Investigation of the Flexibility of Protein Kinases Implicated in the Pathology of Alzheimer’s Disease

Abstract
:
1. Introduction


2. Results and Discussion
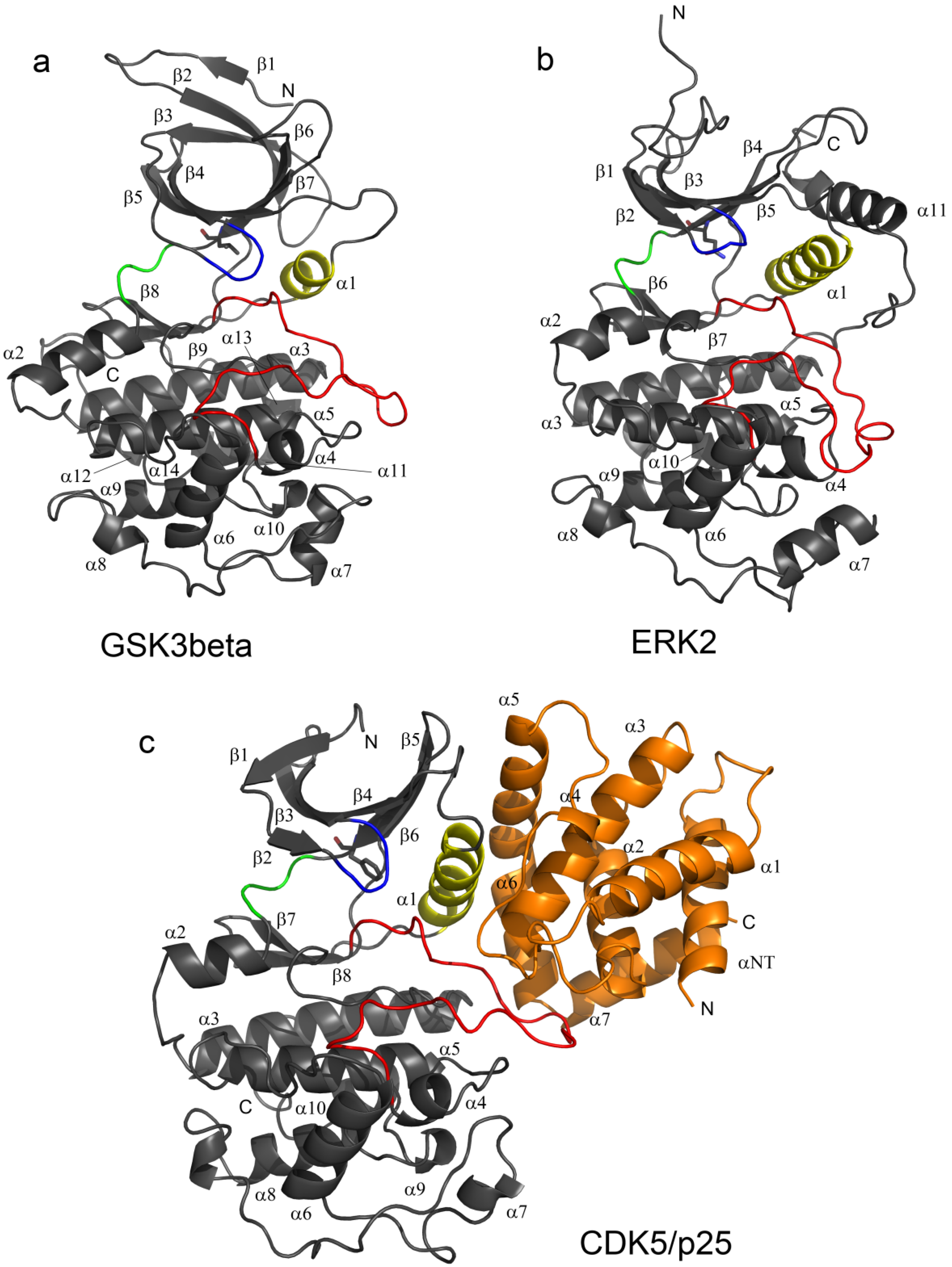
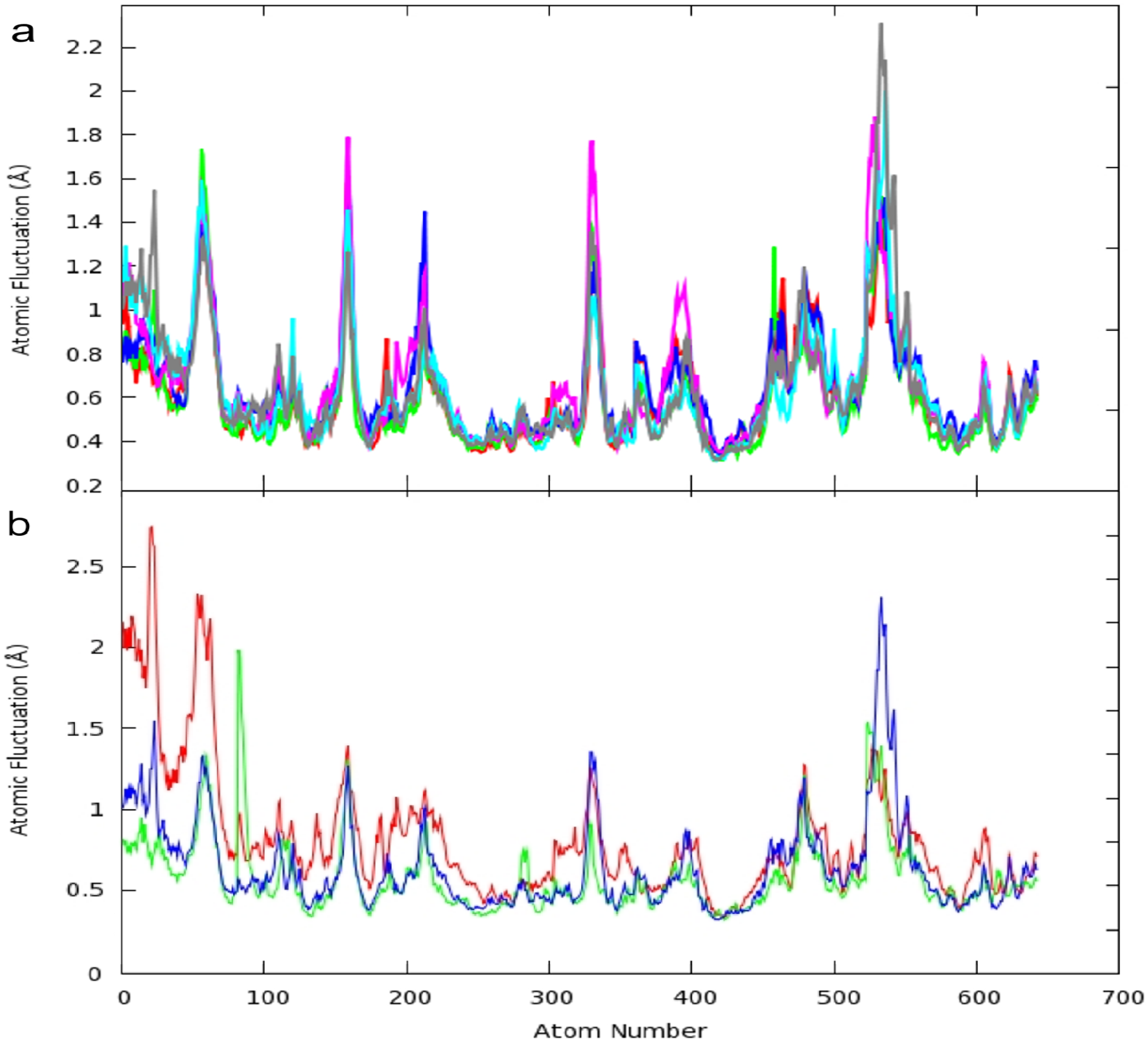
2.1. Structural and Dynamics Analysis


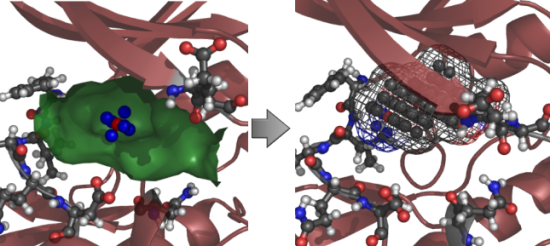
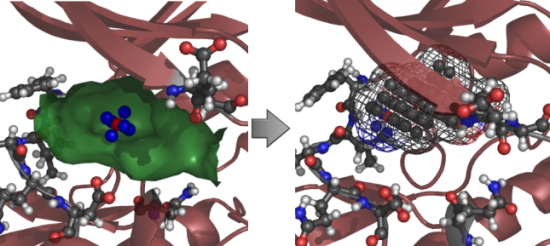{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
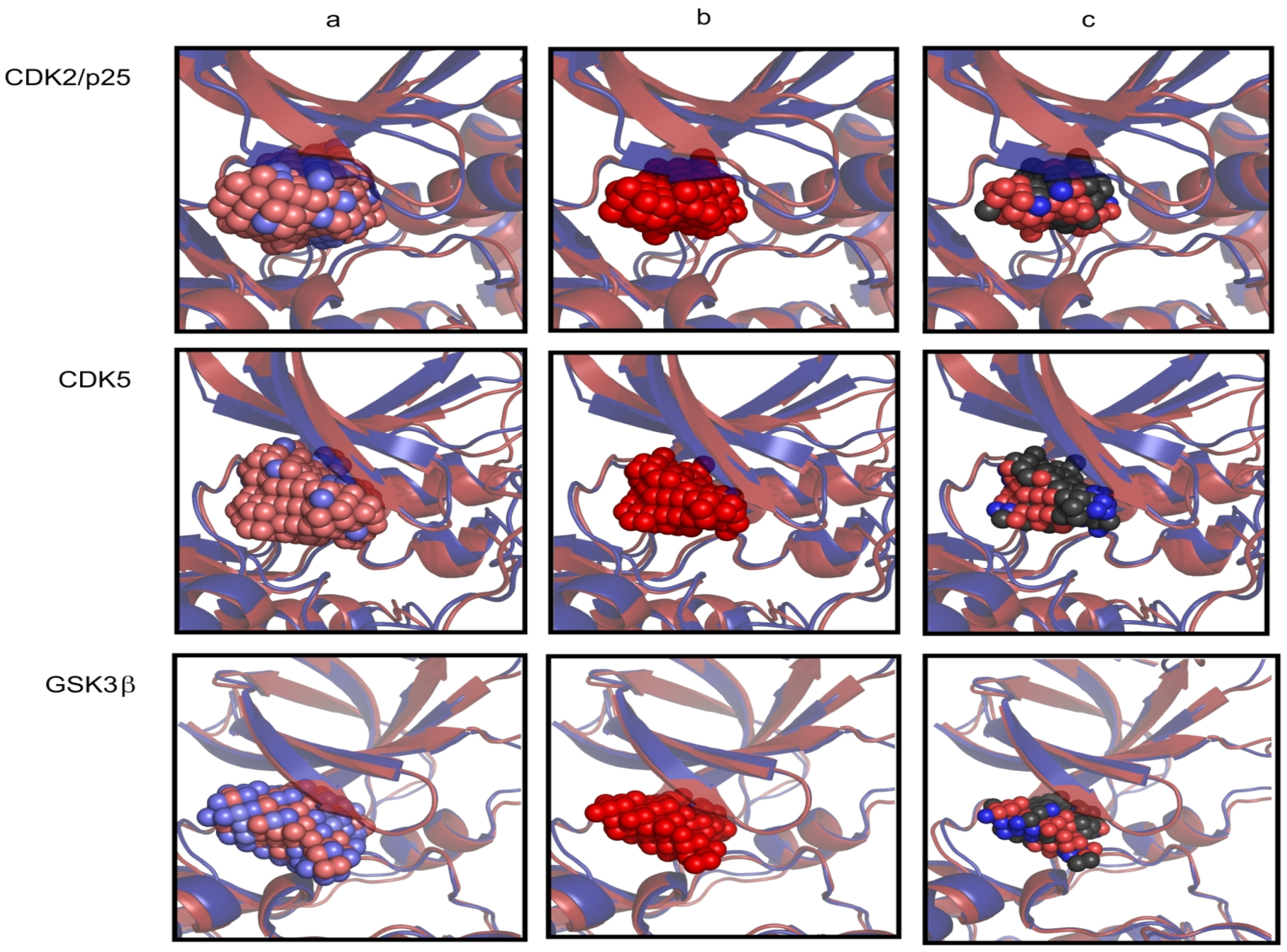{kind=link}
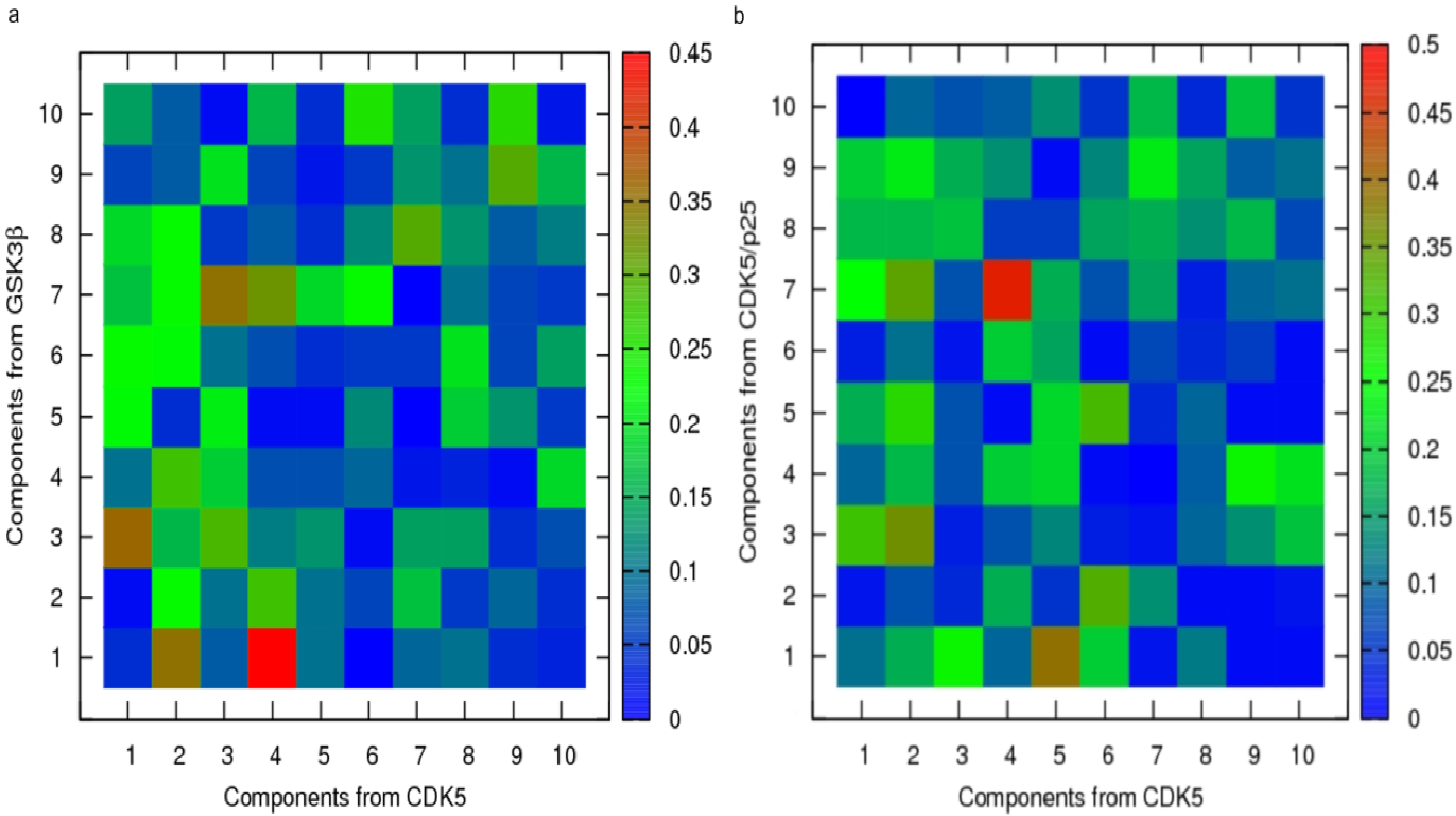{kind=link}
{kind=link}
{kind=link}
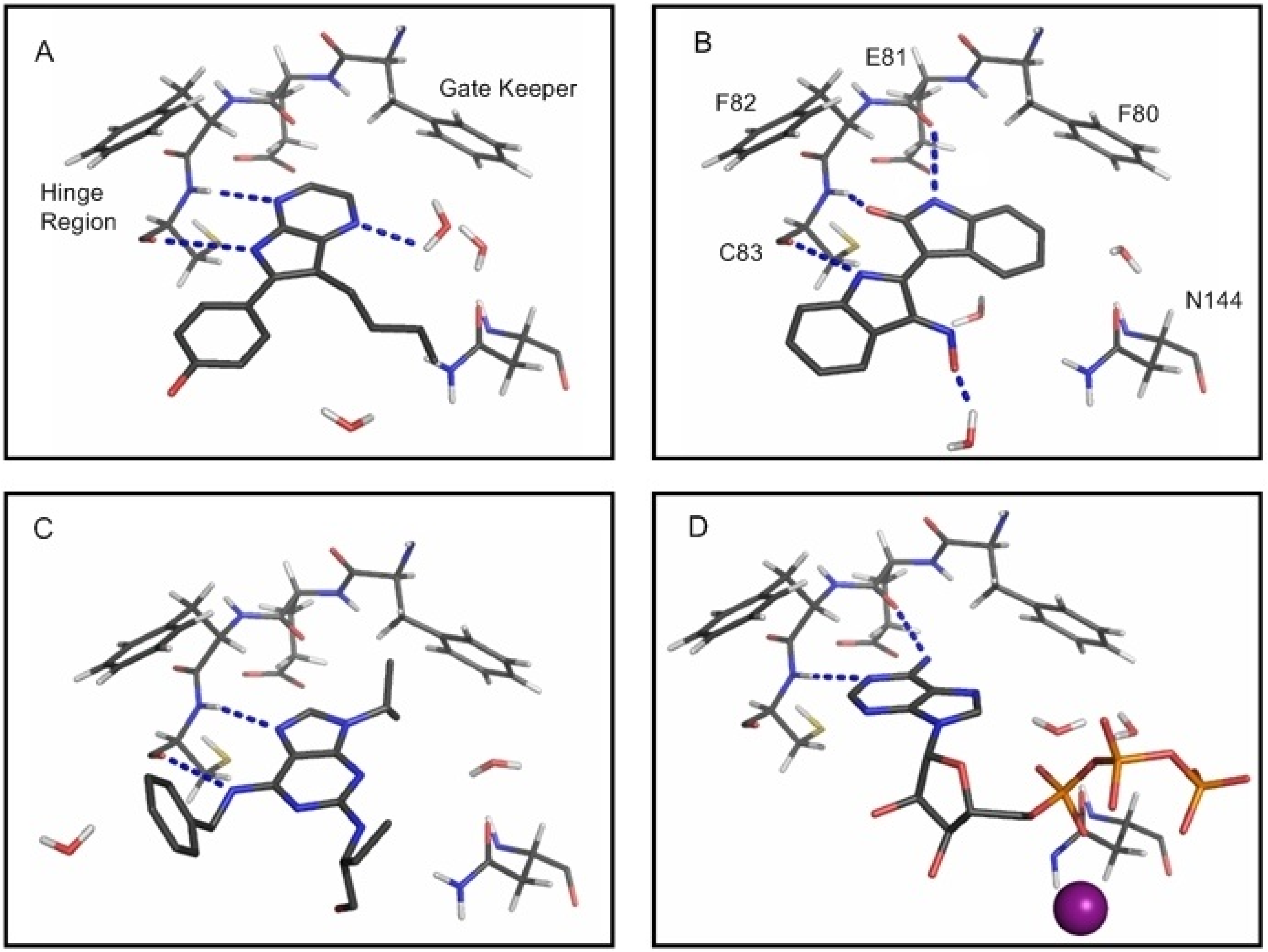{kind=link}
| Residue Numbers | Number of Amino Acids | Core Atom Numbering |
|---|---|---|
| 7L-33K | 27 | 1–81 |
| 47S-72S | 26 | 82–159 |
| 75K-92D | 18 | 160–213 |
| 98L-144D | 47 | 214–354 |
| 147L-148A | 2 | 355–360 |
| 166W-197N | 32 | 361–456 |
| 199G-220G | 22 | 457–522 |
| 241M-250N | 10 | 523–552 |
| 255L-284P | 30 | 553–642 |
| Protein | RMSD (Å) | Residues in core |
|---|---|---|
| CDK5D144N/p25 | 0.000 | 7–33, 47–72, 75–92, 98–144, 147–148, 166–197, 199–220, 241–250, 255–284 |
| GSK3β | 1.164 | 35–61, 69–94, 103–120, 129–167, 169–176, 179–180, 197–250, 270–279, 285–314 |
| ERK2 | 1.184 | 21–47, 59–64, 66–85, 93–110, 114–160, 163–164, 185–238, 260–266, 268–270, 275–304 |
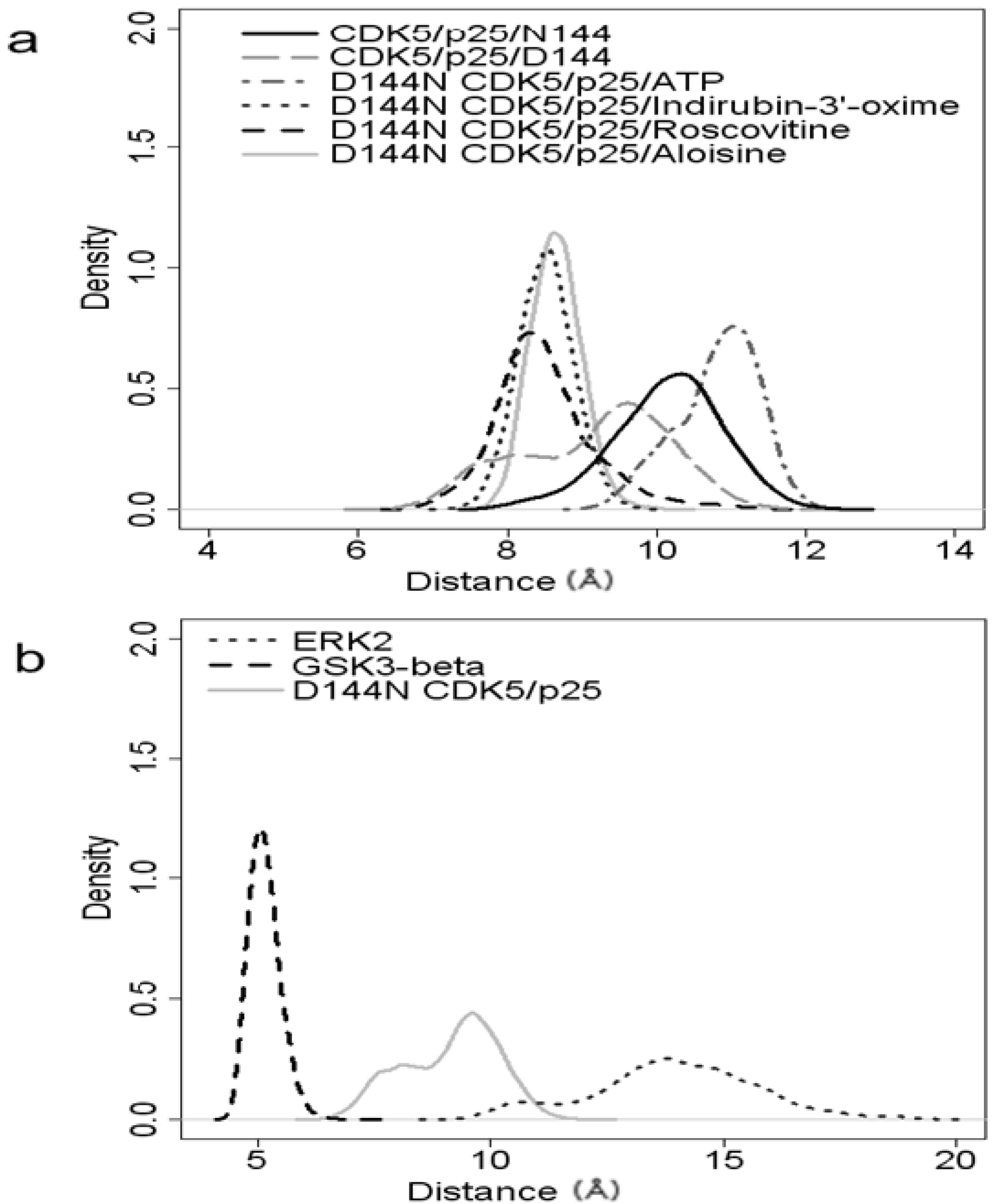
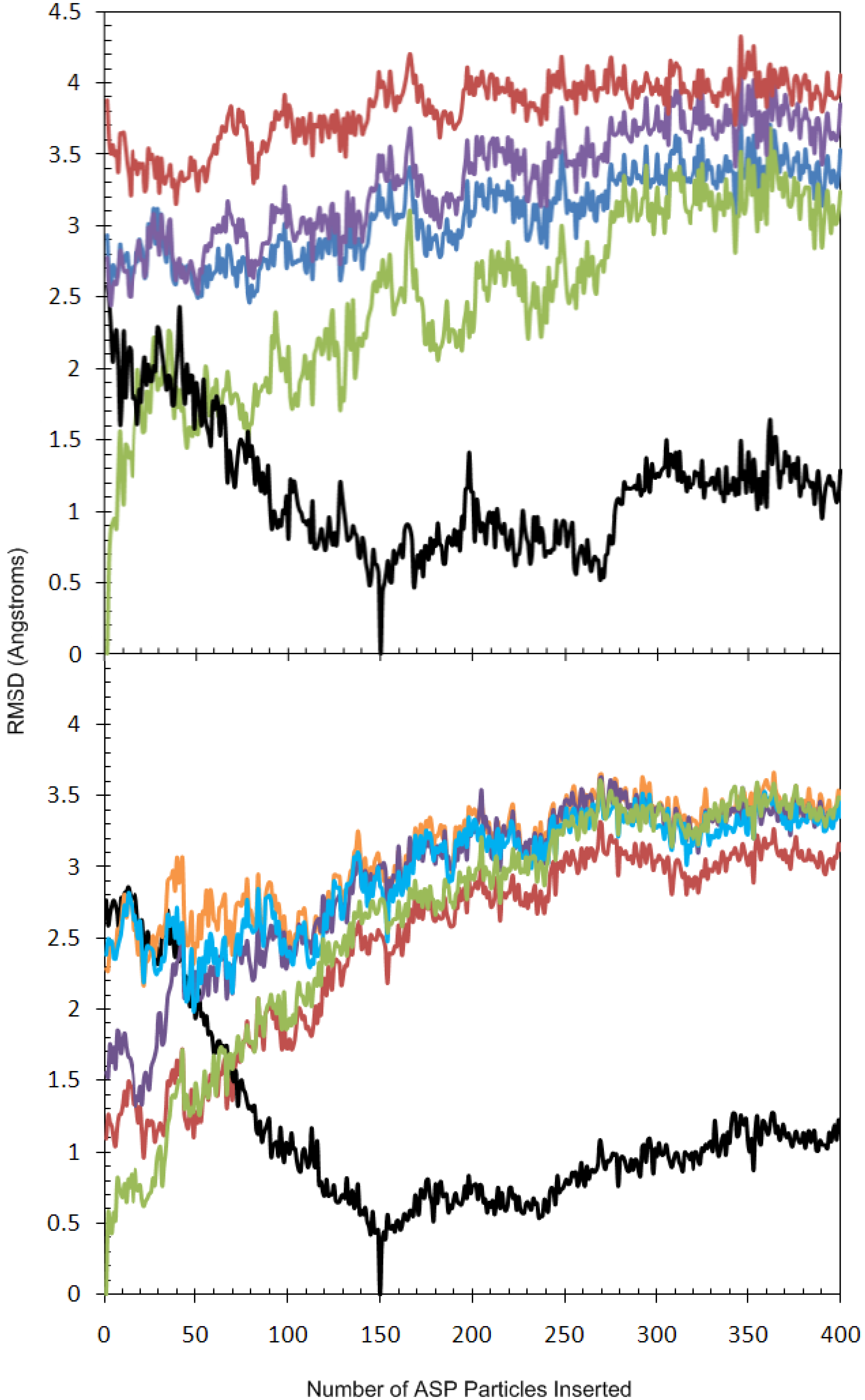
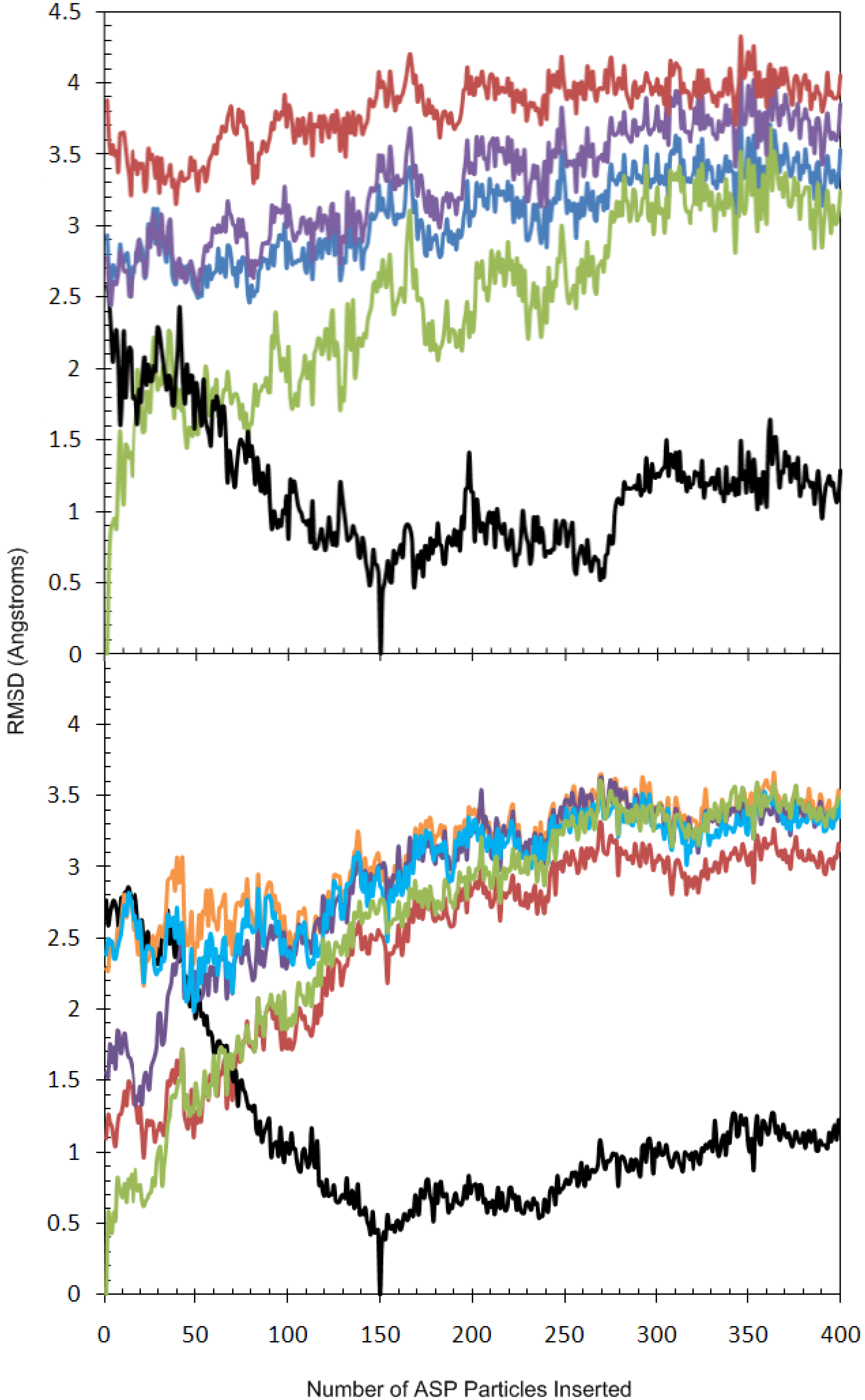
2.2. Analysis of Active Site Pressurisation Dynamics

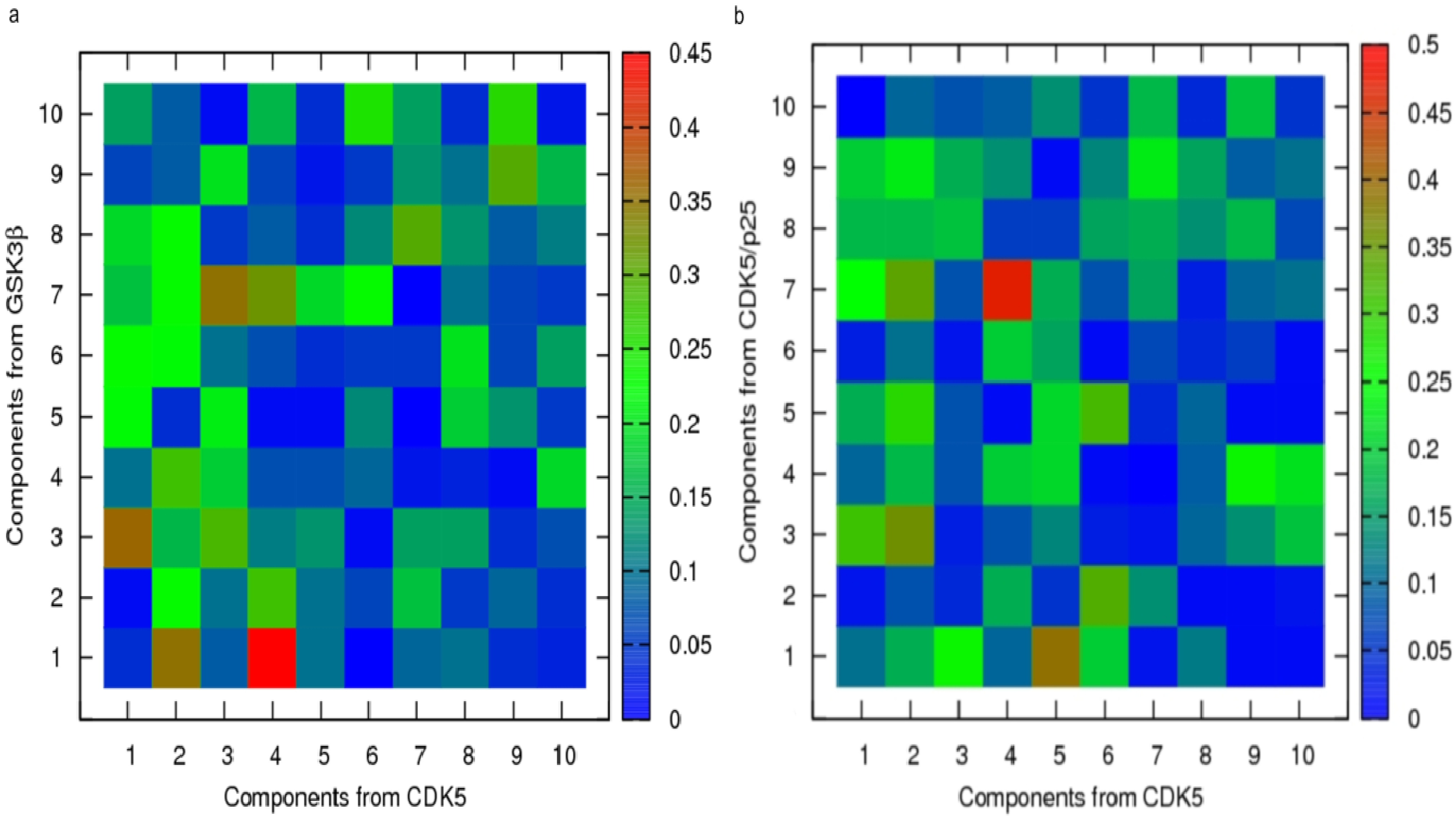
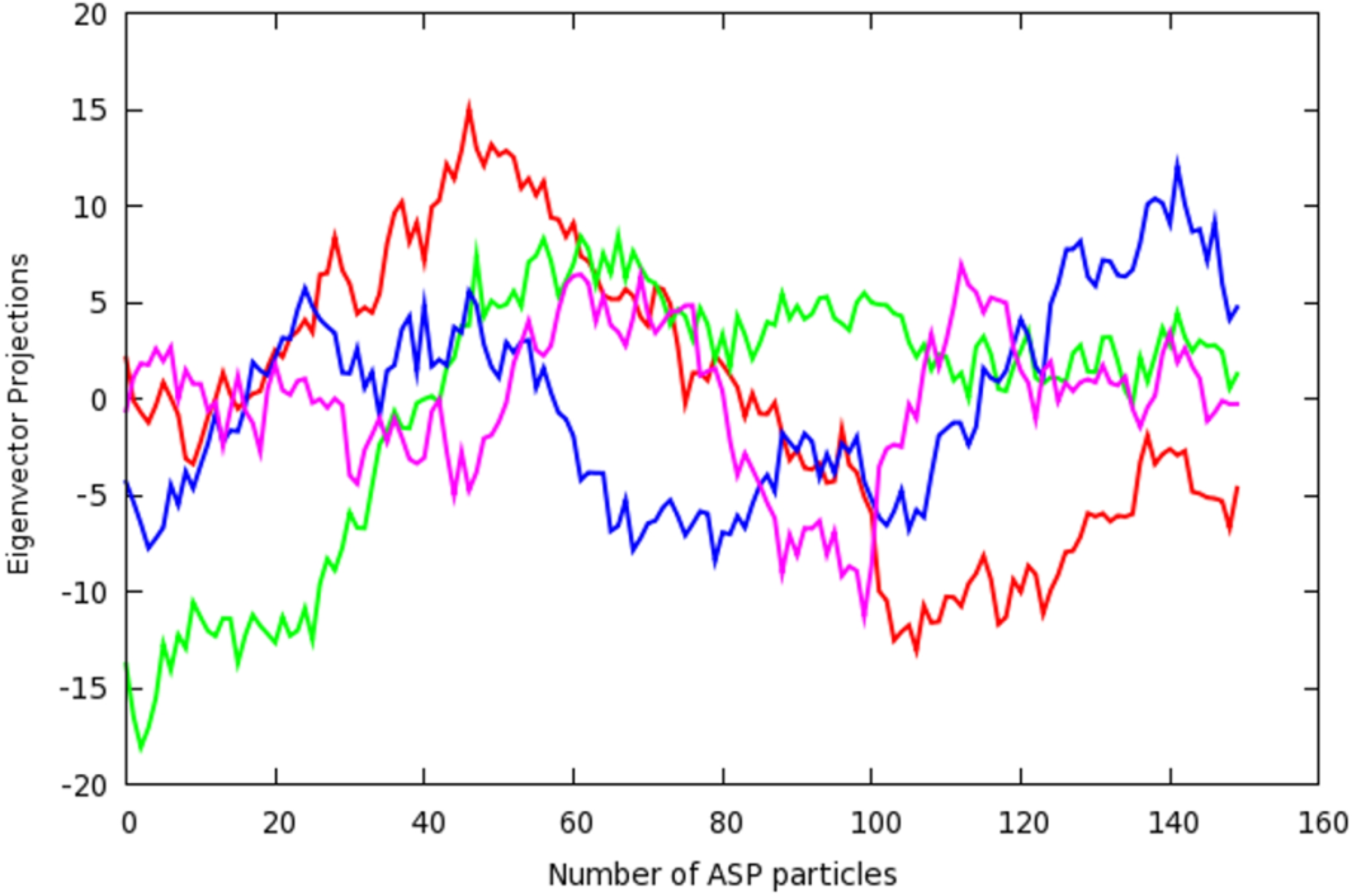
2.3. Principal Component Analysis of Protein Dynamics
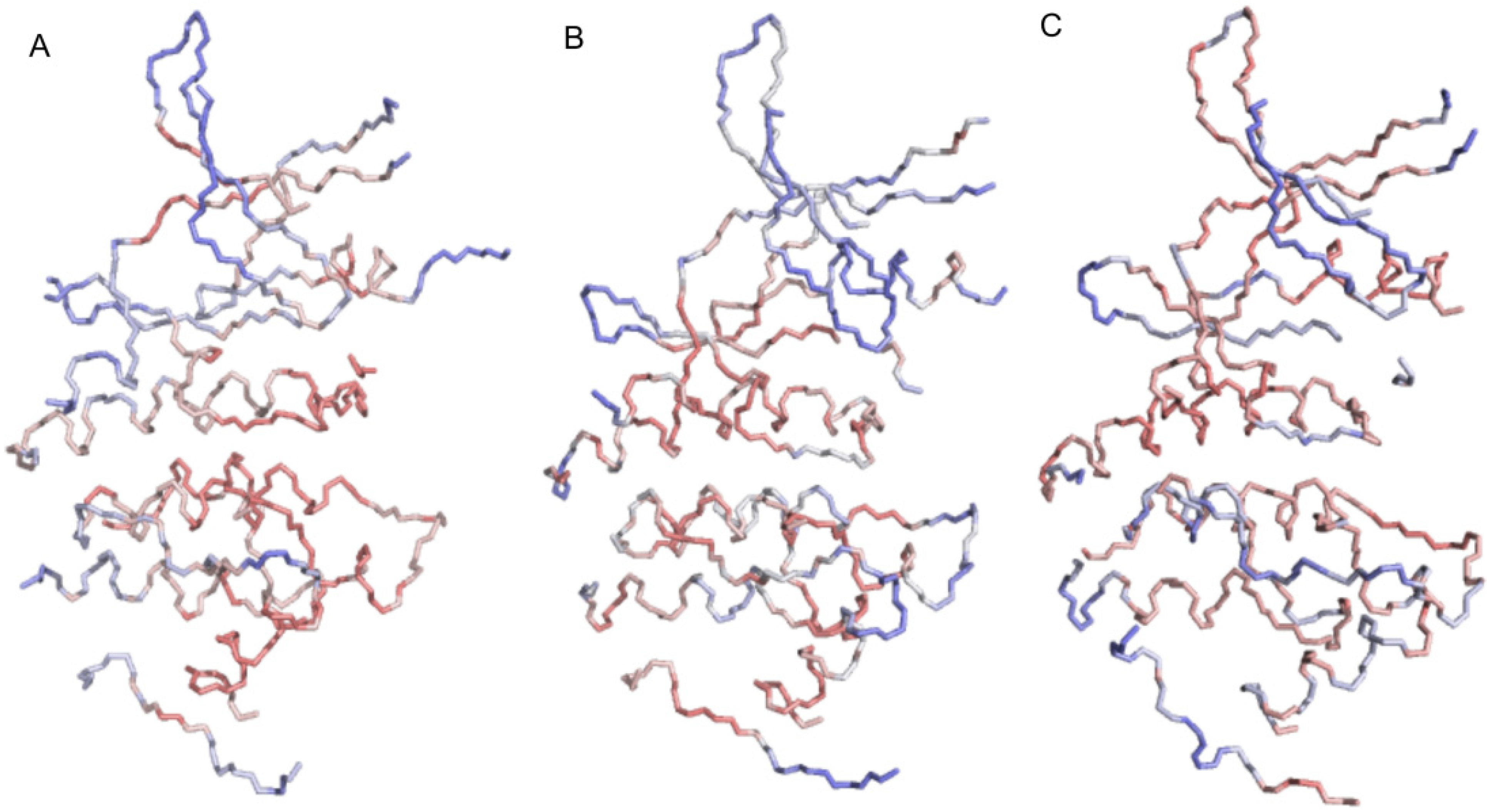
3. Experimental
3.1. Ligand Parameterisation
3.2. Molecular Dynamics Simulations
