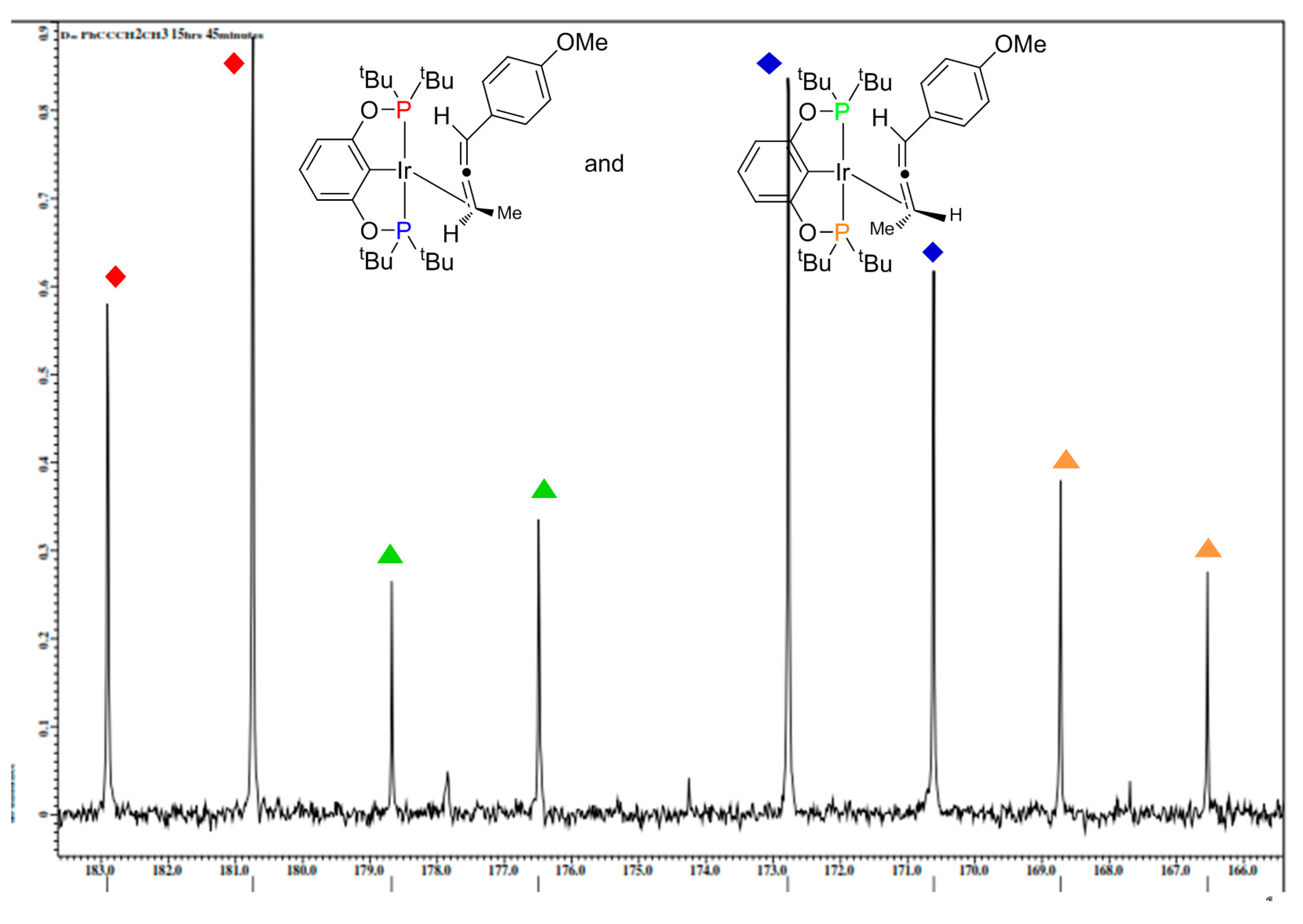
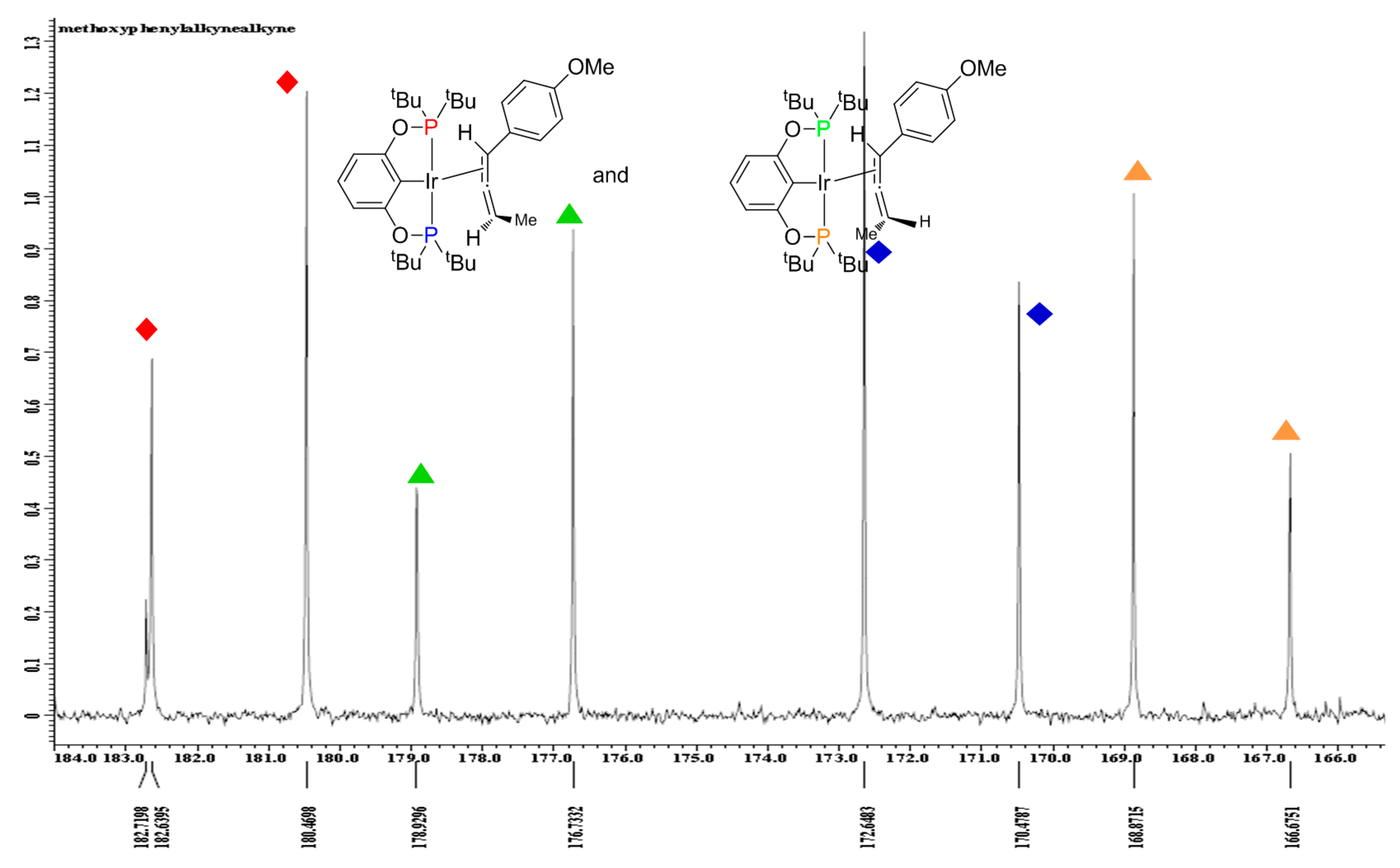
2.4. Liberation of Free Allenes Using Carbon Monoxide, Possibility of a Catalytic Isomerization Methodology
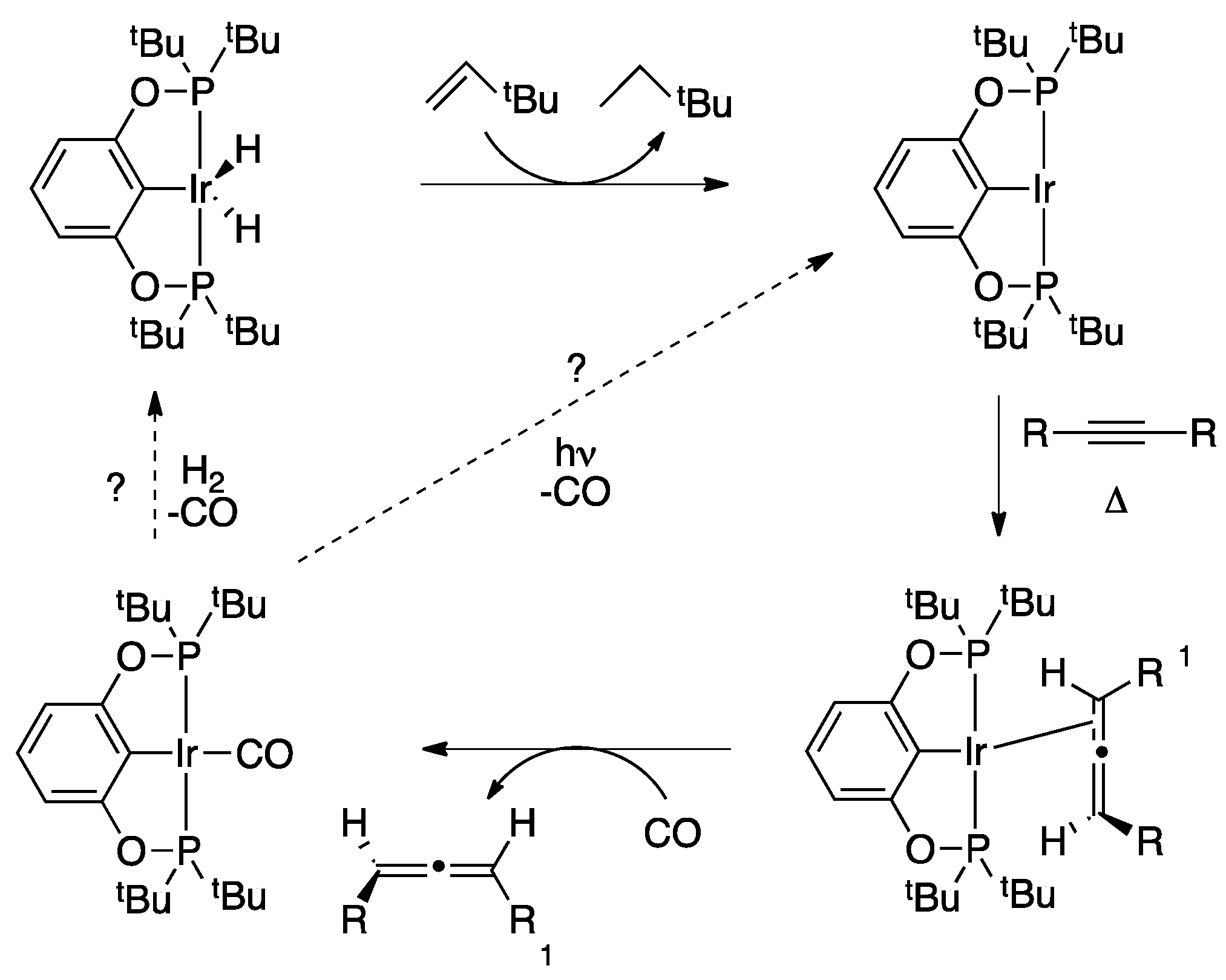
3. Experimental Section
3.1. General Information
3.2. Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- BrÜckl, T.; Baxter, R.D.; Ishihara, Y.; Baran, P.S. Innate and Guided C–H Functionalization Logic. Acc. Chem. Res. 2012, 45, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, W.R.; Baran, P.S. C–H Functionalization Logic in Total Synthesis. Chem. Soc. Rev. 2011, 40, 1976–1991. [Google Scholar] [CrossRef] [PubMed]
- Dobereiner, G.; Crabtree, R. Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition-Metal Catalysis. Chem. Rev. 2010, 110, 681–703. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Organometallic Alkane CH Activation. J. Organomet. Chem. 2004, 689, 4083–4091. [Google Scholar] [CrossRef]
- Findlater, M.; Choi, J.W.; Goldman, A.S.; Brookhart, M. Alkane Dehydrogenation. In Alkane C–H Activation by Single-Site Metal Catalysis; Perez, P.J., Ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Choi, J.; MacArthur, A.H.R.; Brookhart, M.; Goldman, A.S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Balcells, D.; Parent, A.R.; Crabtree, R.H.; Eisenstein, O. Cp* Iridium Precatalysts for Selective C–H Oxidation via Direct Oxygen Insertion: A Joint Experimental/Computational Study. ACS Catal. 2011, 2, 208–218. [Google Scholar] [CrossRef]
- Zhou, M.; Hintermair, U.; Hashiguchi, B.G.; Parent, A.R.; Hashmi, S.M.; Elimelech, M.; Periana, R.A.; Brudvig, G.W.; Crabtree, R.H. Cp* Iridium Precatalysts for Selective C–H Oxidation with Sodium Periodate as the Terminal Oxidant. Organometallics 2013, 32, 957–965. [Google Scholar] [CrossRef]
