
Glutathionylspermidine in the Modification of Protein SH Groups: The Enzymology and Its Application to Study Protein Glutathionylation

{kind=link}
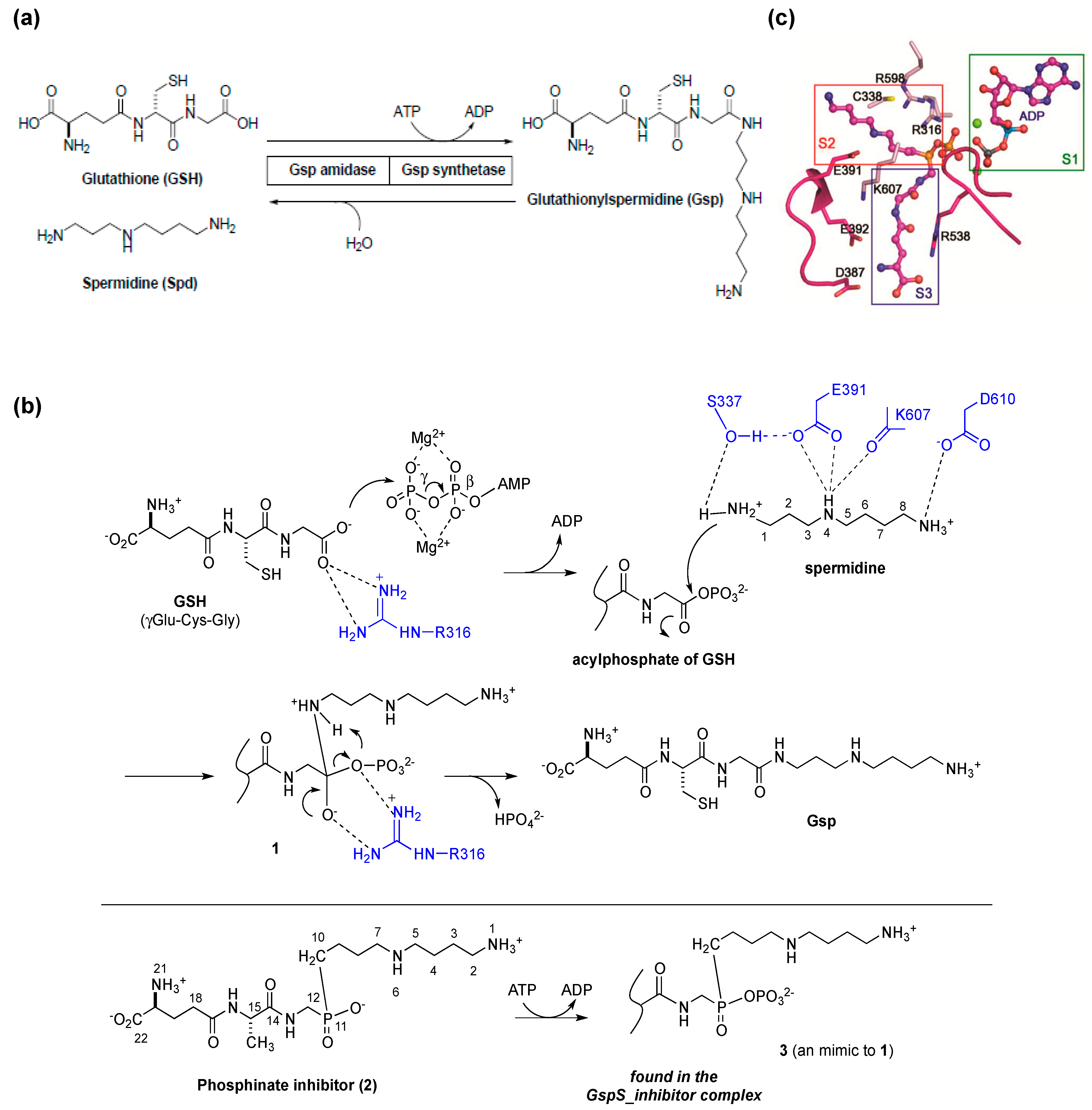{kind=link}
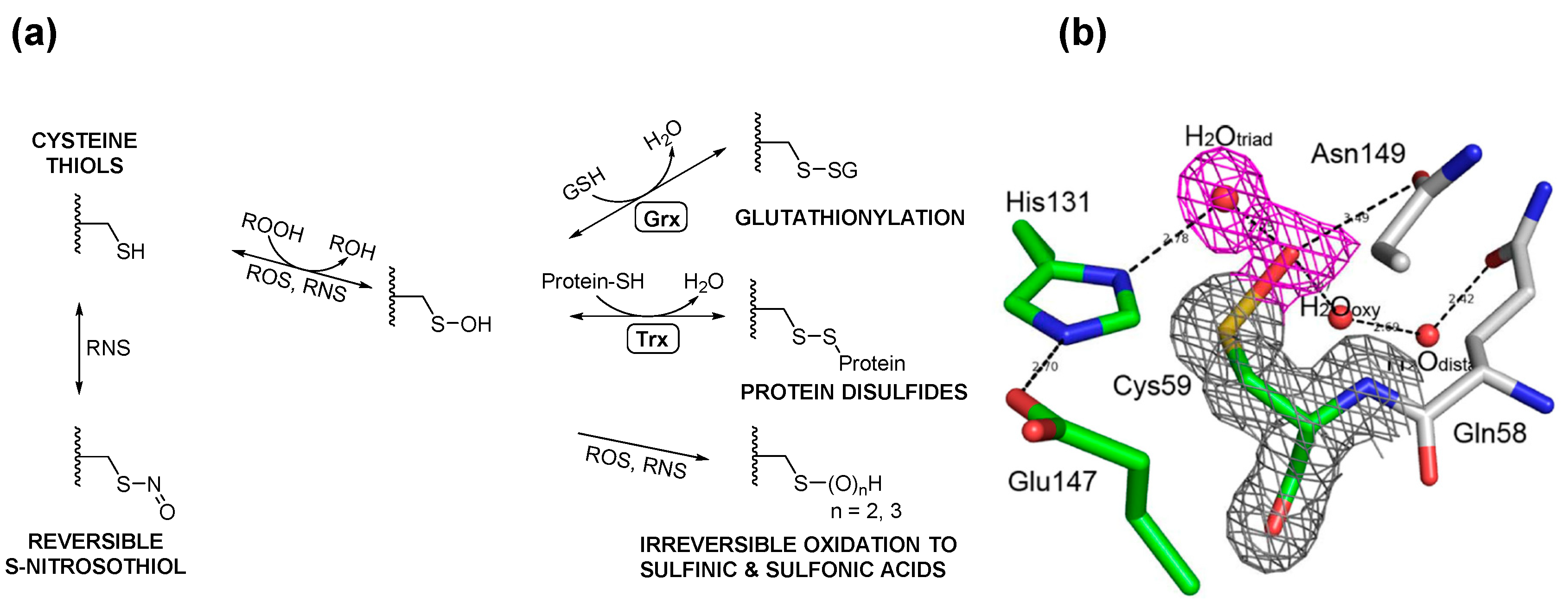{kind=link}
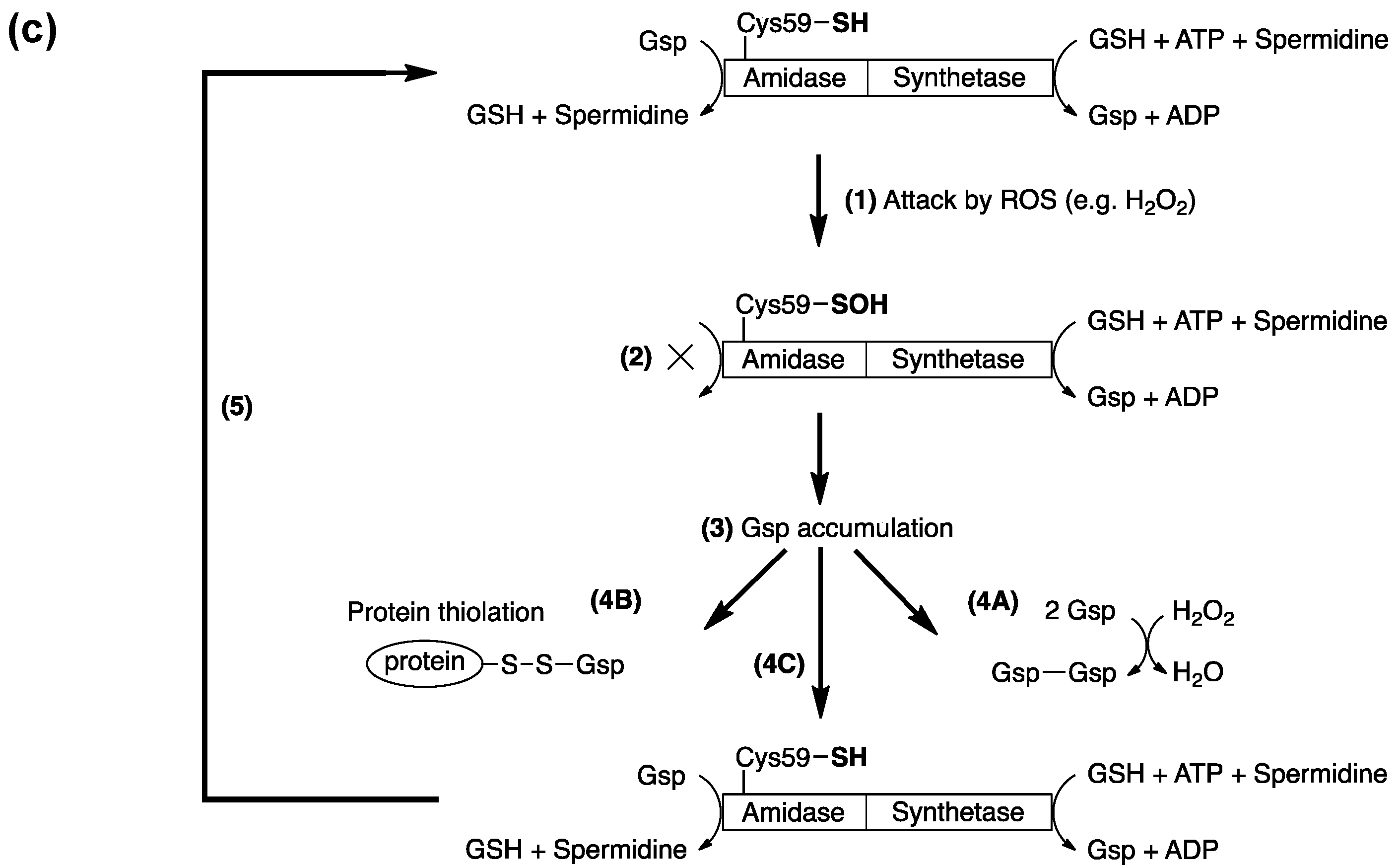{kind=link}
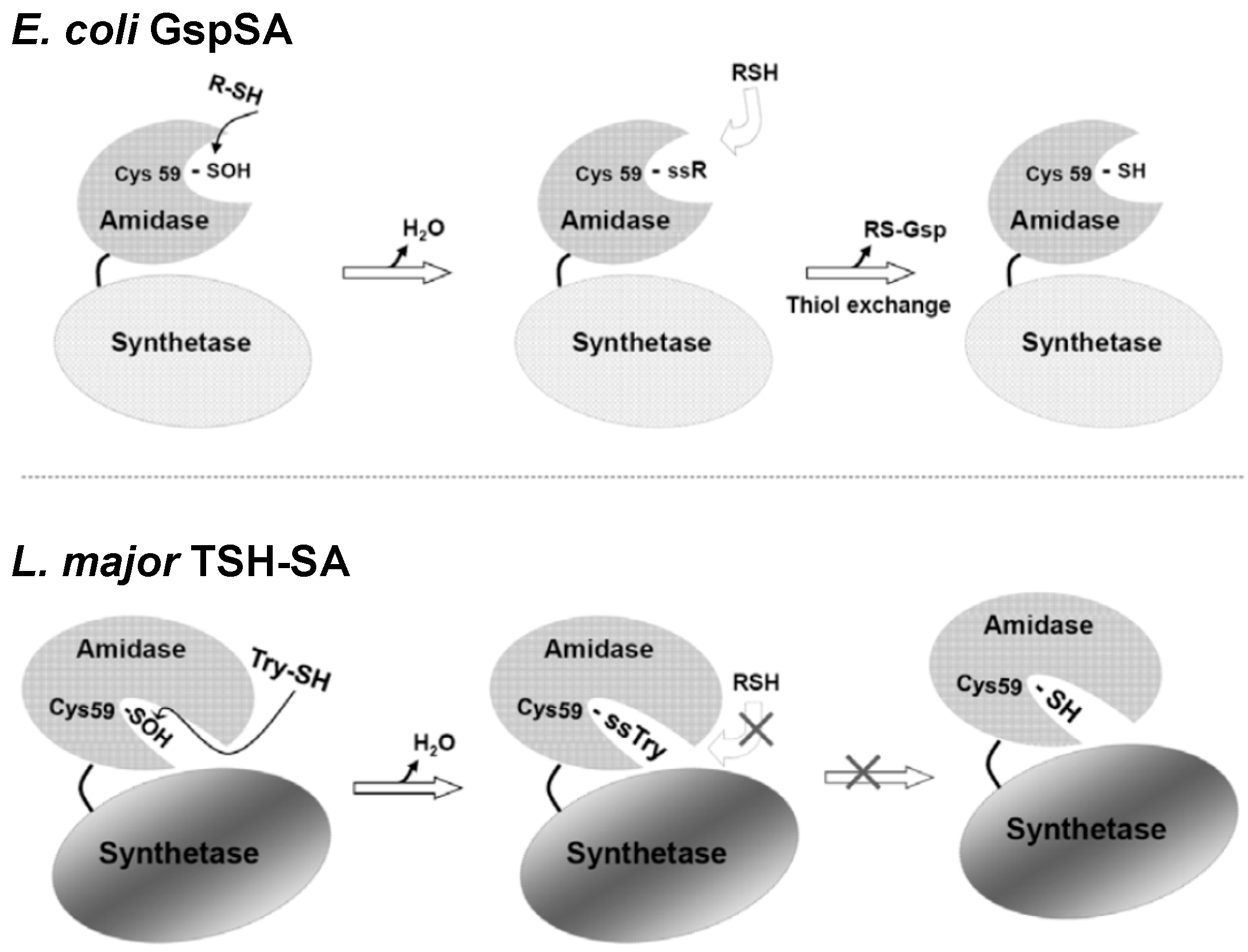{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Roles of Glutathione in Prokaryotes
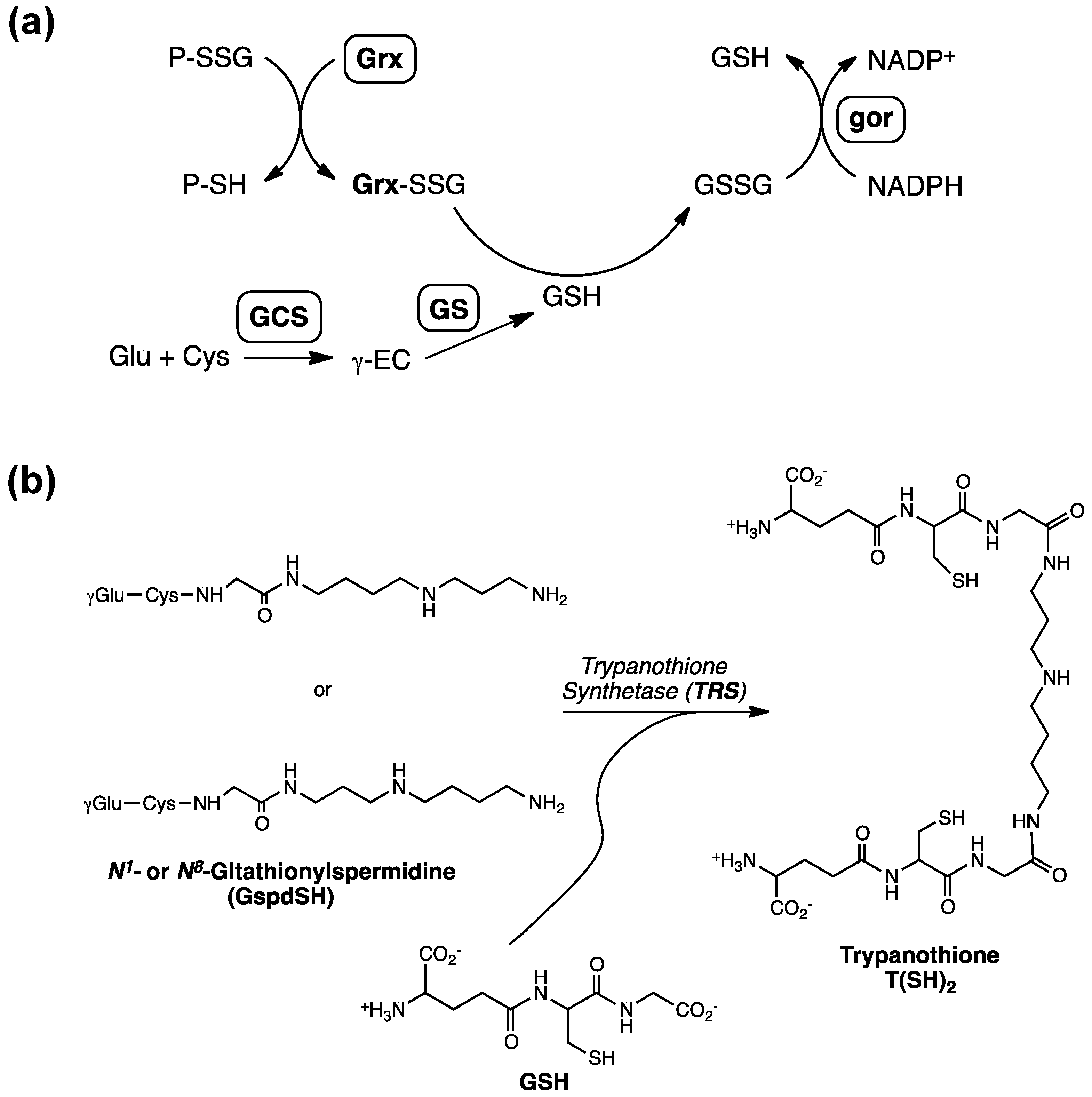
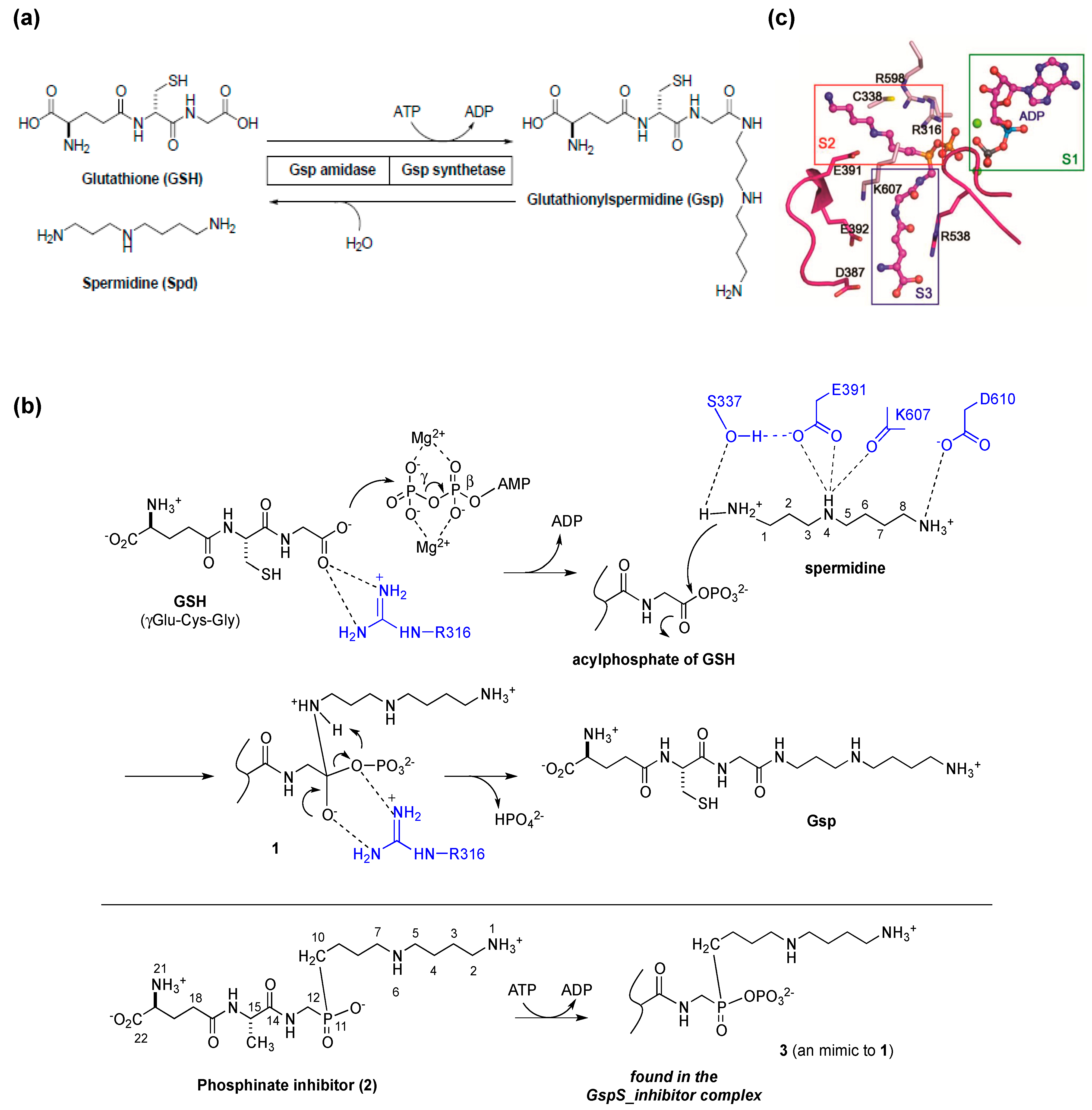
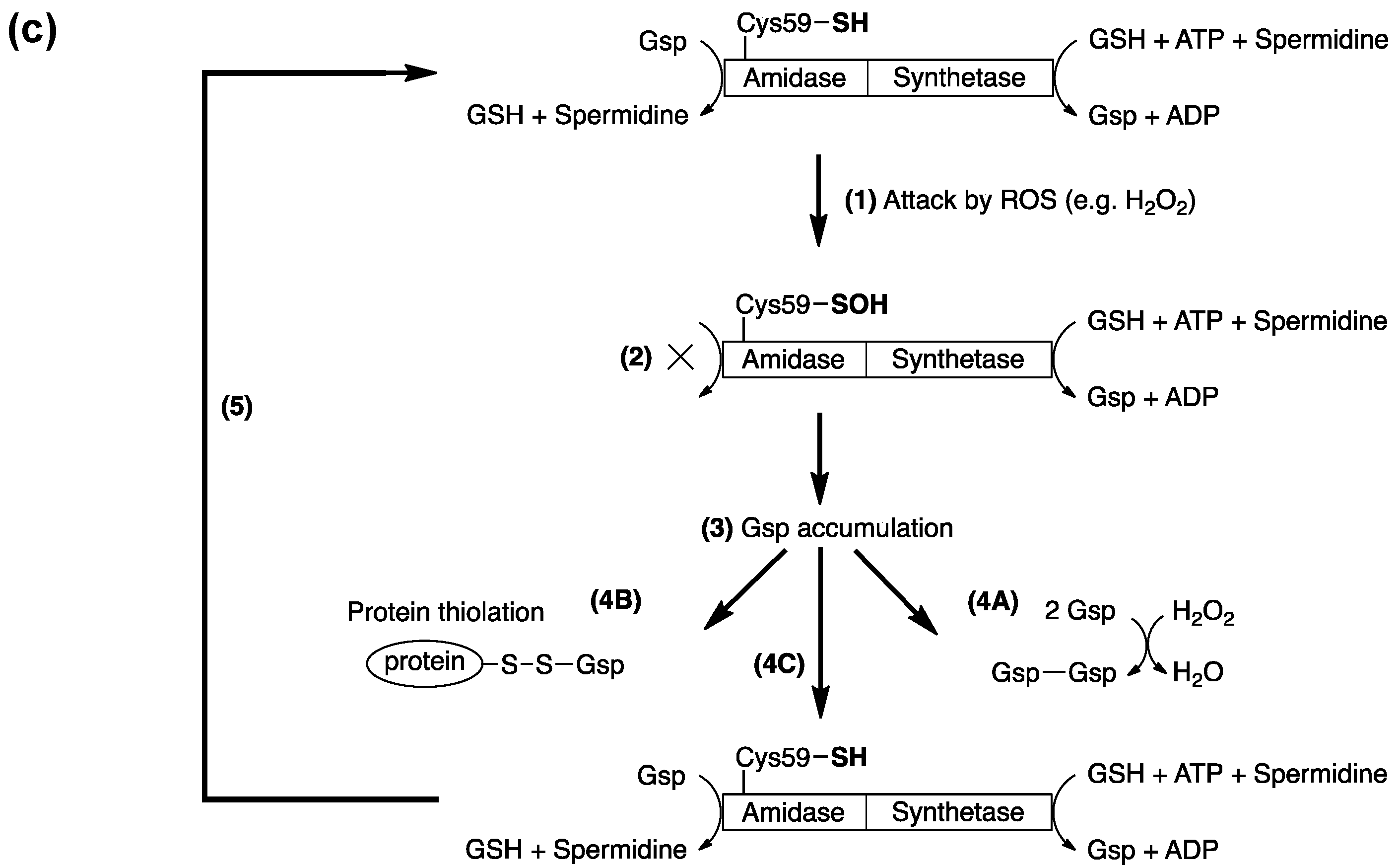
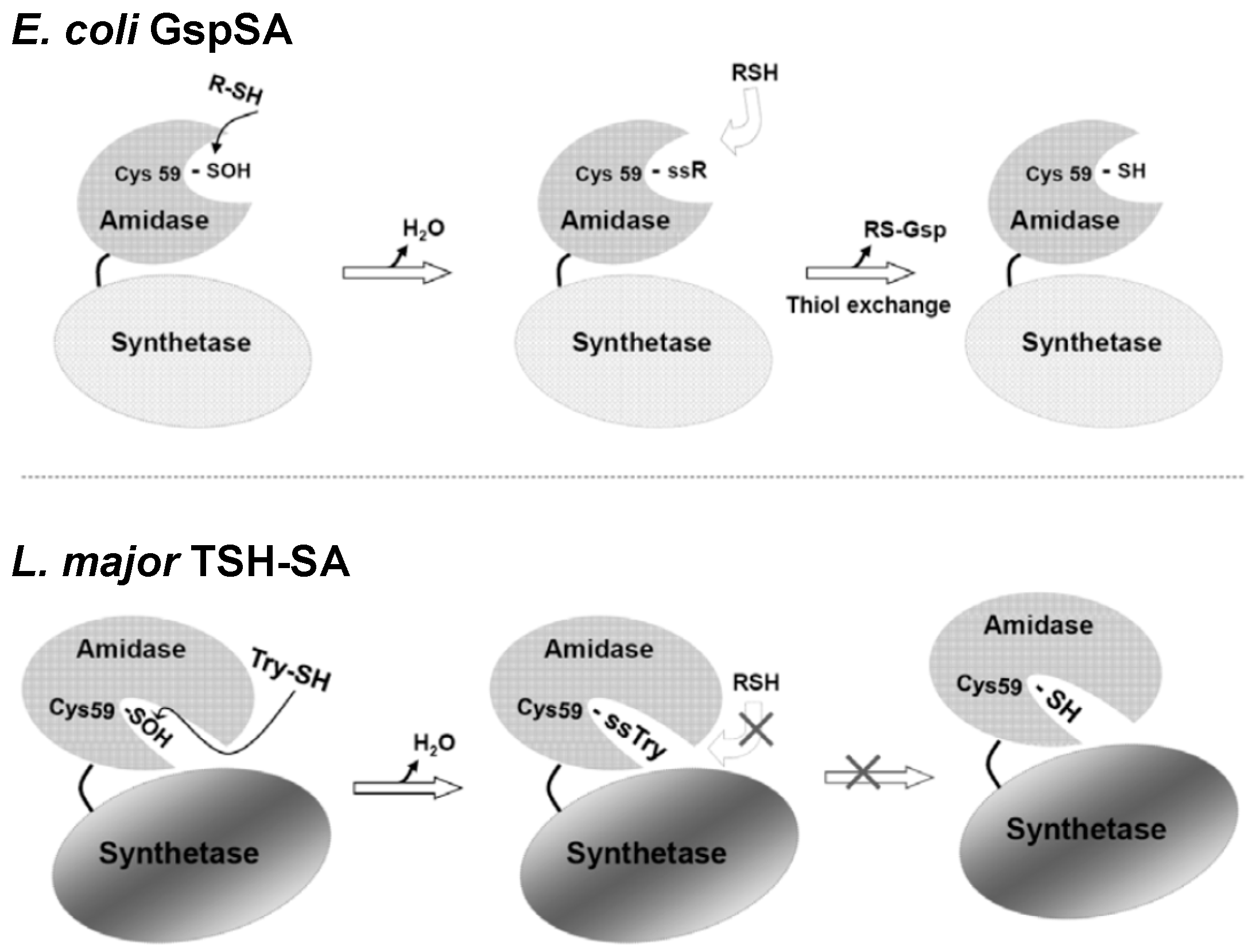
2. Glutathionylspermidine (Gsp) in E. coli and Protozoa
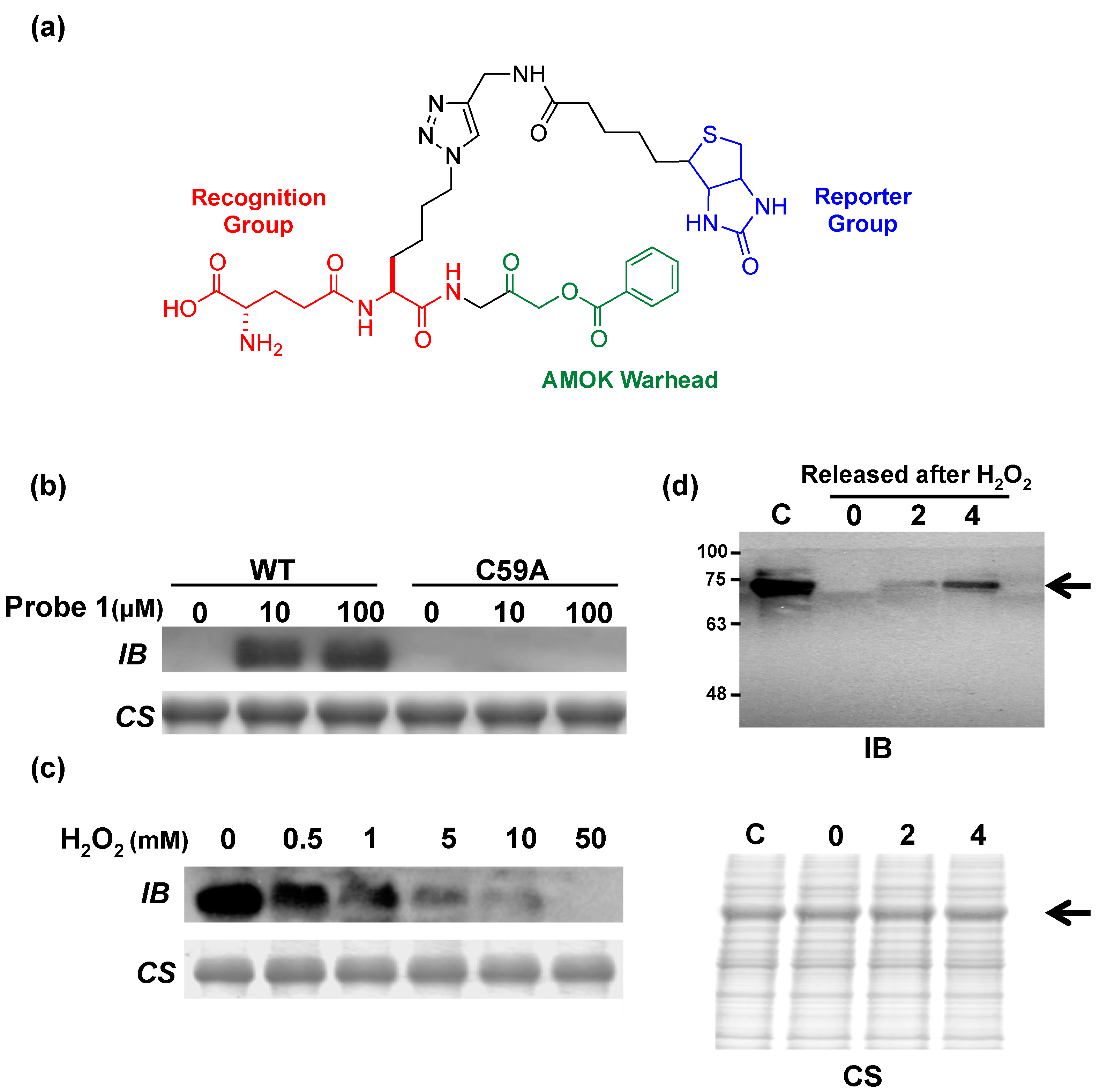
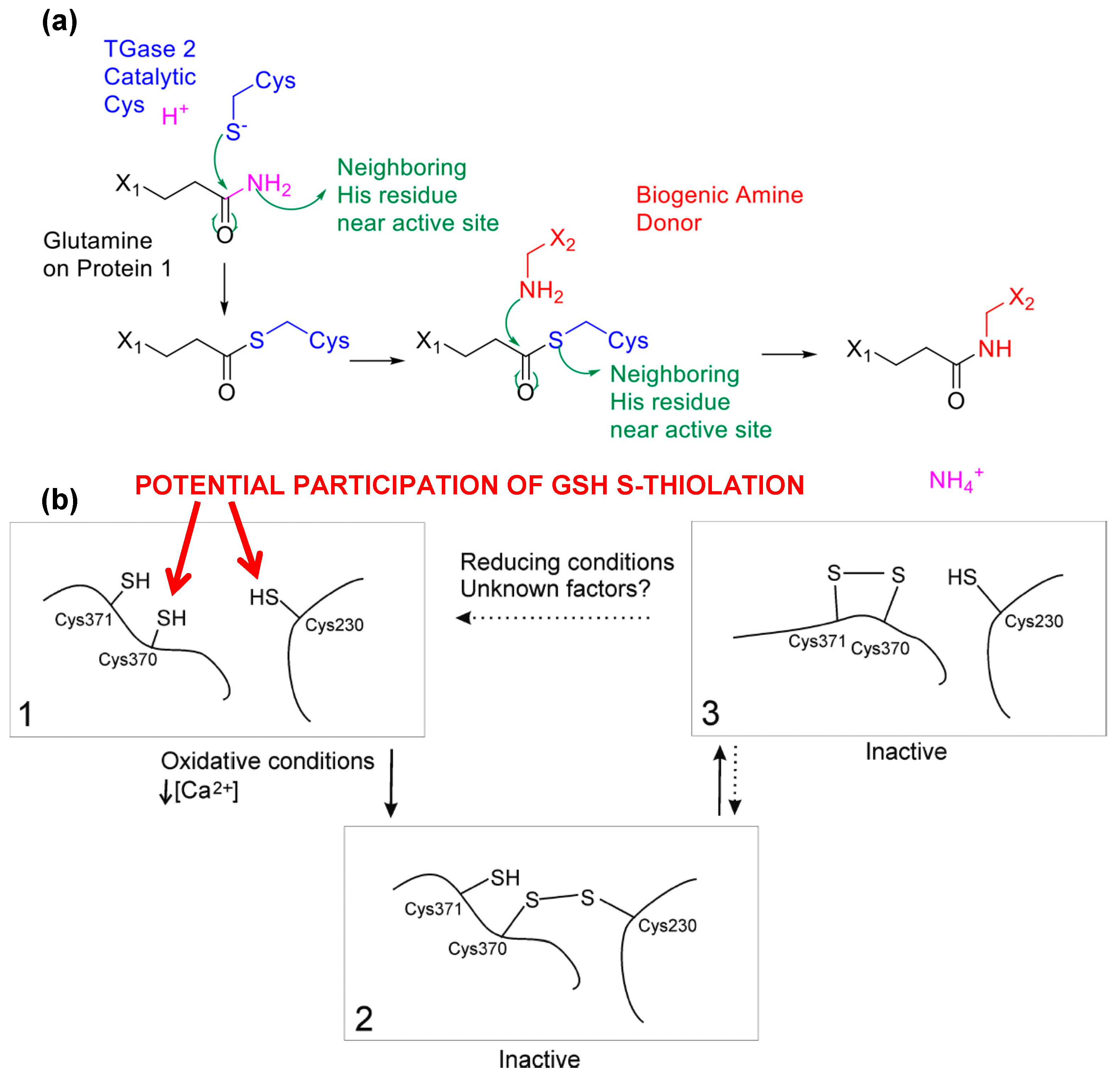
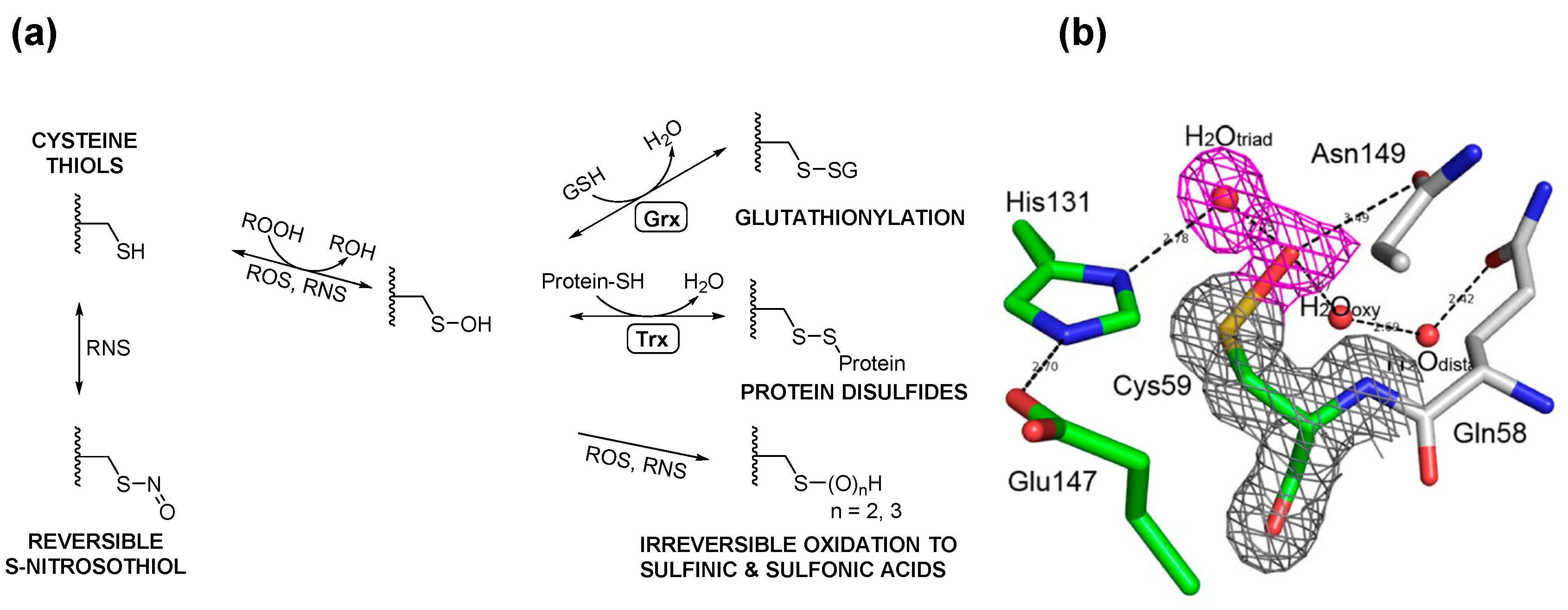
3. Posttranslational Thiol Modifications of Cysteine
4. Protein S-Thiolation with GSH and Gsp
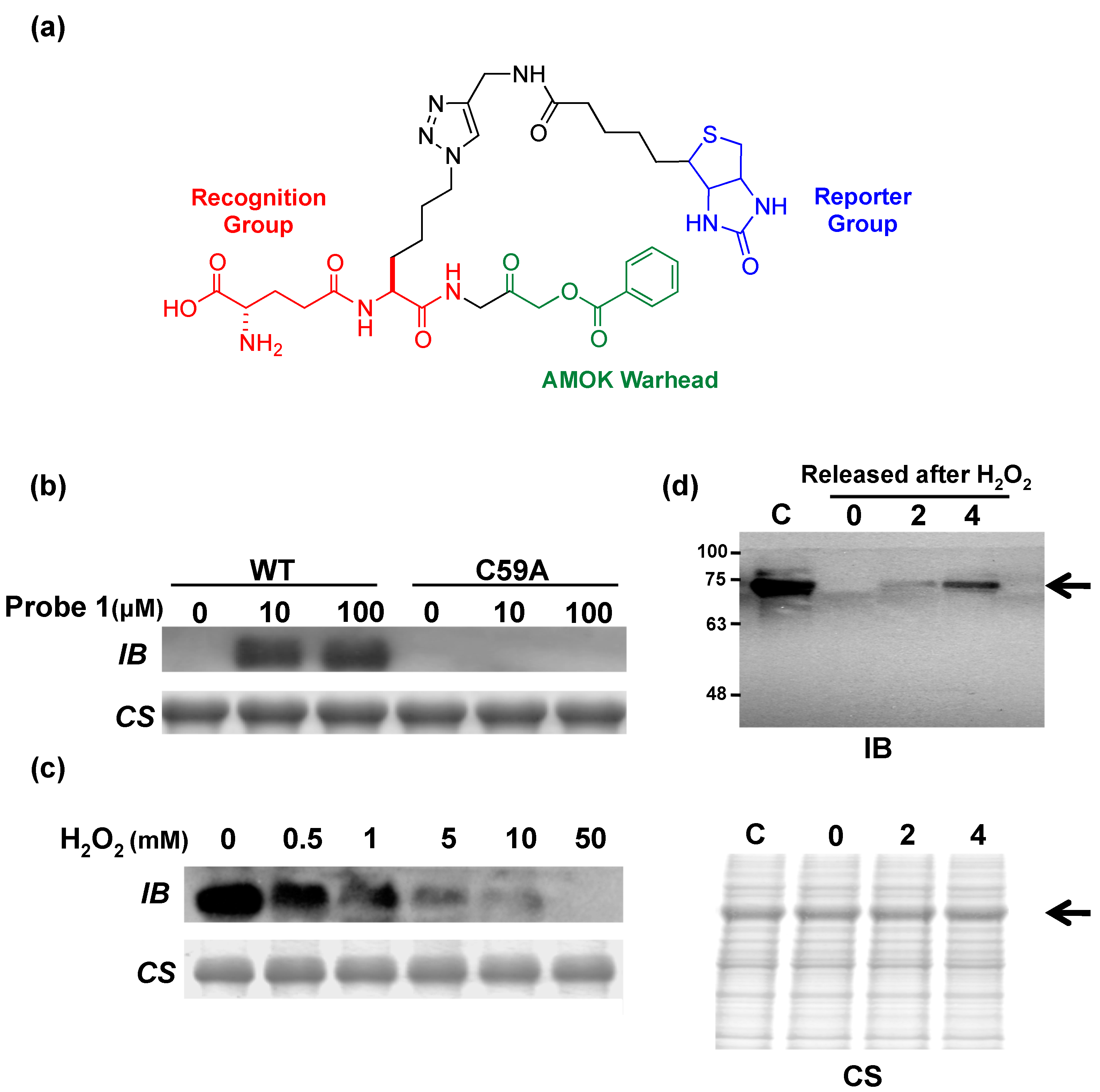
5. Potential Biological Applications
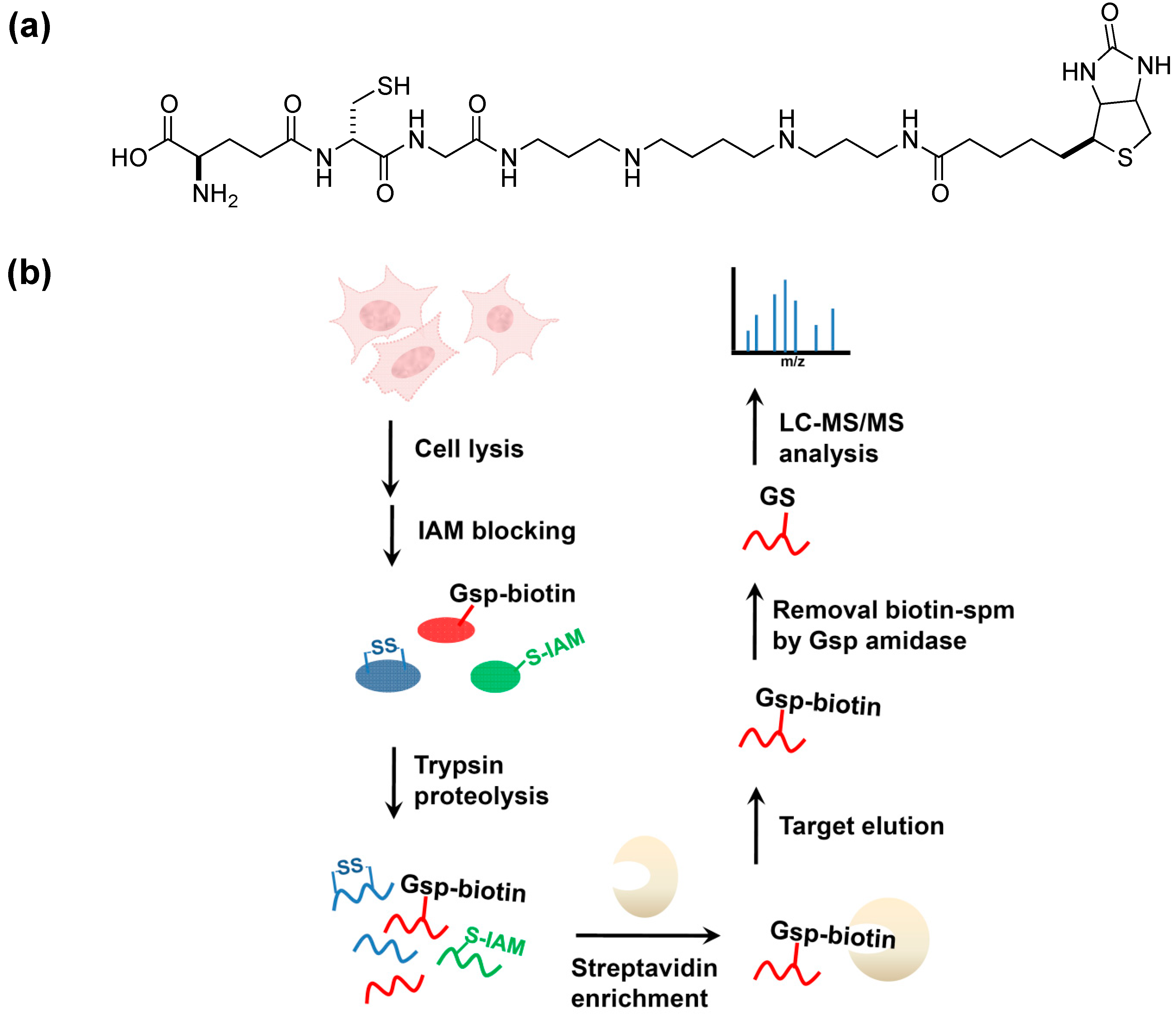
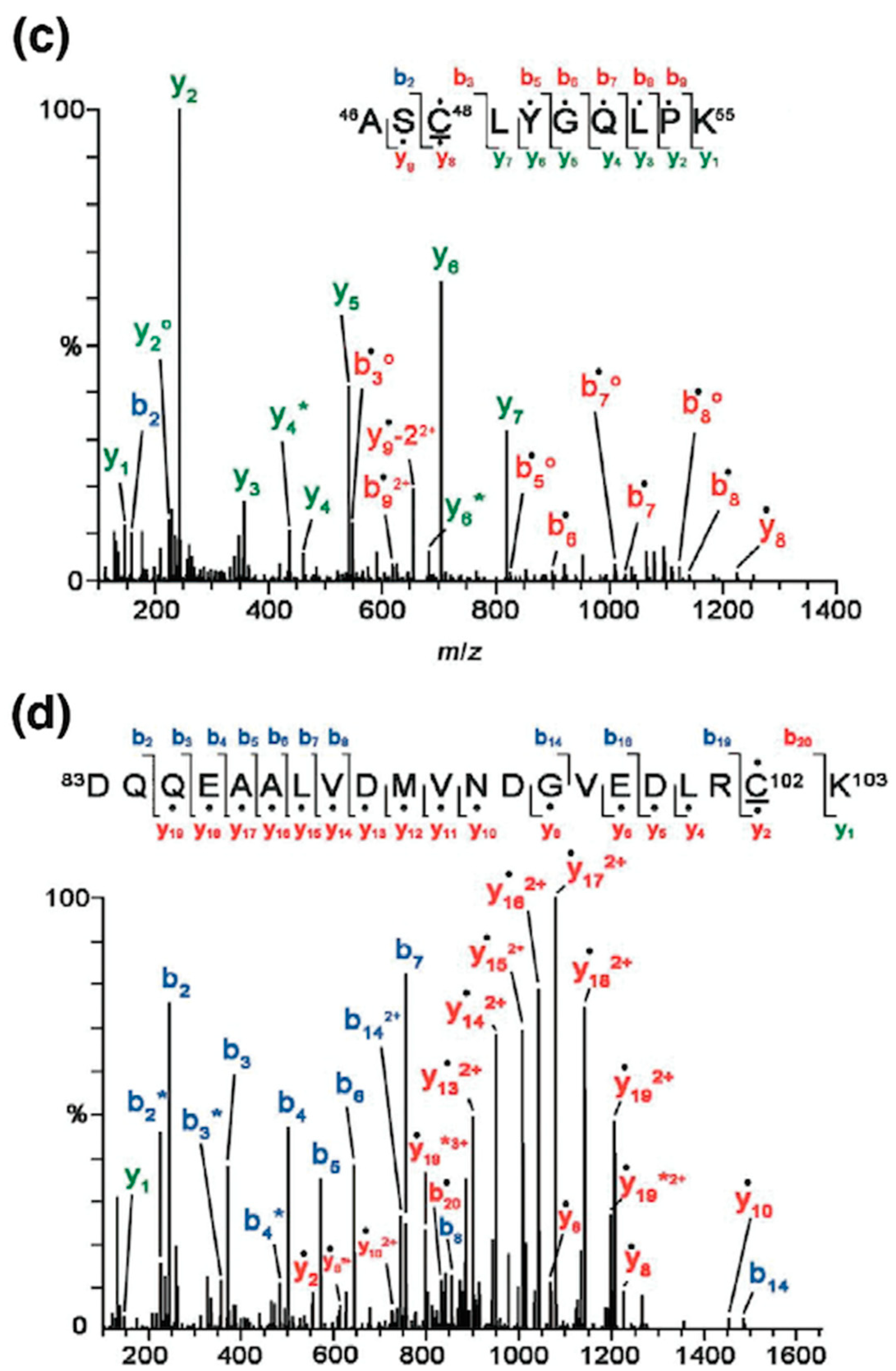
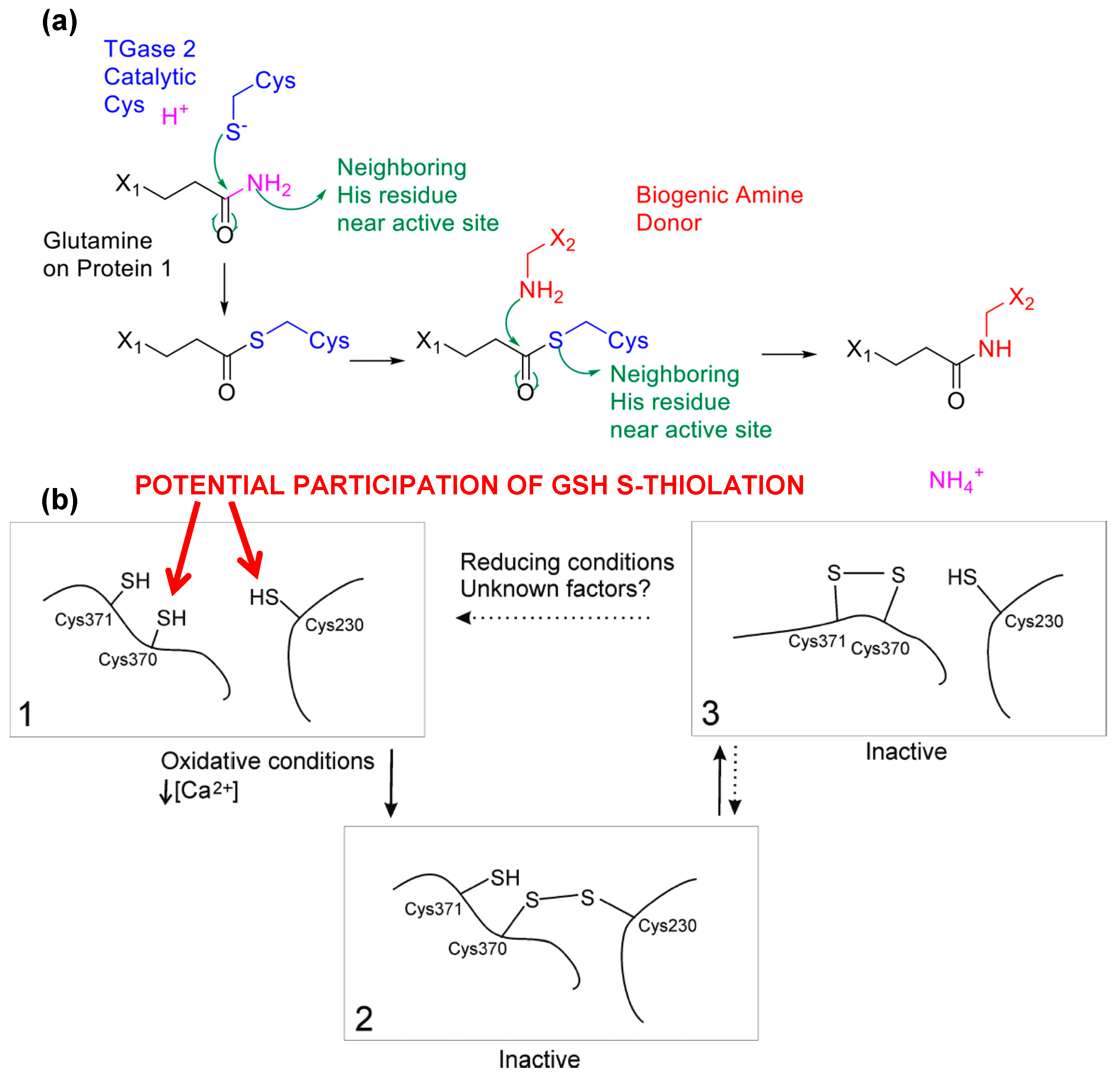
6. Conclusions and Future Aspects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Masip, L.; Veeravalli, K.; Georgiou, G. The many faces of glutathione in bacteria. Antioxid. Redox Signal. 2006, 8, 753–762. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Fahey, R.C.; Brown, W.C.; Adams, W.B.; Worsham, M.B. Occurrence of glutathione in bacteria. J. Bacteriol. 1978, 133, 1126–1129. [Google Scholar] [PubMed]
- Newton, G.L.; Arnold, K.; Price, M.S.; Sherrill, C.; Delcardayre, S.B.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Fahey, R.C.; Davis, C. Distribution of thiols in microorganisms: Mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 1996, 178, 1990–1995. [Google Scholar] [PubMed]
- Fahey, R.C.; Sundquist, A.R. Evolution of glutathione metabolism. Adv. Enzymol. Relat. Areas Mol. Biol. 1991, 64, 1–53. [Google Scholar] [PubMed]
- Fahey, R.C.; Buschbacher, R.M.; Newton, G.L. The evolution of glutathione metabolism in phototrophic microorganisms. J. Mol. Evol. 1987, 25, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Rawat, M.; la Clair, J.J.; Jothivasan, V.K.; Budiarto, T.; Hamilton, C.J.; Claiborne, A.; Helmann, J.D.; Fahey, R.C. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009, 5, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.K.; Roberts, A.A.; Huyen, T.T.; Bäsell, K.; Becher, D.; Albrecht, D.; Hamilton, C.J.; Antelmann, H. S-bacillithiolation protects conserved and essential proteins against hypochlorite stress in firmicutes bacteria. Antioxid. Redox Signal. 2013, 18, 1273–1295. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.K.; Gronau, K.; Mäder, U.; Hessling, B.; Becher, D.; Antelmann, H. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell Proteomics 2011, 10, M111.009506. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.K.; Busche, T.; van Laer, K.; Bäsell, K.; Becher, D.; Clermont, L.; Seibold, G.M.; Persicke, M.; Kalinowski, J.; Messens, J.; et al. Protein S-mycothiolation functions as redox-switch and thiol protection mechanism in Corynebacterium glutamicum under hypochlorite stress. Antioxid. Redox Signal. 2014, 20, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Fahey, R.C. Glutathione analogs in prokaryotes. Biochim. Biophys. Acta 2013, 5, 3182–3198. [Google Scholar] [CrossRef]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.A.; Eaton, J.W.; Mahoney, J.R., Jr. Bacterial glutathione: A sacrificial defense against chlorine compounds. J. Bacteriol. 1996, 178, 2131–2135. [Google Scholar] [PubMed]
- Ferguson, G.P.; Booth, I.R. Importance of glutathione for growth and survival of Escherichia coli cells: Detoxification of methylglyoxal and maintenance of intracellular K+. J. Bacteriol. 1998, 180, 4314–4318. [Google Scholar] [PubMed]
- Carmel-Harel, O.; Storz, G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu. Rev. Microbiol. 2000, 54, 439–461. [Google Scholar] [CrossRef] [PubMed]
- Ritz, D.; Beckwith, J. Roles of thiol-redox pathways in bacteria. Annu. Rev. Microbiol. 2001, 55, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Tetaud, E.; Manai, F.; Barrett, M.P.; Nadeau, K.; Walsh, C.T.; Fairlamb, A.H. Cloning and characterization of the two enzymes responsible for trypanothione biosynthesis in crithidia fasciculata. J. Biol. Chem. 1998, 273, 19383–19390. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.M., Jr.; Kwon, D.S.; Huisman, G.W.; Kolter, R.; Walsh, C.T. Glutathionylspermidine metabolism in Escherichia coli. Purification, cloning, overproduction, and characterization of a bifunctional glutathionylspermidine synthetase/amidase. J. Biol. Chem. 1995, 270, 14031–14041. [Google Scholar] [CrossRef] [PubMed]
- Tabor, H.; Tabor, C.W. Isolation, characterization, and turnover of glutathionylspermidine from Escherichia coli. J. Biol. Chem. 1975, 250, 2648–2654. [Google Scholar] [PubMed]