3.3. Protein Models
| PDB Code | Kinase | R Factor | Rfree Factor a | Resolution (Å) | Ramachandran Analysis | Reference | |
|---|---|---|---|---|---|---|---|
| Favoured Regions (98%) | Allowed Regions (>99.8%) | ||||||
| 1UNH | CDK5 | 0.229 | 0.230 | 2.35 | 93.9 | 98.9 | [70] |
| 1UNL | CDK5 | 0.216 | 0.219 | 2.20 | 95.0 | 98.9 | [70] |
| 1UNG | CDK5 | 0.216 | 0.225 | 2.30 | 91.7 | 98.1 | [70] |
| 1H4L | CDK5 | 0.236 | 0.287 | 2.65 | 89.0 | 97.2 | [71] |
| 1JST | CDK2 | 0.200 | NA | 2.60 | 92.7 | 98.4 | [72] |
| 1PW2 | CDK2 | 0.213 | 0.249 | 1.95 | 97.9 | 99.3 | [73] |
| 1Q4L | GSK3β | 0.212 | 0.251 | 2.77 | 94.3 | 98.2 | [36] |
| 1I09 | GSK3β | 0.242 | 0.274 | 2.70 | 91.9 | 98.8 | [74] |
| 1Q5K | GSK3β | 0.222 | 0.242 | 1.94 | 94.2 | 98.4 | [75] |
| 1TVO | ERK2 | 0.263 | 0.272 | 2.50 | 90.5 | 97.1 | [76] |
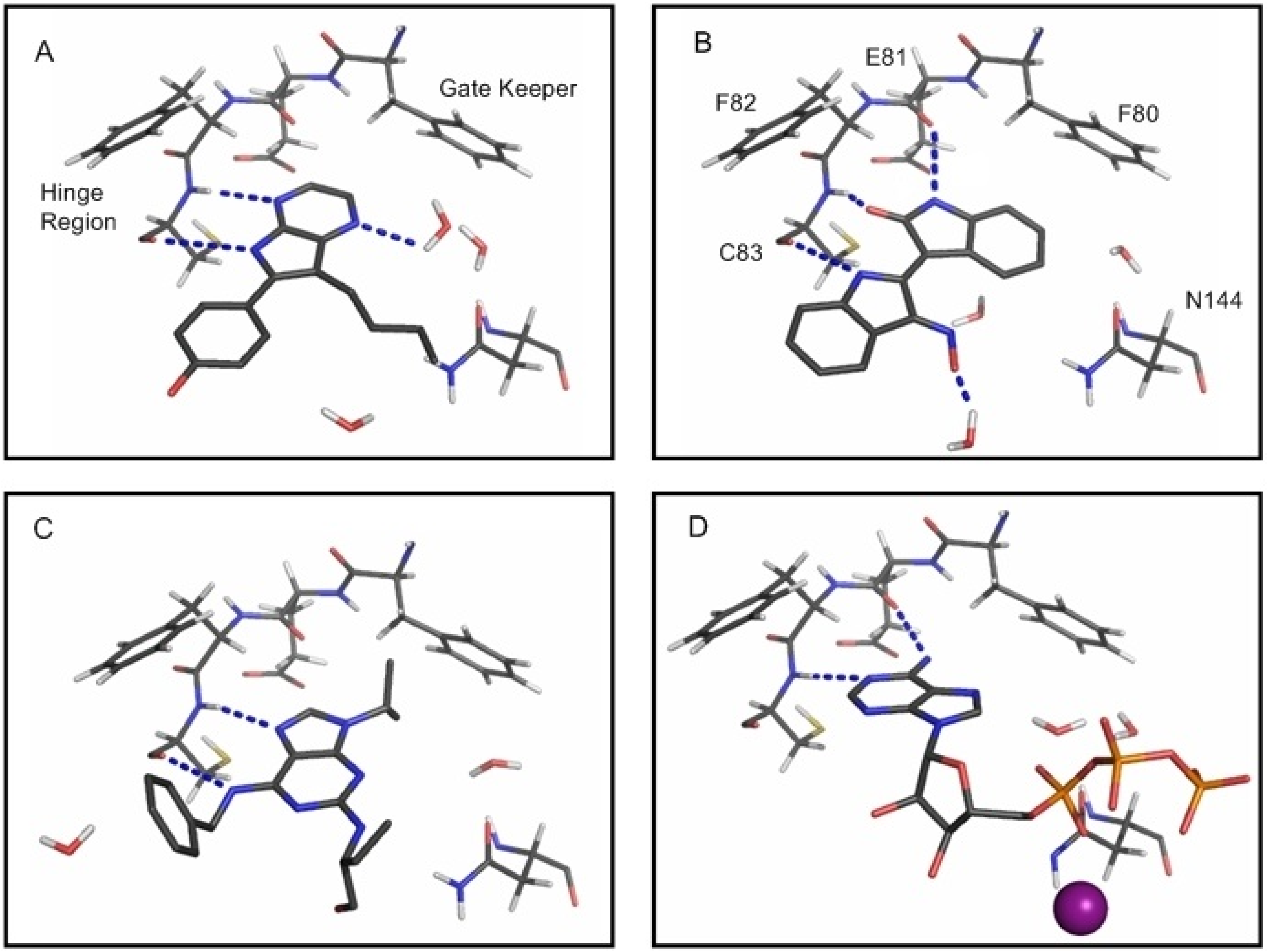
3.4. Active Site Pressurisation
3.5. Grid Orientation and Dimensions
3.6. ASP MD Method
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Semple, D.; Smyth, R.; Burns, J.; Darjee, R.; McIntosh, A. Oxford Handbook of Pyschiatry; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Kuljis, R.O. Alzheimer Disease. Available online: http://www.emedicine.com/neuro/topic13.htm/ (accessed on 1 Jurnary 2013).
- Crowther, R.A.; Olesen, O.F.; Jakes, R.; Goedert, M. The microtubule binding repeats of tau protein assemble into filaments like those found in Alzheimer’s disease. FEBS Lett. 1992, 309, 199–202. [Google Scholar]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 121–131. [Google Scholar] [CrossRef]
- Hardy, J. The relationship between amyloid and tau. J. Mol. Neurosci. 2003, 20, 203–206. [Google Scholar] [CrossRef]