- Zhou, M.; Schley, N.D.; Crabtree, R.H. Cp* Iridium Complexes Give Catalytic Alkane Hydroxylation with Retention of Stereochemistry. J. Am. Chem. Soc. 2010, 132, 12550–12551. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Guironnet, D.; Findlater, M.; Brookhart, M. Synthesis of p-Xylene from Ethylene. J. Am. Chem. Soc. 2012, 134, 15708–15711. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, R.; Punji, B.; Findlater, M.; Supplee, C.; Schinski, W.; Brookhart, M.; Goldman, A.S. Catalytic Dehydroaromatization of n-Alkanes by Pincer-Ligated Iridium Complexes. Nat. Chem. 2011, 3, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.E.; Heinekey, D.M.; Goldman, A.S.; Goldberg, K.I. Regeneration of an Iridium(III) 229 Complex Active for Alkane Dehydrogenation Using Molecular Oxygen. Organometallics 2014, 33, 1337–1340. [Google Scholar] [CrossRef]
- Pahls, D.R.; Allen, K.E.; Goldberg, K.I.; Cundari, T.R. Understanding the Effect of Ancillary Ligands on Concerted Metalation–Deprotonation by (dmPhebox)Ir(OAc)2(H2O) Complexes: A DFT Study. Organometallics 2014, 33, 6413–6419. [Google Scholar] [CrossRef]
- Allen, K.E.; Heinekey, D.M.; Goldman, A.S.; Goldberg, K.I. Alkane Dehydrogenation by C–H Activation at Iridium(III). Organometallics 2013, 32, 1579–1582. [Google Scholar] [CrossRef]
- Owens, C.P.; Varela-Alvarez, A.; Boyarskikh, V.; Musaev, D.G.; Davies, H.M.L.; Blakey, S.B. Iridium(iii)-bis(oxazolinyl)phenyl Catalysts for Enantioselective C-H Functionalization. Chem. Sci. 2013, 4, 2590–2596. [Google Scholar] [CrossRef]
- Phadke, N.; Findlater, M. Formation of Iridium(III) Allene Complexes via Isomerization of Internal Alkynes. Organometallics 2014, 33, 16–18. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Röder, A. Synthesis of Allenes with Organometallic Reagents. Tetrahedron 2004, 60, 11671–11694. [Google Scholar] [CrossRef]