- Krauth-Siegel, R.L.; Ludemann, H. Reduction of dehydroascorbate by trypanothione. Mol. Biochem. Parasitol. 1996, 80, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Ariyanayagam, M.R.; Fairlamb, A.H. Ovothiol and trypanothione as antioxidants in trypanosomatids. Mol. Biochem. Parasitol. 2001, 115, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.; Henderson, G.B.; Cerami, A.; Held, K.D. Effects of trypanothione on the biological activity of irradiated transforming DNA. Int. J. Radiat. Biol. 1992, 62, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Chen, W.; Tabor, H. Escherichia coli glutathionylspermidine synthetase/amidase: Phylogeny and effect on regulation of gene expression. FEMS Microbiol. Lett. 2013, 338, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Ansong, C.; Wu, S.; Meng, D.; Liu, X.; Brewer, H.M.; Kaiser, B.L.D.; Nakayasu, E.S.; Cort, J.R.; Pevzner, P.; Smith, R.D.; et al. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 10153–10158. [Google Scholar] [CrossRef] [PubMed]
- Chiang, B.Y.; Chen, T.C.; Pai, C.H.; Chou, C.C.; Chen, H.H.; Ko, T.P.; Hsu, W.H.; Chang, C.Y.; Wu, W.F.; Wang, A.H.; et al. Protein S-thiolation by glutathionylspermidine (Gsp): The role of Escherichia coli Gsp synthetase/amidase in redox regulation. J. Biol. Chem. 2010, 285, 25345–25353. [Google Scholar] [CrossRef] [PubMed]
- Nusuetrong, P.; Suwannasual, U.; Merksuriyen, D. Spermidine-induced apoptosis via reactive oxygen species generation and Caspase 3 activation in mouse P19 embryonal carcinoma cells. Int. J. Pharmacol. 2010, 6, 903–909. [Google Scholar] [CrossRef]
- Massey, V.; Williams, C.H.; Graham, P. The presence of S0-containing impurities in commercial samples of oxidized glutathione and their catalytic effect on the reduction of cytochrome C. Biochem. Biophys. Res. Commun. 1971, 42, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.H.; Chiang, B.Y.; Ko, T.P.; Chou, C.C.; Chong, C.M.; Yen, F.J.; Chen, S.; Coward, J.K.; Wang, A.H.; Lin, C.H. Dual binding sites for translocation catalysis by Escherichia coli glutathionylspermidine synthetase. EMBO J. 2006, 25, 5970–5982. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Kwon, D.S.; Bollinger, J.M., Jr.; Walsh, C.T. Evidence for a glutathionyl-enzyme intermediate in the amidase activity of the bifunctional glutathionylspermidine synthetase/amidase from Escherichia coli. Biochemistry 1997, 36, 14930–14938. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Nelson, K.J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef]