- Mandelkow, E.; Song, Y.H.; Schweers, O.; Marx, A.; Mandelkow, E.M. On the structure of microtubules, tau, and paired helical filaments. Neurobiol. Aging 1995, 16, 347–354. [Google Scholar] [CrossRef]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell. Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef]
- Mazanetz, M.P.; Fischer, P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef]
- Shelton, S.B.; Johnson, G.V. Cyclin-dependent kinase-5 in neurodegeneration. J. Neurochem. 2004, 88, 1313–1326. [Google Scholar] [CrossRef]
- Spittaels, K.; van den Haute, C.; van Dorpe, J.; Geerts, H.; Mercken, M.; Bruynseels, K.; Lasrado, R.; Vandezande, K.; Laenen, I.; Boon, T.; et al. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 2000, 275, 41340–41349. [Google Scholar] [CrossRef]
- Perry, G.; Roder, H.; Nunomura, A.; Takeda, A.; Friedlich, A.L.; Zhu, X.; Raina, A.K.; Holbrook, N.; Siedlak, S.L.; Harris, P.L.; et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10, 2411–2415. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef]
- Otyepka, M.; Bartova, I.; Kriz, Z.; Koca, J. Different Mechanisms of CDK5 and CDK2 Activation as Revealed by CDK5/p25 and CDK2/Cyclin A Dynamics. J. Biol. Chem. 2006, 281, 7271–7281. [Google Scholar] [CrossRef]
- Pande, V.; Ramos, M.J. Structural basis for the GSK-3beta binding affinity and selectivity against CDK-2 of 1-(4-aminofurazan-3yl)-5-dialkylaminomethyl-1H-[1,2,3] triazole-4-carboxylic acid derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 5129–5135. [Google Scholar] [CrossRef]
- Rastelli, G.; Rosenfeld, R.; Reid, R.; Santi, D.V. Molecular modeling and crystal structure of ERK2-hypothemycin complexes. J. Struct. Biol. 2008, 164, 18–23. [Google Scholar] [CrossRef]