- Yu, S.; Ma, S. How Easy Are the Syntheses of Allenes? Chem. Commun. 2011, 47, 5384. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ma, S. Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses. Angew. Chem. Int. Ed. 2012, 51, 3074–3112. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.K.; Frantz, D.E. Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment. ACS Catal. 2014, 4, 519–528. [Google Scholar] [CrossRef]
- Gotzig, J.; Otto, H.; Werner, H. Bildung Eines Neuartigen C4-Liganden Durch Oxidative Kupplung Zweir Metallgebunder Alkinylgruppen. J. Organomet. Chem. 1985, 287, 247. [Google Scholar] [CrossRef]
- Bai, T.; Ma, S.; Jia, G. Insertion Reactions of Allenes with Transition Metal Complexes. Coord. Chem. Rev. 2009, 253, 423–448. [Google Scholar] [CrossRef]
- Casey, C. Acid-Catalyzed Isomerization of Rhenium Alkyne Complexes to Rhenium Allene Complexes via 1-Metallacyclopropene Intermediates. Organometallics 1998, 17, 4620–4629. [Google Scholar] [CrossRef]
- Werner, H.; Schwab, P.; Mahr, N.; Wolf, J. Synthesis, Reactions, and Molecular Structure of Bis(arsane) and Bis(stibane) Rhodium(I) Complexes trans-[RhCl(L)(EiPr3)2](E=As, Sb) Including the Rhodium–Mediated Rearrangement of Alkynes to the Isomeric Allenes. Chemische Berichte 1992, 125, 2641–2650. [Google Scholar] [CrossRef]
- Coughlan, S. Rearrangement of cyclooctyne to 1,2-cyclooctadiene within the coordination sphere of CpMn(CO)2. J. Organomet. Chem. 1993, 450, 151–155. [Google Scholar] [CrossRef]
- Pinkas, J.; Gyepes, R.; Císařová, I.; Kubišta, J.; Horáček, M.; Mach, K. Displacement of ethene from the decamethyltitanocene-ethene complex with internal alkynes, substituent-dependent alkyne-to-allene rearrangement, and the electronic transition relevant to the back-bonding interaction. Dalton Trans. 2015, 44, 7276–7291. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L.; Pombeiro, A.J.L.; Pickett, C.J.; Richards, R.L. An eta2-allene complex of rhenium formed form an alkyne: X-ray structure of [ReCl(?2-H2C=C=CHPh)(Ph2PCH2CH2PPh2)2]. J. Chem. Soc. Chem. Commun. 1984, 992–993. [Google Scholar] [CrossRef]