- Woo, H.A.; Jeong, W.; Chang, T.S.; Park, K.J.; Park, S.J.; Yang, J.S.; Rhee, S.G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005, 280, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Redox regulation of protein tyrosine phosphatases by hydrogen peroxide: Detecting sulfenic acid intermediates and examining reversible inactivation. Methods Enzymol. 2002, 348, 297–305. [Google Scholar] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Villa, P.; Eberini, I.; Vandekerckhove, J.; Gianazza, E.; Ghezzi, P. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics 2003, 3, 1154–1161. [Google Scholar] [CrossRef]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Rinna, A.; Torres, M.; Forman, H.J. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1b. Free Radic. Biol. Med. 2006, 41, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. Actin S-glutathionylation: Evidence against a thiol-disulphide exchange mechanism. Free Radic. Biol. Med. 2003, 35, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Huang, Y.T.; Hsieh, C.W.; Yang, P.M.; Wung, B.S. Carbon monoxide induces heme oxygenase-1 to modulate STAT3 activation in endothelial cells via S-glutathionylation. PLoS One 2014, 9, e100677. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galougahi, K.K.; Liu, C.C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J. Am. Heart Assoc. 2014, 3, e000731. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.; Netto, L.E.; Silva, G.M.; Hand, A.; de Oliveira, C.L.; Bicev, R.N.; Gozzo, F.; Barros, M.H.; Leme, J.M.; Ohara, E. Redox regulation of the proteasome via S-glutathionylation. Redox Biol. 2013, 2, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Òlaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasí, F.; Gimeno, A.; Pérez-Quilis, C.; Palacios, O.; et al. Histone H3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Fairlamb, A.H. Levels of polyamines, glutathione and glutathione-spermidine conjugates during growth of the insect trypanosomatid Crithidia fasciculata. J. Gen. Microbiol. 1988, 134, 807–817. [Google Scholar] [PubMed]