- Zhang, B.; Tan, V.B.C.; Lim, K.M.; Tay, T.E. Molecular dynamics simulations on the inhibition of Cyclin-Dependent Kinases 2 and 5 in the presence of activators. J. Comput. Aided Mol. Des. 2006, 20, 395–404. [Google Scholar] [CrossRef]
- Zhang, B.; Tan, V.B.; Lim, K.M.; Tay, T.E.; Zhuang, S. Study of the inhibition of cyclin-dependent kinases with roscovitine and indirubin-3'-oxime from molecular dynamics simulations. J. Mol. Model. 2007, 13, 79–89. [Google Scholar]
- Zhang, B.; Tan, V.B.C.; Lim, K.M.; Tay, T.E. Significance of Water Molecules in the Inhibition of Cylin-Dependent Kinase 2 and 5 Complexes. J. Chem. Inf. Model. 2007, 47, 1877–1885. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Gaspar, R.T.; Canduri, F.; Camera, J.C.; da Silveira, N.J.F. Molecular model of cyclin-dependent kinase 5 complexed with roscovitine. Biochem. Biophys. Res. Commun. 2002, 297, 1154–1158. [Google Scholar] [CrossRef]
- Hilser, V.J.; Garcia-Moreno, E.B.; Oas, T.G.; Kapp, G.; Whitten, S.T. A statistical thermodynamic model of the protein ensemble. Chem. Rev. 2006, 106, 1545–1558. [Google Scholar]
- Jacobs, D.J.; Rader, A.J.; Kuhn, L.A.; Thorpe, M.F. Protein flexibility predictions using graph theory. Proteins 2001, 44, 150–165. [Google Scholar]
- Ming, L.; Maria, I.Z.; Leslie, A.K.; Thorpe, M.F. Sampling protein conformations and pathways. J. Comput. Chem. 2004, 25, 1133–1148. [Google Scholar]
- Seeliger, D.; Haas, J.; de Groot, B.L. Geometry-based sampling of conformational transitions in proteins. Structure 2007, 15, 1482–1492. [Google Scholar] [CrossRef]
- Hayward, S.; de Groot, B.L. Normal modes and essential dynamics. Methods Mol. Biol. 2008, 443, 89–106. [Google Scholar]