- Wen, T.B.; Zhou, Z.Y.; Lau, C.-P.; Jia, G. Isomerization of CH3C⋮CPh to Phenylallene Promoted by an Osmium Hydride Complex. Organometallics 2000, 19, 3466–3468. [Google Scholar] [CrossRef]
- Doyle, M.P.; Devora, G.A.; Nefedov, A.O.; High, K.G. Addition/elimination in the rhodium (II) perfluorobutyrate catalyzed hydrosilylation of 1-alkenes. Rhodium hydride promoted isomerization and hydrogenation. Organometallics 1992, 11, 549–555. [Google Scholar] [CrossRef]
- Lee, W.-C.; Wang, C.-H.; Lin, Y.-H.; Shih, W.-C.; Ong, T.-G. Tandem Isomerization and C–H Activation: Regioselective Hydroheteroarylation of Allylarenes. Org. Lett. 2013, 15, 5358–5361. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J.; O’Riordan, T.J.C.; Rosa, C.P. Ruthenium-Catalyzed Isomerization of Terminal Olefins: Applications to Synthesis. Angew. Chem. Int. Ed. 2009, 48, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Seayad, A.; Ahmed, M.; Klein, H.; Jackstell, R.; Gross, T. Internal olefins to linear amines. Science 2002, 297, 1676–1678. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.S.; Roy, A.H.; Huang, Z.; Ahuja, R.; Schinski, W.; Brookhart, M. Catalytic alkane metathesis by tandem alkane dehydrogenation-olefin metathesis. Science 2006, 312, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Parshall, G.W.; Ittel, S.D. Homogeneous Catalysis. The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes, 2nd ed.; Wiley: Hoboken, NJ, USA, 1992. [Google Scholar]
- Cornils, B.; Herrmann, W.A. Applied Homogeneous Catalysis with Organometallic Compounds; Wiley-VCH: Weinheim, Germany, 1996; Volume 2. [Google Scholar]
- Biswas, S. Mechanistic Understanding of Transition-Metal-Catalyzed Olefin Isomerization: Metal-Hydride Insertion-Elimination vs. π-Allyl Pathways. Comments Inorg. Chem. 2015, 35, 301–331. [Google Scholar] [CrossRef]