- Pai, C.H.; Wu, H.J.; Lin, C.H.; Wang, A.H. Structure and mechanism of Escherichia coli glutathionylspermidine amidase belonging to the family of cysteine, histidine-dependent amidohydrolase/peptidases. Protein Sci. 2011, 20, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Gao, S.; Chiang, B.Y.; Hsu, W.H.; Lin, T.C.; Chen, T.C.; Lin, C.H. An acyloxymethyl ketone-based probe to monitor the activity of glutathionylspermidine amidase in Escherichia coli. ChemBioChem 2011, 12, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Kidd, D.; Liu, Y.; Cravatt, B.F. Profiling serine hydrolase activities in complex proteomes. Biochemistry 2001, 40, 6107–6115. [Google Scholar] [CrossRef] [PubMed]
- Jessani, N.; Liu, Y.; Humphrey, M.; Cravatt, B.F. Enzyme activity profiles of the secreted and membrane proteome that depict cancer invasiveness. Proc. Natl. Acad. Sci. USA 2002, 99, 10335–10340. [Google Scholar] [CrossRef] [PubMed]
- Fratelli, M.; Gianazza, E.; Ghezzi, P. Redox proteomics: Identification and functional role of glutathionylated proteins. Expert Rev. Proteomics 2004, 1, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Lind, C.; Gerdes, R.; Hamnell, Y.; Schuppe-Koistinen, I.; von Lowenhielm, H.B.; Holmgren, A.; Cotgreave, I.A. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch. Biochem. Biophys. 2002, 406, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.P.; Miller, J.I.; Fuller, W.; Wait, R.; Begum, S.; Dunn, M.J.; Eaton, P. The utility of n,n-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell. Proteomics 2006, 5, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473. [Google Scholar] [CrossRef] [PubMed]
- Chiang, B.Y.; Chou, C.C.; Hsieh, F.T.; Gao, S.; Lin, J.C.; Lin, S.H.; Chen, T.C.; Khoo, K.H.; Lin, C.H. In vivo tagging and characterization of S-glutathionylated proteins by a chemoenzymatic method. Angew. Chem. Int. Ed. 2012, 51, 5871–5875. [Google Scholar] [CrossRef]
- Lancel, S.; Zhang, J.; Evangelista, A.; Trucillo, M.P.; Tong, X.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res. 2009, 104, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Hamnell-Pamment, Y.; Lind, C.; Palmberg, C.; Bergman, T.; Cotgreave, I.A. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem. Biophys. Res. Commun. 2005, 332, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, S.; Bonetto, V.; Fratelli, M.; Gianazza, E.; Eberini, I.; Massignan, T.; Salmona, M.; Chang, G.; Holmgren, A.; Ghezzi, P. Glutathionylation of human thioredoxin: A possible crosstalk between the glutathione and thioredoxin systems. Proc. Natl. Acad. Sci. USA 2002, 99, 9745–9749. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Jones, A.D.; Cross, C.E.; Wong, P.S.; van Der Vliet, A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem. J. 2000, 347 Pt 3, 821–827. [Google Scholar] [CrossRef]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Oh, C.J.; Kil, I.S.; Park, J.W. Glutathionylation regulates cytosolic NADP+-dependent isocitrate dehydrogenase activity. Free Radic. Res. 2009, 43, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Mieyal, J.J.; Rhee, S.G.; Chock, P.B. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 2009, 284, 23364–23374. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, K.C.; Zhou, L.; Jordon, J.K.; Huang, Y.; Yu, Y.; Redler, R.L.; Chen, X.; Caplow, M.; Dokholyan, N.V. Modifications of superoxide dismutase (SOD1) in human erythrocytes: A possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 2009, 284, 13940–13947. [Google Scholar] [CrossRef] [PubMed]
- Cotton, N.J.; Stoddard, B.; Parson, W.W. Oxidative inhibition of human soluble catechol-O-methyltransferase. J. Biol. Chem. 2004, 279, 23710–23718. [Google Scholar] [CrossRef] [PubMed]
- Caplan, J.F.; Filipenko, N.R.; Fitzpatrick, S.L.; Waisman, D.M. Regulation of Annexin A2 by reversible glutathionylation. J. Biol. Chem. 2004, 279, 7740–7750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific glcnacylation of human erythrocyte proteins: Potential biomarker(s) for diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Jeong, J.; Na, S.; Lee, H.S.; Kim, H.Y.; Lee, K.J.; Paek, E. New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 2010, 9, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Ward, N.E.; O’Brian, C.A. Potent inactivation of representative members of each pkc isozyme subfamily and pkd via S-thiolation by the tumor-promotion/progression antagonist glutathione but not by its precursor cysteine. Carcinogenesis 2001, 22, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.J.; Peter, J.U.; Winter, S.; Holtje, M.; Paulmann, N.; Grohmann, M.; Vowinckel, J.; Alamo-Bethencourt, V.; Wilhelm, C.S.; Ahnert-Hilger, G.; et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 2003, 115, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Choi, S.S.; Ha, K.S. Transglutaminase 2: A multi-functional protein in multiple subcellular compartments. Amino Acids 2010, 39, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Orru, S.; Caputo, I.; D’Amato, A.; Ruoppolo, M.; Esposito, C. Proteomics identification of acyl-acceptor and acyl-donor substrates for transglutaminase in a human intestinal epithelial cell line. Implications for celiac disease. J. Biol. Chem. 2003, 278, 31766–31773. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, L.; Laskin, D.L.; Fanburg, B.L. Role of protein transamidation in serotonin-induced proliferation and migration of pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Karpuj, M.V.; Garren, H.; Slunt, H.; Price, D.L.; Gusella, J.; Becher, M.W.; Steinman, L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in huntington’s disease brain nuclei. Proc. Natl. Acad. Sci. USA 1999, 96, 7388–7393. [Google Scholar] [CrossRef] [PubMed]
- Hartley, D.M.; Zhao, C.; Speier, A.C.; Woodard, G.A.; Li, S.; Li, Z.; Walz, T. Transglutaminase induces protofibril-like amyloid beta-protein assemblies that are protease-resistant and inhibit long-term potentiation. J. Biol. Chem. 2008, 283, 16790–16800. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gao, H.; Xu, J.; Reuben, J.; Yu, D.; Mehta, K. Evidence that aberrant expression of tissue transglutaminase promotes stem cell characteristics in mammary epithelial cells. PLoS One 2011, 6, e20701. [Google Scholar] [CrossRef] [PubMed]
- Rossin, F.; D’Eletto, M.; Macdonald, D.; Farrace, M.G.; Piacentini, M. TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 2012, 42, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehta, K. Tissue transglutaminase, inflammation, and cancer: How intimate is the relationship? Amino Acids 2013, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Marquet, R.; Colson, P.; Houssier, C. The condensation of chromatin and histone h1-depleted chromatin by spermine. J. Biomol. Struct. Dyn. 1986, 4, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Petrov, A.S.; Vitko, J.R.; Santai, C.T.; Harvey, S.C.; Mukerji, I.; Hud, N.V. Integration host factor (IHF) dictates the structure of polyamine-DNA condensates: Implications for the role of IHF in the compaction of bacterial chromatin. Biochemistry 2009, 48, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A. Oxidative stress: A dead end or a laboratory hypothesis? Biochem. Biophys. Res. Commun. 