- Ahmed, A.; Gohlke, H. Multiscale modeling of macromolecular conformational changes combining concepts from rigidity and elastic network theory. Proteins 2006, 63, 1038–1051. [Google Scholar]
- Hegler, J.A.; Latzer, J.; Shehu, A.; Clementi, C.; Wolynes, P.G. Restriction versus guidance in protein structure prediction. Proc. Natl. Acad. Sci. USA 2009, 106, 15302–15307. [Google Scholar]
- Withers, I.M.; Mazanetz, M.P.; Wang, H.; Fischer, P.M.; Laughton, C.A. Active Site Pressurization: A New Tool for Structure-Guided Drug Design and Other Studies of Protein Flexibility. J. Chem. Inf. Model. 2008, 48, 1448–1454. [Google Scholar]
- Mazanetz, M.P.; Withers, I.M.; Laughton, C.A.; Fischer, P.M. Exploiting glycogen synthase kinase 3β flexibility in molecular recognition. Biochem. Soc. Trans. 2008, 36, 55–58. [Google Scholar]
- Mazanetz, M.P.; Withers, I.M.; Laughton, C.A.; Fischer, P.M. A Study of CDK2 inhibitors using a novel 3D-QSAR method exploiting receptor flexibility. QSAR Comb. Sci. 2009, 28, 878–884. [Google Scholar]
- Case, D.A.; Pearlman, D.A.; Caldwell, J.W.; Cheatham, T.E., III; Wang, J.; Ross, W.S.; Simmerling, C.L.; Darden, T.A.; Merz, K.M.; Stanton, R.V.; et al. AMBER 7; University of California: San Francisco, CA, USA, 2002. [Google Scholar]
- The Molecular Operating Environment (MOE); Version 2009.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2009.
- DeLano, W. The PyMOL Molecular Graphics System; Version v0.99rc6; DeLano: Palo Alto, CA, USA, 2006. [Google Scholar]