- Ting, C.-M.; Hsu, Y.-L.; Liu, R.-S. Gold-catalyzed isomerization of unactivated allenes into 1,3-dienes under ambient conditions. Chem. Commun. 2012, 48, 6577–6579. [Google Scholar] [CrossRef]
- Knapp, S.M.M.; Shaner, S.E.; Dimitar, D.K.; Shopov, Y.; Tendler, J.A.; Pudalov, D.M.; Chianese, A.R. Mechanistic Studies of Alkene Isomerization Catalyzed by CCC-Pincer Complexes of Iridium. Organometallics 2014, 33, 473–484. [Google Scholar] [CrossRef]
- Chianese, A.R.; Shaner, S.E.; Tendler, J.A.; Pudalov, D.M.; Shopov, D.Y.; Kim, D.; Rogers, S.L.; Mo, A. Iridium Complexes of Bulky CCC-Pincer N-Heterocyclic Carbene Ligands: Steric Control of Coordination Number and Catalytic Alkene Isomerization. Organometallics 2012, 31, 7359–7367. [Google Scholar] [CrossRef]
- Brown, T.J.; Sugie, A.; Leed, M.G.D.; Widenhoefer, R.A. Structures and Dynamic Solution Behavior of Cationic, Two-Coordinate Gold(I) π-Allene Complexes. Chem. Eur. J. 2012, 18, 6959–6971. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Peng, T.-S.; Arif, A.M.; Gladysz, J.A. Synthesis, Structure, and reactivity of allene complexes of the chiral rhenium fragment [(eta5-C5H5)Re(NO)(PPh3)]+. Organometallics 1992, 11, 3232. [Google Scholar] [CrossRef]
- Omrcen, T.; Conti, N.J.; Jones, W.M. Synthesis and ring-opening reactions of alpha-fluoro-substituted cyclopropanes sigma-bonded to iron. Organometallics 1991, 10, 913–917. [Google Scholar] [CrossRef]
- Chacon, S.T.; Chisholm, M.C.; Folting, K.; Huffman, J.C.; Hampden-Smith, M.J. Allene adducts of ditungsten hexaalkoxides. Three modes of allene coordination to dinuclear centers as seen in the structures of W2(OBu-tert)6(C3H4), W2(OBu-tert)6(C3H4)2, and W2(Obu-tert)6(C3H4)(CO)2. Organometallics 1991, 10, 3722–3735. [Google Scholar] [CrossRef]