2007, 362, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Fesus, L.; Szondy, Z. Transglutaminase 2 in the balance of cell death and survival. FEBS Lett. 2005, 579, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; Macgregor, A.M.; et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.S.; Hausladen, A.; Slaughter, T.F.; Eu, J.P.; Stamler, J.S.; Greenberg, C.S. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 2001, 40, 4904–4910. [Google Scholar] [CrossRef] [PubMed]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef] [PubMed]
- Connellan, J.M.; Folk, J.E. Mechanism of the inactivation of guinea pig liver transglutaminase by 5,5'-dithiobis-(2-nitrobenzoic acid). J. Biol. Chem. 1969, 244, 3173–3181. [Google Scholar] [PubMed]
- Liu, S.; Cerione, R.A.; Clardy, J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Helmann, J.D. Bacillithiol, a new player in bacterial redox homeostasis. Antioxid. Redox Signal. 2011, 15, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Antelmann, H.; Hamilton, C.J. Bacterial mechanisms of reversible protein S-thiolation: Structural and mechanistic insights into mycoredoxins. J. Mol. Microbiol. 2012, 86, 759–764. [Google Scholar] [CrossRef]
- Sousa, A.F.; Gomes-Alves, A.G.; Benítez, D.; Comini, M.A.; Flohé, L.; Jaeger, T.; Passos, J.; Stuhlmann, F.; Tomás, A.M.; Castro, H. Genetic and chemical analyses reveal that trypanothione synthetase but not glutathionylspermidine synthetase is essential for Leishmania infantum. Free Radic. Biol. Med. 2014, 73, 229–238. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.C.-Y.; Chiang, B.-Y.; Chou, C.-C.; Chen, T.-C.; Chen, Y.-J.; Chen, Y.-J.; Lin, C.-H. Glutathionylspermidine in the Modification of Protein SH Groups: The Enzymology and Its Application to Study Protein Glutathionylation. Molecules 2015, 20, 1452-1474. https://doi.org/10.3390/molecules20011452
Lin JC-Y, Chiang B-Y, Chou C-C, Chen T-C, Chen Y-J, Chen Y-J, Lin C-H. Glutathionylspermidine in the Modification of Protein SH Groups: The Enzymology and Its Application to Study Protein Glutathionylation. Molecules. 2015; 20(1):1452-1474. https://doi.org/10.3390/molecules20011452
Chicago/Turabian StyleLin, Jason Ching-Yao, Bing-Yu Chiang, Chi-Chi Chou, Tzu-Chieh Chen, Yi-Ju Chen, Yu-Ju Chen, and Chun-Hung Lin. 2015. "Glutathionylspermidine in the Modification of Protein SH Groups: The Enzymology and Its Application to Study Protein Glutathionylation" Molecules 20, no. 1: 1452-1474. https://doi.org/10.3390/molecules20011452