- Bertrand, J.A.; Thieffine, S.; Vulpetti, A.; Cristiani, C.; Valsasina, B.; Knapp, S.; Kalisz, H.M.; Flocco, M. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J. Mol. Biol. 2003, 333, 393–407. [Google Scholar] [CrossRef]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Goldsmith, E.J.; Cobb, M.H. Protein kinases. Curr. Opin. Struct. Biol. 1994, 4, 833–840. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar]
- Amadei, A.; Ceruso, M.A.; di Nola, A. On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins’ molecular dynamics simulations. Proteins 1999, 36, 419–424. [Google Scholar] [CrossRef]
- Garcia, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef]
- Aschi, M.; Roccatano, D.; di Nola, A.; Gallina, C.; Gavuzzo, E.; Pochetti, G.; Pieper, M.; Tschesche, H.; Mazza, F. Computational study of the catalytic domain of human neutrophil collagenase. Specific role of the S3 and S'3 subsites in the interaction with a phosphonate inhibitor. J. Comput. Aided Mol. Des. 2002, 16, 213–225. [Google Scholar] [CrossRef]
- Hess, B. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E 2000, 62, 8438. [Google Scholar] [CrossRef]
- Merlino, A.; Vitagliano, L.; Ceruso, M.A.; Mazzarella, L. Subtle functional collective motions in pancreatic-like ribonucleases: From ribonuclease A to angiogenin. Proteins 2003, 53, 101–110. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98; Gaussian, Inc.: Pittsburgh, PA, USA, 2001. [Google Scholar]
- LEaP; University of California: San Francisco, CA, USA, 1995.
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Modell. 2006, 25, 247–260. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef]