- Lundquist, E.G.; Folting, K.; Streib, W.E.; Huffman, J.C.; Eisenstein, O.; Caulton, K.G. Reactivity of the molecular hydrogen complex [IrH4(PMe2Ph)3]BF4 towards olefins. The origin of stereochemical rigidity of M(PR3)3(olefin)2 species. J. Am. Chem. Soc. 1990, 112, 855–863. [Google Scholar] [CrossRef]
- Winchester, W.R.; Jones, W.M. Bis(triphenylphosphine)platinum complexes of cycloheptatetraene benzocycloheptatetraene and dibenzocycloheptatetraene. Organometallics 1985, 4, 2228–2230. [Google Scholar] [CrossRef]
- Okamoto, K.; Kai, Y.; Yasuoka, N.; Kasai, N. The molecular structure of bis(triphenylphosphine)allene-palladium. J. Organomet. Chem. 1974, 65, 427–441. [Google Scholar] [CrossRef]
- Göttker-Schnetmann, I.; White, P.; Brookhart, M. Synthesis and Properties of Iridium Bis(phosphinite) Pincer Complexes (p-XPCP)IrH2, (p-XPCP)Ir(CO), (p-XPCP)Ir(H)(aryl), and {(p-XPCP)Ir}2{μ-N2} and Their Relevance in Alkane Transfer Dehydrogenation. Organometallics 2004, 23, 1766–1776. [Google Scholar] [CrossRef]
- Zhang, W.; Kraft, S.; Moore, J. Highly active trialkoxymolybdenum(VI) alkylidyne catalysts synthesized by a reductive recycle strategy. J. Am. Chem. Soc. 2004, 126, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phadke, N.; Findlater, M. Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology. Molecules 2015, 20, 20195-20205. https://doi.org/10.3390/molecules201119686
Phadke N, Findlater M. Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology. Molecules. 2015; 20(11):20195-20205. https://doi.org/10.3390/molecules201119686
Chicago/Turabian StylePhadke, Neha, and Michael Findlater. 2015. "Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology" Molecules 20, no. 11: 20195-20205. https://doi.org/10.3390/molecules201119686
APA StylePhadke, N., & Findlater, M. (2015). Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology. Molecules, 20(11), 20195-20205. https://doi.org/10.3390/molecules201119686