- Crowley, M.; Darden, T.; Cheatham, T.; Deerfield, D. Adventures in improving the scaling and accuracy of a parallel molecular dynamics program. J. Supercomput. 1997, 11, 255–278. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. 2004, 25, 1605–1612. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Revised TIPS for simulations of liquid water and aqueous solutions. J. Chem. Phys. 1982, 77, 4156–4163. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald—An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1998, 98, 10089–10092. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Wang, B.; Pearlman, D.A.; et al. AMBER 8.0; University of California: San Francisco, CA, USA, 2004. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynmaics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. NCBI-BLAST. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi/ (accessed on 10 April 2006).
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar]
- Meng, E.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339–348. [Google Scholar]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- SDSC. Biology Workbench. Available online: http://seqtool.sdsc.edu/ (accessed on 10 April 2006).
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Mapelli, M.; Massimiliano, L.; Crovace, C.; Seeliger, M.A.; Tsai, L.H.; Meijer, L.; Musacchio, A. Mechanism of CDK5/p25 binding by CDK inhibitors. J. Med. Chem. 2005, 48, 671–679. [Google Scholar] [CrossRef]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.H.; Musacchio, A. Structure and regulation of the CDK5-p25(NCK5a) complex. Mol. Cell. 2001, 8, 657–669. [Google Scholar] [CrossRef]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef]
- Wu, S.Y.; McNae, I.; Kontopidis, G.; McClue, S.J.; McInnes, C.; Stewart, K.J.; Wang, S.; Zheleva, D.I.; Marriage, H.; Lane, D.P.; et al. Walkinshaw, M.D. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: Structural basis for ligand-induced disordering of the activation loop. Structure 2003, 11, 399–410. [Google Scholar] [CrossRef]
- Ter Haar, E.; Coll, J.T.; Austen, D.A.; Hsiao, H.M.; Swenson, L.; Jain, J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001, 8, 593–596. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormo, M.; Nilsson, Y.; Radesater, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef]
- Ohori, M.; Kinoshita, T.; Okubo, M.; Sato, K.; Yamazaki, A.; Arakawa, H.; Nishimura, S.; Inamura, N.; Nakajima, H.; Neya, M.; et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005, 336, 357–363. [Google Scholar] [CrossRef]
- SYBYL; Tripos: St. Louis, MO, USA, 2006.
- Meyer, T.; Ferrer-Costa, C.; Perez, A.; Rueda, M.; Bidon-Chanal, A.; Luque, F.J.; Laughton, C.A.; Orozco, M. Essential Dynamics: A Tool for Efficient Trajectory Compression and Management. J. Chem. Theory Comput. 2006, 2, 251–258. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotech. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazanetz, M.P.; Laughton, C.A.; Fischer, P.M. Investigation of the Flexibility of Protein Kinases Implicated in the Pathology of Alzheimer’s Disease. Molecules 2014, 19, 9134-9159. https://doi.org/10.3390/molecules19079134
Mazanetz MP, Laughton CA, Fischer PM. Investigation of the Flexibility of Protein Kinases Implicated in the Pathology of Alzheimer’s Disease. Molecules. 2014; 19(7):9134-9159. https://doi.org/10.3390/molecules19079134
Chicago/Turabian StyleMazanetz, Michael P., Charles A. Laughton, and Peter M. Fischer. 2014. "Investigation of the Flexibility of Protein Kinases Implicated in the Pathology of Alzheimer’s Disease" Molecules 19, no. 7: 9134-9159. https://doi.org/10.3390/molecules19079134